

Abstract
The present study uses a novel approach to gene therapy in which plasmid DNA is targeted to the pancreas in vivo using ultrasound targeted microbubble destruction (UTMD) to achieve islet regeneration. Intravenous microbubbles carrying plasmids are destroyed within the pancreatic microcirculation by ultrasound, achieving local gene expression that is further targeted to beta-cells by a modified rat insulin promoter (RIP3.1). A series of genes implicated in endocrine development were delivered to rats 2 days after streptozotocin-induced diabetes. The genes PAX4, Nkx2.2, Nkx6.1, Ngn3, and Mafa produced alpha cell hyperplasia, but no significant improvement in beta cell mass or blood glucose 30 days after UTMD. In contrast, RIP3.1-NeuroD1 promoted islet regeneration from surviving beta-cells, with normalization of glucose, insulin, and C-peptide at 30 days. In a longer-term experiment, 4 of 6 rats had return of diabetes at 90 days, accompanied by beta cell apoptosis on Tunel staining. Pre-treatment with the JNK inhibitor SP600125 successfully blocked beta-cell apoptosis and resulted in restoration of beta cell mass and normalization of blood glucose for up to 90 days. This technique allows in vivo islet regeneration, restoration of beta cell mass, and normalization of blood sugar, insulin, and C-peptide in rats without viruses.
Keywords: islets transcriptional factors, diabetes, pancreatic islets, regeneration, gene therapy, microbubble, ultrasound
Introduction
Blood glucose control is often inadequate in diabetes because drug therapy, including insulin replacement, is not able to replicate the glucose regulatory function of normal islets. Accordingly, new treatment strategies have focused on replenishing the deficiency of beta cell mass common to both major forms of diabetes by islet transplantation or beta-cell regeneration. (1,2). Islet transplantation has been limited by the supply of donor islets and need for immunosuppressive therapy (3). Islet regeneration has been successful in animal models, primarily targeting liver tissue using viral vectors (4-7), which are not likely to be used in humans for safety reasons. A novel approach to gene therapy is to target delivery of DNA to pancreatic islets using ultrasound targeted microbubble destruction (UTMD)(8,9). Intravenous microbubbles carrying plasmid DNA are destroyed within the pancreatic microcirculation by ultrasound, achieving local gene expression that can be further targeted to beta-cells by using a modified rat insulin-I promoter. UTMD has been used to deliver betacellulin and PDX1 in streptozotocin (STZ)-induced diabetes in rats with reversal of diabetes for up to 15 days by reprogramming pancreatic acinar cells into insulin-producing cells (10). Similarly, direct injection of adenoviri encoding a combination of genes PDX1, neurogenin3, and Mafa into mouse pancreata has been shown to reprogram exocrine cells into small insulin-producing cells without formation of islets (11). However, regeneration of islets within the pancreas has remained an elusive goal of gene therapy. This study sought to evaluate whether islet regeneration could be achieved within the adult rat pancreas by gene therapy using UTMD.
The embryological development of the endocrine pancreas is associated with activation of a number of genes, encoding various transcription factors and other proteins (12,13). Recognizing that there may be important differences between neonatal endocrine development and islet regeneration in an adult animal with diabetes, we sought to evaluate whether the latter could be accomplished by gene therapy targeted to the pancreas by UTMD. We specifically constructed plasmids encoding cDNA for Ngn3, Pax4, Nkx2.2, Nkx6.1, MafA, and NeuroD1, all under control of the modified rat insulin promoter (RIP3.1). Microbubbles containing these genes were infused intravenously over a 10-minute period while ultrasound was used to destroy the microbubbles within the pancreatic microcirculation. UTMD was performed 48 hours after induction of diabetes by intraperitoneal STZ (60mg/kg). Controls included UTMD with a marker gene (RIP3.1-DsRed), STZ only without gene therapy, and normal controls that did not receive STZ. Three different repetitions of the experiments were carried out, with sacrifice at 10 days, 20 days, and 30 days to evaluate islet morphology and gene expression. Results of the 30-day experiment are shown in the figures. A long-term experiment was also performed using RIP3.1-NeuroD1 after this gene was shown to be optimal for islet regeneration in this model.
Results
Effects of endocrine proliferative genes on islet structure
Figure 1 shows representative histological samples stained with FITC-labeled anti-insulin antibodies (green) and CY5-labeled anti-glucagon antibodies (red). These sections were obtained from a group of rats sacrificed 30 days after UTMD. Normal islets contain a central core of beta-cells (green) surrounded by a smaller number of alpha-cells (red) at the periphery of the islet. After STZ, the normal islet architecture is virtually abolished with occasional isolated beta-cells or small clusters of beta-cells remaining. Gene therapy with Ngn3, Pax4, Nkx2.2, Nkx6.1, and MafA resulted regeneration of islets, but with abnormal islet architecture, in which alpha cells were predominant. In contrast, gene therapy with NeuroD1 resulted in regeneration of islets with relatively normal morphology. Interestingly, there were a few cells that co-stained with anti-insulin and anti-glucagon in the NeuroD1 group (Figure 2), but not in normal islets. This finding has been previously reported to be a marker for endocrine proliferation (13). Confocal microscopy also demonstrated colocalization of anti-insulin and anti-Ki67, indicating beta cell cell proliferation (data not shown). Compared to normal, and STZ-treated control groups, Ki67-positive cells were statistically significantly more numerous (10 ± 2% of insulin positive cells, p<0.0001) in the NeuroD1 group than in controls (only rare Ki67 positive cells). Because NeuroD1 was under the control of the RIP3.1 promoter, we hypothesized that the observed islet regeneration was due to replication of beta-cells that survived STZ. This was supported by three observations. First, there was no evidence of islet regeneration when RIP3.1-NeuroD1 was delivered by UTMD 7 days after STZ, a timepoint in which no residual beta cells were identified in the STZ controls. Second, none of the islets induced by RIP3.1-NeuroD1 expressed CK19, indicating that the new islets were not derived from ductal cells. Third, the colocalization of Ki67 and insulin suggested beta-cell proliferation.
Fig. 1.
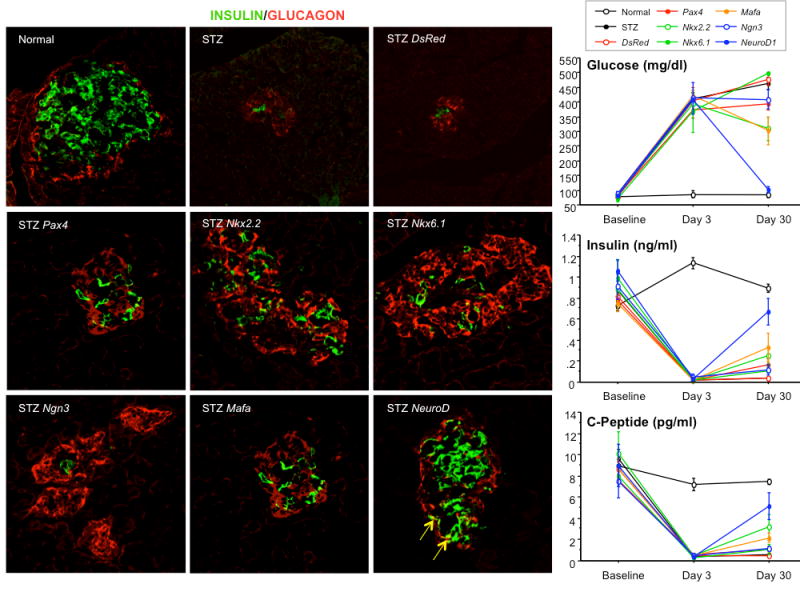
Results of immunofluorescent microscopy. The group of imaging panels at left show representative examples of islets from the 30-day experiments. Beta cells are stained with anti-insulin (green), whereas alpha cells are stained with anti-glucagon (red). The top left panel shows a normal islet with a central core of beta cells surrounded by alpha cells at the islet periphery. Controls treated with STZ, but without active gene therapy, show loss of islet integrity with few very beta cells surrounded by alpha cells (top middle and right). Ultrasound targeted gene therapy with Nkx2.2, Nkx6.1, Pax4, Ngn3, and MafA resulted in formation of alpha-cell dominant islets. In contrast, NeuroD1-treated rats had nearly normal islet architecture with central beta-cells surrounded by peripheral alpha cells (bottom right image). The graphs depict blood glucose (right upper panel), blood insulin (right middle panel), C-peptide (right lower panel) in the 30-day experiment. At day 3, all STZ-treated rats had markedly elevated blood glucose and decreased insulin and C-peptide relative to normal controls (p<0.0001). However, by day 30, only the NeuroD1-treated rats had restoration of blood glucose, insulin, and C-peptide to normal or nearly normal levels.
Fig. 2.
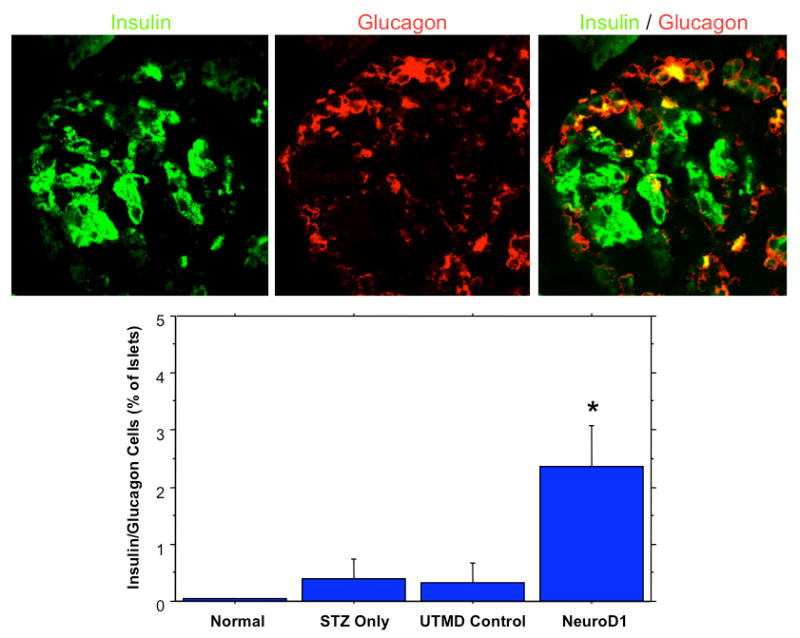
The top panels show a representative islet from a NeuroD1-treated rat. Anti-insulin (green) is shown in the left panel, along with anti-glucagon (middle) and confocal (right) imaging. The confocal image shows colocalization of insulin and glucagon is some cells (yellow), indicating endocrine proliferation. The graph shows that colocalization of insulin and glucagon is far more common in the NeuroD1-treated rats than in controls (p<0.0001).
Effects of endocrine proliferative genes on blood glucose, insulin, and C-peptide
Figure 1 also shows plots of blood glucose (top right), insulin (middle right), and C-peptide (bottom right) at baseline, 3 days after STZ, and 30 days after STZ. Blood glucose increased dramatically by day 3 in all rats treated with STZ (approximately 400 mg/dl), and remained elevated at day 30 except in the NeuroD1-treated rats. At day 30, blood glucose was 101 ± 11 mg/dl in the NeuroD1-treated rats, which was statistically significantly lower than all other STZ-treated groups (p<0.0001), but not from the normal controls. In the shorter-term experiments, blood glucose was also normal at days 10 and 20 in the NeuroD1-treated rats (data not shown). Insulin and C-peptide levels were markedly depressed at day 3 in all STZ-treated rats. By day 30, insulin and C-peptide levels were nearly normal in the NeuroD1-treated rats, being statistically significantly higher than at day 3 or than all other STZ-treated groups (p<0.0001).
Effects of endocrine proliferative genes on islet count and beta cell mass
Islet morphometry 30 days after UTMD is shown in Figure 3. The top panels are representative low power sections stained with hematoxylin (blue) and anti-insulin (brown) from normal controls (left), Ngn3–treated rats (middle), and NeuroD1–treated rats (right). The number of islets in the various treatment groups is shown in the bottom left graph of Figure 3. In normal controls, there were 61 ± 6 islets per slide, compared to only 3 ± 2 after STZ treatment alone. NeuroD1 resulted in 37 ± 4 islets per slide, a number that was statistically significantly higher than all other groups except normal controls (p<0.0001). Beta-cell mass is shown in the bottom right graph of Figure 3. Beta cell mass was 1.49 ± 0.17% in normal controls, and was dramatically reduced in the STZ-treated controls (0.04 ± 0.01%) and DsRed controls (0.05 ± 0.01%). In the groups treated with PAX4, Nkx2.2, Nkx6.1, Ngn3, and Mafa, beta-cell mass was similar (0.20 ± 0.04%). However, in the rats treated with NeuroD1 gene therapy, beta-cell mass was restored to roughly half that of normal controls (0.76 ± 0.07%). Beta-cell mass was statistically significantly higher for NeuroD1-treated rats than for all other genes and controls (p<0.0001), but less than that of normal controls (p=0.0006).
Fig. 3.
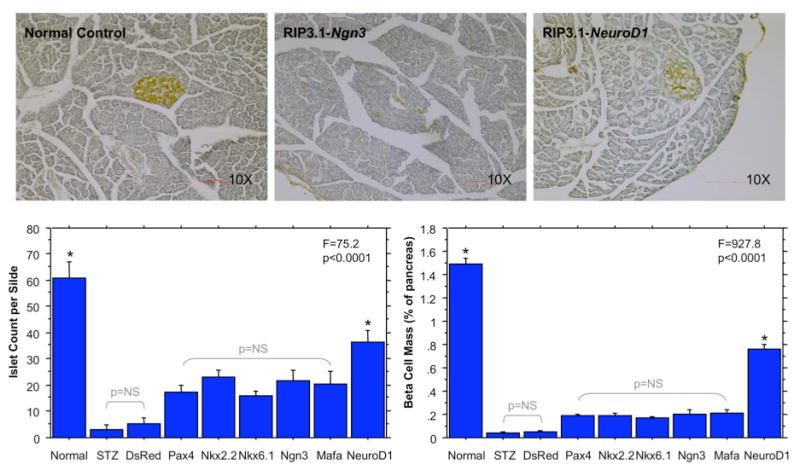
Results of Islet Morphometry. The top panel show representative islets at low power after staining with hematoxylin and anti-insulin (brown color). Normal control islets (left) show dense well-formed beta cell cores stained with anti-insulin (brown). After Ngn3 gene therapy (middle), scattered beta cells are present but without well-formed islet cores. After NeuroD1 gene therapy, islet cores are nearly normal in appearance. The bottom left panel shows the islet count per slide for the different groups. Both normal controls and NeuroD1-treated rats had significantly more islets per slide than all other groups (*p<0.0001 by ANOVA). The bottom right panel shows beta cell mass. NeuroD1-treated rats had a beta cell mass that was half that of normal controls (p<0.0001). Both normal controls and NeuroD1-treated rats had significantly higher beta cell mass than all other groups (*p<0.0001 by ANOVA).
Duration of islet regeneration after RIP3.1-NeuroD1 gene therapy
Figure 4 shows islets stained with anti-insulin (green), anti-NeuroD1 (red) and confocal images in islets from normal controls, STZ-treated controls, and the NeuroD1 group at two timepoints, 2 and 4 weeks after UTMD. No NeuroD1 signal is present in the islets of normal or STZ-treated rats. In rats treated with NeuroD1 gene therapy, NeuroD1 is present at 2 weeks, but not 4 weeks after UTMD. This is consistent with our previous report that plasmid DNA expression by UTMD peaks 4-7 days after treatment, and is virtually gone by 4 weeks. Thus, transient gene expression by UTMD triggers islet regeneration that persists after clearance of the delivered gene and the expressed protein. To determine the duration of islet regeneration and blood glucose control after UTMD, a group of 6 different rats were treated with NeuroD1 gene therapy 2 days after STZ and euthanized 90 days later. The results of this experiment are shown in Figure 5. All 6 rats were profoundly hyperglycemic at baseline (2 days after STZ). At days 20 and 30, all 6 rats had normal blood glucose levels (graph, left panel). However, by day 90, 4 of the 6 rats had return of severe hyperglycemia, whereas 2 rats remained normoglycemic. Confocal microscopy demonstrated substantial loss of beta cells in the islet center of the hyperglycemia rats (top right), compared to the normoglycemic rats (bottom right), which had preserved islet architecture.
Fig. 4.
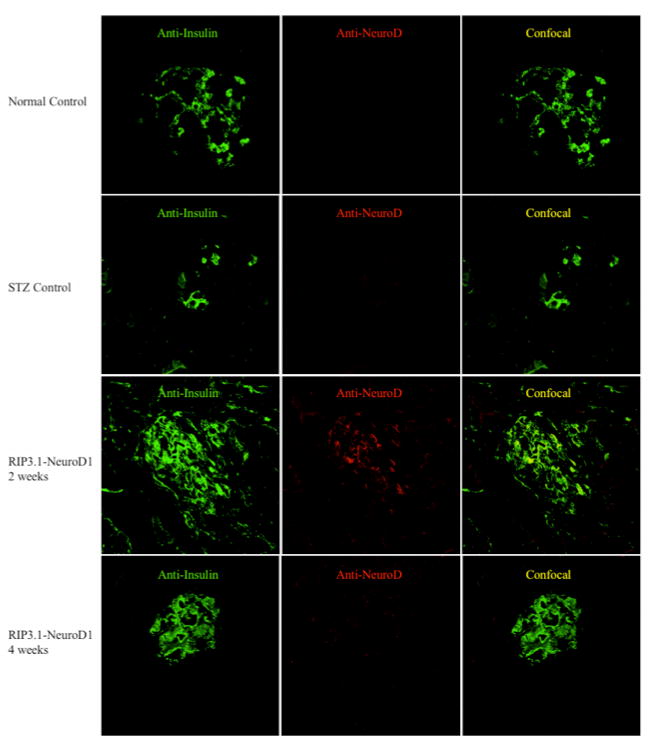
Immunostaining for the presence of NeuroD1 protein in normal controls (top panels), STZ-treated rats (second panels), two weeks after NeuroD1 gene therapy (third panels), and 4 weeks after NeuroD1 gene therapy (bottom panels). Left panels are stained with anti-insulin FITC (green), center panels with anti-NeuroD1 CY5 (red), and right panels are confocal. Detectable levels of NeuroD1 are only seen in beta cells 2 weeks, but not 4 weeks after UTMD.
Fig. 5.
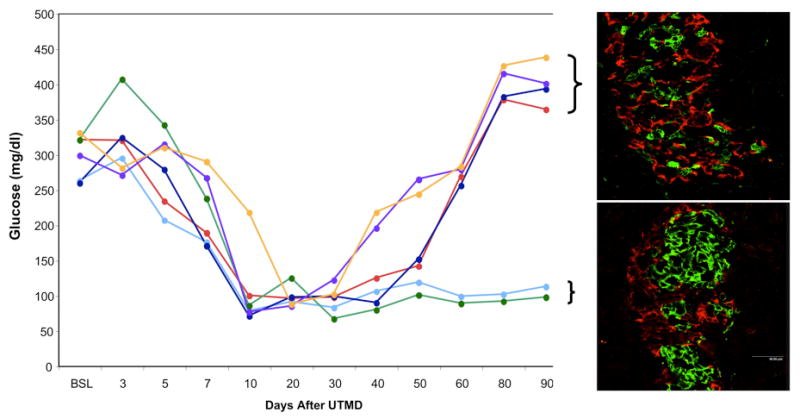
Duration of NeuroD1 gene therapy after STZ-induced diabetes. Graph shows individual blood glucose levels over time in 6 rats treated with UTMD 2 days after STZ (BSL). Profound hyperglycemia is restored to normal in all 6 rats by NeuroD1 gene therapy at days 20 and 30. By day 90, 2 rats remain normoglycemic and 4 rats have returned to severe diabetes. Confocal microscopic images from representative islets are shown in the right panels, stained with anti-insulin (green) and anti-glucagon (red). The diabetic rats have few beta cells present (top right), whereas the normoglycemic rats have preserved islet architecture (bottom right).
Prolongation of Islet Viability by JNK Inhibition
We hypothesized that this late loss of islets was due to apoptosis. Therefore, the experiment was repeated using pre-treatment with SP600125, a JNK inhibitor which has been shown to block JNK-mediated apoptosis in islets.28 Figure 6 shows the results of Tunel staining in normal controls (n-=3), STZ-treated diabetic rats (n=3), STZ rats treated with NeuroD1 gene therapy with (n=6) and without SP600125 (n=6). Apoptosis (green signal) was abolished in the NeuroD1 UTMD group that received pre-treatment with SP600125. Figure 7 shows islet morphometry and blood glucose measurements at 90 days after NeuroD1 gene therapy with and without SP600125. The left panels show representative islets in the rats that did not receive SP600125 (top) compared to those that were re-treated with SP600125 (bottom). Large well-formed islets were seen in the SP600125 group, compared to small clusters of beta cells are present in the controls (no SP600125). Beta cell mass was 0.61 ± 0.07% in the SP600125 group compared to 0.37 ± 0.07% in the controls (p<0.0001) (top right graph). Blood glucose levels out to 90 days are shown in the bottom right graph of Fig 7. Blood sugar was significant lower in the NeuroD1-treated SP600125 group than in the NeuroD1-group that did not receive SP600125 (p<0.05), but was higher than the normal controls (p<0.05). This was due to return of hyperglycemia in 2 of 6 rats treated with SP600125.
Fig. 6.
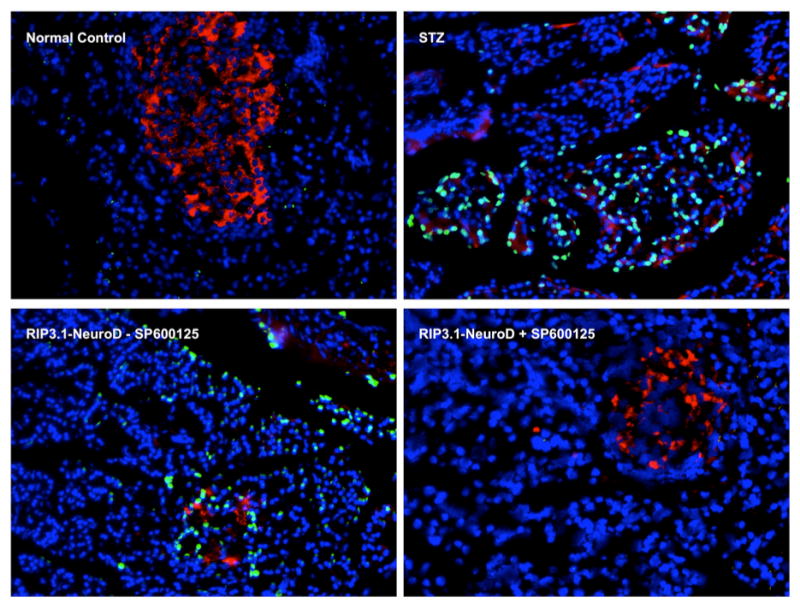
Tunel staining (green) from normal controls (top left), STZ only controls (top right), and NeuroD1-treated rats without SP600125 pretreatment (bottom left), and with SP600125 pretreatment (right). Nuclei are stained with DAPI (blue), beta cells with anti-insulin (red), and apopotic cells are green. Pretreatment with SP600125 protects the regenerated islets from apoptosis (bottom right).
Fig. 7.
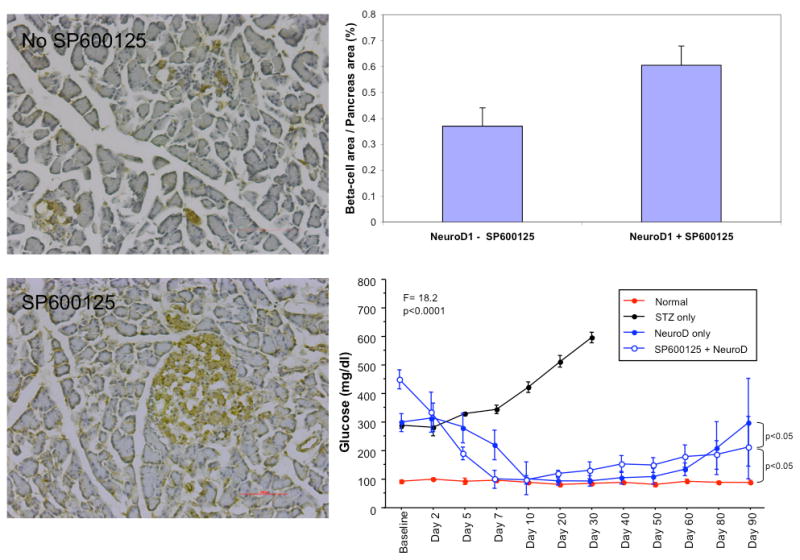
Results of the SP600125 experiments after 90 days. Representative histologic sections from rats treated with NeuroD1 gene therapy without SP600125 (top left) and with SP600125 (bottom left). Scattered beta cells stained with anti-insulin (brown) are present 90 days after UTMD in the absence of SP600125. Well-formed islet cores are present 90 days after UTMD in the presence of SP600125. Beta cell mass is significantly greater in the SP600125 group (p<0.0001)(top right graph). Blood glucose is lower in the NeuroD1-treated rats that received SP600125 compared to those that did not receive SP600125 (p<0.05), but is higher than in normal controls (p<0.05).
Discussion
To our knowledge, this is the first study to demonstrate in vivo regeneration of pancreatic islets with recovery from diabetes without using a viral vector. Specifically, NeuroD1 was targeted to the pancreas of STZ-treated rats with subsequent regeneration of nearly normal appearing islets and restoration of normal blood levels of glucose, insulin, and C-peptide at 30 days. The peak expression of reporter genes delivered as plasmids by UTMD is 4 days, with rapid degradation thereafter (8). Thus, transient gene expression by this method is capable of inducing islet regeneration, while theoretically minimizing the risk of insertional mutagenesis that might be associated with prolonged expression of exogenous gene therapy. By pretreating the rats with SP600125, a JNK inhibitor that has been shown to block beta cell apoptosis, we were able to extend the duration of islet regeneration and normoglycemia to 90 days in the rat model of STZ-induced diabetes.
NeuroD1 is a basic helix-loop-helix transcription factor that is found in the pancreas, intestine, and central nervous system (15). NeuroD1 is present at pancreatic bud development and remains detectable in all mature islet cell types. In NeuroD1 knockout mice, all endocrine cell types develop, but there is decreased numbers of islets and increased beta-cell apoptosis (16). It is thought that NeuroD1 is not essential for early differentiation, but plays an important role in later stage differentiation and maintenance of beta cells, and in cell fate determination (17,18). This view of the role of NeuroD1 in endocrine development, though based primarily on transgenic mouse studies, is consistent with the observed finding of islet regeneration in adult rats in the present study.
Other transcription factors, specifically Ngn3, Pax4, Nkx2.2, Nkx6.1, and MafA, also resulted in islet regeneration, but the islets were comprised predominantly of alpha cells, and blood glucose, insulin, and C-peptide were not normalized. Interestingly, transgenic mice expressing Ngn3 under regulation of a Pdx1 promoter show mostly glucagon-positive cells (19), similar to our findings after in vivo delivery of Ngn3. When Ngn3 is overexpressed in developing chicken gut, it also produces predominantly alpha cells (20). It seems likely that the role of these transcription factors when delivered exogenously to adult animals with diabetes, may differ from their role in embryological development. It is also possible that various combinations of transcription factor genes or other genes implicated in pancreatic development or cell cycling, would result in even more robust islet regeneration than that observed in this study. For example, Chen, et al, showed that insulin production by acinar cells could be induced in STZ-treated rats by the combination of betacellulin and Pdx1 plasmids delivered in vivo with restoration of normal blood glucose and insulin for up to 15 days (10). More recently, Zhou, et al, reported that the combination of Ngn3, MafA, and Pdx1, delivered by direct injection of adenovirus into the pancreas of immune-deficient mice, resulted in reprogramming of exocrine cells to a beta-cell phenotype (11). These new beta-cells were isolated into single cells or small clusters of only a few cells, rather than aggregating into islets. Blood glucose, insulin, and C-peptide were improved but not restored to normal levels. When single transcription factors were delivered, new beta cells were not seen in significant numbers. The present study differs in two potentially important respects. First, we used a non-viral gene delivery method that, unlike direct injection, targets the entire pancreas. Second, we used the RIP3.1 promoter to selectively target gene expression to the endocrine pancreas. The more commonly used CMV promoter is more efficient in exocrine than endocrine pancreas (21). Similarly, adenovirus is more robust in exocrine than endocrine pancreas (22). Islet regeneration has been achieved in STZ-mediated diabetes by using adenovirus to deliver various genes to liver, with resultant restoration of normal blood glucose (4-5). However, adenovirus is not likely to be used in humans due to safety considerations. Although liver is a suitable organ for islet regeneration and islet transplantation, our technique uses ultrasound-mediated microbubble destruction to deliver plasmid cDNA to the whole pancreas with relatively potent organ specificity. This offers a theoretical advantage for islet regeneration and maintenance since the pancreas is the normal physiologic milieu for islets. As in our previous studies of gene delivery by UTMD (8-10), we did not find any evidence of pancreatic damage by UTMD, either by serum amylase or lipase, or by histology.
We did not perform complex studies of cell lineage. However, several observations suggest that the regenerated islets observed after RIP3.1-NeuroD1 result from replication of scattered beta-cells that survived STZ. Immunofluorescent staining with CK19, a ductal cell marker, did not show any significant uptake in the regenerated islets (data not shown). Islet regeneration was only seen to occur when gene delivery was administered immediately after STZ treatment or within 48 hours afterward. When the experiments were repeated with gene delivery 7 days after STZ, a time period in which there were no detectable insulin-staining cells in STZ control rats, islet regeneration was not seen and there was progression of severe hyperglycemia and weight loss. Colocalization of Ki67 and insulin was present in the regenerated islets, suggesting beta-cell proliferation. We used a modification of the rat insulin 1 promoter, which is strongly beta-cell specific, particularly under conditions of hyperglycemia (8,9), and would not be expected to have substantial activity in exocrine pancreas. These considerations are consistent with the prevailing view that beta-cell replication is the predominant mechanism for increasing beta-cell mass (13,23,24).
The concept of using gene therapy to promote islet regeneration in diabetic humans is plausible. Meier, et al, showed that 88% of autopsy specimens from adult humans with long-standing Type 1 diabetes had substantial numbers of beta-cells, as well as beta-cell apoptosis and T-lymphyocyte infiltration (25). Thus, existing beta-cells are a potential target for regenerative gene therapy with NeuroD1, alone or in combination with other transcription factors. However, any strategy to regenerate islets will have to also account for potential destruction of the new islets by apoptosis, inflammation, or autoimmunity (2). We used SP600125, a JNK inhibitor, to reduce beta cell apoptosis and extend the duration of efficacy after NeuroD1 gene therapy. It should be possible to combine regenerative genes with genes or drugs that inhibit apoptosis (26-29), and/or the autoimmune response. With regard to the latter, recent evidence suggests that tacrolimus and sirolimus might have direct toxic effects on beta cells (14). Thus, the optimal immunosuppressive regimen for islet protection may not yet be established. Finally, there are important differences between rodent and human islet biology (2), so these findings need to be confirmed in higher order animals before human trials can be contemplated.
Materials and Methods
Rat insulin promoters and plasmids constructs
Sprague-Dawley rat genomic DNA was extracted from rat peripheral blood with a QIAamp Blood kit (Qiagen Inc, Valencia, CA) according to the manufacturer's instructions. Rat insulin gene 1 promoter fragments (-412 to +165), was amplified from SD rat genomic DNA using PCR. The resulting DNA fragments were subcloned into the pDsRed1-1 reporter vector (Clonetech, CA). hMafA cDNA and hamster Nkx6.1 cDNA were donated/cordially gifts from the Olson lab at Michigan State University and the Newgard lab at Duke University Medical Center. Rat Ngn3, NeuroD1, Pax4, and Nkx2.2 cDNAs were PCR products from Sprague-Dawley new-born rat pancreas cDNA pool that were reversed from their total RNA according to the manufacturer's instructions. Newborn rat pancreatic samples were flash frozen in liquid nitrogen and stored at -86°C. The frozen samples were thawed in 1 ml of RNA-STAT solution and immediately homogenized using a polytron homogenizer at 10,000 rpm for 30s. Total RNA (30 ng) was reverse transcribed in 20 μl by using a Sensiscript RT kit (Qiagen Inc, Valencia, CA) with oligo(dT)16. The reaction mixture was incubated at 42°C for 50 min, followed by a further incubation at 70°C for 15 min. PCR was performed for all samples using a GeneAmp PCR System 9700 (PE ABI, Foster City, CA, USA) in 50 ul volume containing 2 ul cDNA, 25 ul of HotStarTaq Master Mix (Qiagen Inc, Valencia, CA), and 20 pmol of each primer. The corresponding PCR products were verified by agarose gel electrophoresis and purified by QIAquick Gel Extraction kit (Qiagen Inc, Valencia, CA). To confirm the sequences, direct sequencing of PCR products was performed with dRhodamine Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) on an ABI 3100 Genomic Analyzer. All transcriptional factor gene cDNAs were subcloned to RIP3.1 driving vector. The plasmids digestion, ligation, subclone, isolation and purification were performed by standard procedures, and once again sequenced to confirm that no artifactual mutations were present.
Animal protocols and UTMD
All animal studies were performed in accordance with National Institute of Health (NIH) recommendations and the approval of our local IACUC. Male Sprague–Dawley rats (230–270g) anesthetized with intraperitoneal ketamine (60 mg/kg) and xylazine (5 mg/kg) were shaved from left abdomen and neck, and a polyethylene tube (PE 50, Becton Dickinson, Franklin Lakes, TN, USA) was inserted into the right internal jugular vein by cut-down.
A total of 45 rats received one of nine treatments: (1) no treatment (normal control rats, n=3); (2) STZ (60 mg/kg/i.p., Sigma, St Louis, MO, USA) alone without UTMD (N=3); (3) STZ and UTMD with DsRed (n=3); (4) STZ and UTMD with ngn3 (n=6); (5) STZ and UTMD with NeuroD1 (n=6); (6) STZ and UTMD with Pax4 (n=6). (7) STZ and UTMD with Nkx2.2 (n=6); (8) STZ and UTMD with Nkx6.1 (n=6); (9) STZ and UTMD with MafA (n=6). All genes were delivered as plasmid cDNA under the control of the RIP3.1 promoter. Blood glucose was measured 12 hours after STZ injection. Animals with fasting blood glucose over 250 mg/dl were considered as successful diabetes type 1 model and subsequently underwent UTMD within 48 hours of STZ treatment. Microbubble or control solutions (0.5 ml diluted with 0.5 ml phosphate-buffered solution (PBS)) were infused over 10 min via pump (Genie, Kent Scientific, Torrington, CT, USA). During the infusion, ultrasound was directed to the pancreas using a commercially available ultrasound transducer (S3, Sonos 5500, Philips Ultrasound, Bothell, WA, USA). The probe was clamped in place. Ultrasound was then applied in ultraharmonic mode (transmit 1.3 MHz/receive 3.6 MHz) at a mechanical index of 1.4. Four bursts of ultrasound were triggered to every fourth end-systole by electrocardiogram using a delay of 45–70 ms after the peak of the R wave. These settings have shown to be optimal for plasmid delivery by UTMD using this instrument (8). Bubble destruction was visually apparent in all rats. After UTMD, the jugular vein was tied off, the skin closed, and the animals allowed to recover. Blood samples were drawn after an overnight 12-h fast at baseline, and at different days after treatment. This protocol was repeated 3 times with 135 rats sacrificed at days 10 (n=45), 20 (n=45), and 30 (n=45) using an overdose of sodium pentobarbital (120 mg/kg). Pancreas, liver, spleen, and kidney were harvested for histology. Blood glucose level was measured with blood glucose test strip (Precision, Abbott, Abbott Park, IL, USA); blood insulin and C-peptide were measured with RIA kit (Linco Research, Radioimmunoassay, Billerica, MA, USA).
For the 90-day experiments, only RIP3.1-NeuroD1 was given by UTMD (n=6), along with 3 normal controls, in which STZ was not given, and 3 STZ controls. This experiment was then repeated with pretreatment of rats with SP600125 (n=6) prior to UTMD with RIP3.1-NeuroD1 and without such pretreatment (n=6).
Manufacture of Plasmid-Containing Lipid-Stabilized Microbubbles
Lipid-stabilized microbubbles were prepared as previously described in our laboratory. Briefly, a stock solution is prepared containing 270 mg of 1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine, (Sigma, St. Louis, MO), 30 mg of 1,2-dipalmitoyl-sn-glycero-3-phosphatidylethanolamine, (Sigma, St. Louis, MO), and 1 g of glucose. These ingredients are dissolved in a boiling water bath for 20-30 minutes, with pipetting of contents up and down until no visible particles remain. This stock solution is stored at 4°C. Plasmid-containing microbubbles are prepared hy mixing 2 mg of dried plasmid with 50 ul of lipefectamine 2000 (Invitrogen, Carlsbad, CA) and incubating at room temperature for 15 minutes. This liposome/plasmid DNA mixture is added to 250 ml of lipid stock solution, 50 ml of pure glycerol, and 5 ul of 10% albumin solution, mixed well with a pipette, and then placed in ice. Aliquots of 0.5 ml of this phospholipid-plasmid solution were placed in 1.5 ml clear vials; the air in the headspace of the vials is replaced with perfluoropropane gas (Air Products, Inc, Allentown, PA). Each vial was incubated at 4°C for 30 min and then mechanically shaken for 30 seconds by a dental amalgamator (VialmixTM, Bristol-Myers Squibb Medical Imaging, N. Billerica, MA). The lipid-stabilized microbubbles appear as a milky white suspension floating on the top of a layer of liquid containing unattached plasmid DNA. The subnatant was discarded and the microbubbles washed three times with PBS to removed unattached plasmid DNA. The mean diameter and concentration of the microbubbles in the upper layer were measured by a particle counter (Beckman Coulter Multisizer III).
Immunohistochemistry
Cryostat sections 5-8 μm in thickness were fixed in 4% paraformaldehyde for 15 min at 4 °C and quenched for 5 min with10 mM glycine in PBS. Sections were then rinsed in PBS 3 times, and permeabilized with 0.5% Triton X-100 in PBS for 10 min. Sections were blocked with 10% goat serum at 37°C for 1hr and washed with PBS 3 times. The primary antibody (mouse anti-insulin antibody, 1:500 dilution (Sigma, St. Louis, MO); rabbit anti-glucagon, 1:500; rabbit anti-somatostatin, 1:500; rabbit anti-pancreatic polypeptide, 1:500, rabbit anti-NeuroD1, 1:500, rabbit anti- Ki-67, rabbit anti-BrDu, 1:500, mouse anti-ck19, 1:2000 dilution (Chemicon, Temecula, CA) were added and incubated for 2 hrs at RT. After washing with PBS three times for 5 min, the secondary antibody (Sigma, St; Louis, MO) anti-mouse lgG conjugated with FITC; anti-rabbit lgG conjugated with Cy5) (1:500 dilution in block solution) were added and incubated for 1 hr at RT. Sections were rinsed with PBS for 10 min, 5 times, and then mounted.
Tunel staining
Tunel staining for detection and quantification of apoptosis in rat pancreas slides was performed using a commercially available kit (Roche Diagnostics, Indianapolis, IN) according to a standard protocol. DNA strand breaks at the single cell level, were labeled with fluorescein and imaged by fluorescence microscopy.
Beta Cell Mass Calculation
Beta cell mass was evaluated by the method of Terauchi et al (30). Sections of rat pancreas were immunostained with monoclonal mouse anti-insulin (1:500 dilution) and a second antibody, peroxidase-conjugated goat anti-mouse IgG (1:500 dilution) and developed in liquid DAB. Images from 6 consecutive pancreas sections were captured on a monitor screen of a digital imaging system (Olympus BX60, Nikon digital camera DXM1200C, and NIS-Elements F2.30). The total areas of islet beta cells and whole pancreas were measured using NIH ImageJ software (NIH, Bethesda, MD). Beta cell mass was calculated as the ratio of beta cells to whole pancreas for 6 consecutive pancreatic slides from each rat.
Data analysis
Data was analyzed with Statview software (SAS, Cary, NC, USA). The results are expressed as mean one standard deviation. Differences were analyzed by repeated measures ANOVA with Fisher's post hoc test and considered significant at P<0.05.
Acknowledgments
This work was supported by the Mark and Mary Alice Shepherd endowment of the Baylor Foundation, and by NIDDK grant P02 DK58398 (Newgard, PI).
Footnotes
Conflict of Interest: The authors have no conflicts of interest regarding this work.
References
- 1.Bonner-Weir S, Wier GC. New sources of pancreatic β-cells. Nat Biotechnol. 2005;23:857–861. doi: 10.1038/nbt1115. [DOI] [PubMed] [Google Scholar]
- 2.Butler PC, Meier JJ, Butler AE, Bhushan A. The replication of β cells in normal physiology, in disease, and for therapy. Nat Clin Pract Endocrinol Metab. 2007;3:758–768. doi: 10.1038/ncpendmet0647. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro AMJ, Ricordi C, Hering BJ, Auchincloss H, Lindblad R, Robertson RP, Secchi A, Brendel MD, Berney T, Brennan DC, Cagliero E, et al. International trial of the Edmonton protocol for islet transplantation. New Engl J Med. 2006;355:1318–1330. doi: 10.1056/NEJMoa061267. [DOI] [PubMed] [Google Scholar]
- 4.Ferber S, Halkin A, Cohen H, Ber I, Einav Y, Goldberg I, et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6:568–572. doi: 10.1038/75050. [DOI] [PubMed] [Google Scholar]
- 5.Kojima H, Fujimiya M, Matsumura K, Younan P, Imaeda H, Maedaet M, et al. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9:596–603. doi: 10.1038/nm867. [DOI] [PubMed] [Google Scholar]
- 6.Kaneto H, Nakatani Y, Miyatsuka T, Matsuoka TA, Matsuhisa M, Hori M, et al. PDX-1/VP16 fusion protein, together with NeuroD or Ngn3, markedly induces insulin gene transcription and ameliorates glucose tolerance. Diabetes. 2005;54:1009–102. doi: 10.2337/diabetes.54.4.1009. [DOI] [PubMed] [Google Scholar]
- 7.Wang AY, Ehrhardt A, Xu H, Kay MA. Adenovirus transduction is required for the correction of diabetes using Pdx-1 or Neurogenin-3 in the liver. Mol Ther. 2007;15:255–263. doi: 10.1038/sj.mt.6300032. [DOI] [PubMed] [Google Scholar]
- 8.Chen S, Ding JH, Bekeredjian R, Yang BZ, Shohet RV, Johnston SA, et al. Efficient gene delivery to pancreatic islets with ultrasonic microbubble destruction technology. Proc Natl Acad Sc USA. 2006;103:8469–8474. doi: 10.1073/pnas.0602921103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chai R, Chen SY, Ding JH, Grayburn PA. Efficient, glucose responsive, and islet-specific transgene expression by a modified rat insulin promoter. Gene Ther. 2009;16:1202–1209. doi: 10.1038/gt.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen S, Ding J, Yu C, Yang B, Wood DR, Grayburn PA. Reversal of streptozotocin-induced diabetes in rats by gene therapy with betacellulin and pancreatic duodenal homeobox-1. Gene Ther. 2007;14:1102–1110. doi: 10.1038/sj.gt.3302963. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to β cells. Nature. 2008;455:627–32. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scharfman R. Control of early development of the pancreas in rodents and humans: implications of signals from the mesenchyme. Diabetologia. 2000;43:1083–1092. doi: 10.1007/s001250051498. [DOI] [PubMed] [Google Scholar]
- 13.Bouwens L, Rooman I. Regulation of pancreatic beta-cell mass. Physiol Rev. 2005;85:1255–1270. doi: 10.1152/physrev.00025.2004. [DOI] [PubMed] [Google Scholar]
- 14.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by β cell regeneration. J Clin Invest. 2007;117:2553–2561. doi: 10.1172/JCI32959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naya FJ, Stellrecht CM, Tsai MJ. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev. 1995(9):1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- 16.Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhawan S, Georgia S, Bhushan A. Formation and regeneration of the endocrine pancreas. Curr Opin Cell Biol. 2007;19:634–645. doi: 10.1016/j.ceb.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chao CS, Loomis ZL, Lee JE, Sussel L. Genetic identification of a novel NeuroD1 function in the early differentiation of islet alpha, PP and epsilon cells. Dev Biol. 2007;312:523–532. doi: 10.1016/j.ydbio.2007.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, et al. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400:877–881. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 20.Grapin-Botton A, Majithia AR, Melton DA. Key events of pancreas formation are triggered in gut endoderm by ectopic expression of pancreatic regulatory genes. Genes Dev. 2001;15:444–454. doi: 10.1101/gad.846001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhan Y, Brady JL, Johnston AM, Lew AM. Predominate transgene expression in exocrine pancreas directed by the CMV promoter. DNA Cell Biol. 2000;19:639–645. doi: 10.1089/10445490050199045. [DOI] [PubMed] [Google Scholar]
- 22.Wang AY, Peng PD, Ehrhardt A, Storm TA, Kay MA. Comparison of adenoviral and adeno-associated viral vectors for pancreatic gene delivery in vivo. Hum Gene Ther. 2004;15:405–413. doi: 10.1089/104303404322959551. [DOI] [PubMed] [Google Scholar]
- 23.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 24.Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult β cells does not involve specialized progenitors. Dev Cell. 2007;12:817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 25.Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing type I diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48:2221–2228. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]
- 26.Dror V, Nguyen V, Walia P, Kalynyak TB, Hill JA, Johnson JD. Notch signaling suppresses apoptosis in adult human and mouse pancreatic islet cells. Diabetologia. 2007;50:2504–2515. doi: 10.1007/s00125-007-0835-5. [DOI] [PubMed] [Google Scholar]
- 27.Drucker DJ. Glucagon-like peptide-1 and the islet beta-cell: augmentation of cell proliferation and inhibition of apoptosis. Endocrinology. 2003;144:5145–5148. doi: 10.1210/en.2003-1147. [DOI] [PubMed] [Google Scholar]
- 28.Noguchi H, Nakai Y, Ueda M, Masui Y, Futaki S, Kobayashi N, et al. Activation of c-Jun NH2-terminal kinase (JNK) pathway during islet transplantation and prevention of islet graft loss by intraportal injection of JNK inhibitor. Diabetologia. 2007;50:612–619. doi: 10.1007/s00125-006-0563-2. [DOI] [PubMed] [Google Scholar]
- 29.Lin CY, Gurlo T, Haataja L, Hsueh WA, Butler PC. Activation of peroxisome proliferator-activated receptor-gamma by rosiglitazone protects human islet cells against human islet amyloid polypeptide toxicity by a phosphatidylinositol 3′-kinase-dependent pathway. J Clin Endocrinol Metab. 2005;90:6678–6686. doi: 10.1210/jc.2005-0079. [DOI] [PubMed] [Google Scholar]
- 30.Terauchi Y, Takamoto I, Kubota N, Matsui J, Suzuki R, Komeda K, et al. Glucokinase and IRS-2 are required for compensatory β cell hyperplasia in response to high-fat diet–induced insulin resistance. J Clin Invest. 2007;117:246–257. doi: 10.1172/JCI17645. [DOI] [PMC free article] [PubMed] [Google Scholar]