

Abstract
The fatty acid ethanolamides (FAEs) are a family of bioactive lipid mediators that include the endogenous agonist of peroxisome proliferator-activated receptor-α, palmitoylethanolamide (PEA). FAEs are hydrolyzed intracellularly by either fatty acid amide hydrolase or N-acylethanolamine-hydrolyzing acid amidase (NAAA). Selective inhibition of NAAA by (S)-N-(2-oxo-3-oxetanyl)-3-phenylpropionamide [(S)-OOPP, 7a] prevents PEA degradation in mouse leukocytes and attenuates responses to proinflammatory stimuli. Starting from the structure of 7a a series of β-lactones was prepared and tested on recombinant rat NAAA to explore structure-activity relationships (SARs) for this class of inhibitors and improve their in vitro potency. Following the hypothesis that these compounds inhibit NAAA by acylation of the catalytic cysteine, we identified several requirements for recognition at the active site and obtained new potent inhibitors. In particular, (S)-N-(2-oxo-3-oxetanyl)biphenyl-4-carboxamide (7h) was more potent than 7a at inhibiting recombinant rat NAAA activity (7a, IC50 = 420 nM; 7h, IC50 = 115 nM) in vitro and at reducing carrageenan-induced leukocyte infiltration in vivo.
Introduction
The fatty-acid ethanolamides (FAEs) are a family of bioactive lipid mediators that have stimulated pharmaceutical interest because drugs that block their deactivating metabolism may offer new ways to treat pain and inflammation.1 For example, inhibitors of the enzyme fatty acid amide hydrolase (FAAH),2 which catalyzes the hydrolysis of the endocannabinoid anandamide, are undergoing clinical investigations for the treatment of pain.3 Another endogenous FAE that has attracted considerable attention is palmitoylethanolamide (PEA), which exerts potent antiinflammatory4 and analgesic5 effects, which are due to a large extent to activation of the nuclear receptor peroxisome proliferator-activated receptor-α (PPAR-α)6,7 even though complementary mechanisms might also be involved. 8,9
PEA is produced in many mammalian tissues, including innate immune cells,10 where a selective phospholipase D releases it from its membrane precursor, N-palmitoylphosphatidylethanolamine.11 PEA is deactivated by two intracellular amidases: FAAH and N-acylethanolamine-hydrolyzing acid amidase (NAAA).12,13,14 Potent FAAH inhibitors, such as the compound URB597,15 have been utilized to unmask the functions of the preferred FAAH substrate, anandamide,16 but the search for potent and selective NAAA inhibitors is still at its beginning.17,18,19
NAAA preferentially hydrolyzes PEA over other FAEs and is localized to lysosomes. Despite its functional similarity with FAAH, NAAA shares no homology with this enzyme or other enzymes of the same ‘amidase signature’ family.14 Rather, NAAA is an N-terminal nucleophile hydrolase (Ntn) and belongs to the choloylglycine hydrolase family of hydrolases, which are characterized by the ability to cleave non-peptide amide bonds.13,20 Like other Ntn enzymes, NAAA is converted by proteolysis into a shorter active form upon incubation at acidic pH21. Processing of rodent NAAA renders cysteine 131 (Cys131) the N-terminal amino acid and putative catalytic residue. Site-directed mutagenesis experiments have confirmed the importance of Cys131 for catalysis by NAAA.22,23
Compounds incorporating a β-lactone ring are known to interact covalently with biological nucleophiles, and some have been reported to inhibit enzymes that contain a catalytic cysteine, such as 3-hydroxy-3-methylglutaryl-CoA synthase (HMGS),24 or threonine-based Ntn enzymes, like the proteasome.25 Screening a series of molecules that contain cysteine-reactive warheads, we found that 2-oxo-3-oxetanylcarbamic acid benzyl ester (1, Table 1), which is known for its ability to inhibit a viral cysteine hydrolase,26 was also a weak NAAA inhibitor. Structural modification of this compound allowed us to identify (S)-N-(2-oxo-3-oxetanyl)-3-phenylpropanamide [(S)-OOPP, 7a, Table 1] as a potent non-competitive inhibitor of intracellular NAAA activity.23 Pharmacological experiments in vitro and in vivo have shown that 7a prevents PEA hydrolysis in activated inflammatory cells and dampens tissue reactions to various proinflammatory triggers.23 This lactone derivative had revealed remarkable selectivity, showing no inhibition of several hydrolase enzymes that use lipids as substrates, including FAAH, monoacylglycerol lipase and diacylglycerol lipase type-α.23
Table 1.
Inhibitory Potencies (IC50) of Tested Compounds 1,7a-d,8a,10,11a,12,15,20a,b23, on rat NAAA Activity.
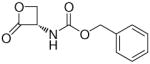
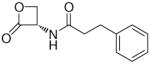
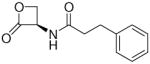
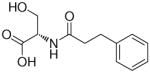
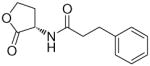
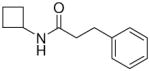
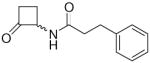
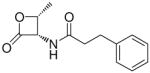
In the present study, we utilized the structures of the α-acylamino-β-lactones 1 and 7a23 (Table 1) as starting points to explore structure-activity relationships (SARs) for compounds belonging to the chemical class of N-(2-oxo-3-oxetanyl)amides with the objective of discovering NAAA inhibitors with improved potency. In particular, we explored the role of the β-lactone ring and amide side chain, first by assessing stereoelectronic requirements in close proximity to the lactone ring, and next by optimizing the size and shape of the lipophilic tail of the amide moiety.
Chemistry
N-tert-butyloxycarbonyl-L-serine (2a) and N-tert-butyloxycarbonyl-D-serine (2b) were cyclized by means of a modified Mitsunobu reaction employing dimethyl azodicarboxylate (DMAD) and triphenylphosphine (Scheme 1).27 Deprotection of β-lactone derivatives 3a27 and 3b with trifluoroacetic acid in the presence of p-toluensulfonic acid gave the tosylate salts (S)- and (R)-2-oxo-3-oxetanylammonium toluene-4-sulfonate (4a28 and 4b29), respectively.28,29 Alternatively, 4a can be obtained from the commercially available (S)-3-(tritylamino)oxetan-2-one (Scheme 1).30
Scheme 1.

a Reagents and conditions: (a) PPh3, DMAD, THF, −78 °C, 20 min, then room temperature, 2.5 h; (b) CF3COOH, p-TsOH, 0 °C, 10–15 min; (c) CH2Cl2/CF3COOH, p-TsOH, 0 °C, 0.5 h.
The amides 7a,23b,31d–f,i,j and 8a,23j were prepared by reaction of the corresponding acyl chloride 6a,b,d–f,i,j, commercially available, and the salts 4a or 4b in the presence of triethylamine (Scheme 2). Epimerization of the stereogenic α-centre was not observed.
Scheme 2.

a Reagents and conditions: (a) 4a or 4b, Et3N, CH2Cl2, 0 °C, 0.5 h then room temperature, 2 h.
The amides 7c,g,h,k–n were prepared by a two steps procedure starting from the corresponding carboxylic acid 9c,g,h,k–n (Scheme 3). These acids, synthesized as reported below if not commercially available, were converted to acyl chlorides with oxalyl chloride and a catalytic amount of dimethylformamide.32 The acyl chlorides were then reacted with 4a.
Scheme 3.

a Reagents and conditions: (a) (COCl)2, DMF, CH2Cl2, 0 °C, 0.5 h then room temperature, 3–16 h; (b) 4a, Et3N, CH2Cl2 (or THF), 0 °C, 0.5 h then room temperature, 3 h.
Compounds 10, 11a,j, 12 and 15 were synthesized as reported in Scheme 4. Compound 1033 was obtained by reacting L-serine with 3-phenylpropionyl chloride (6a) in aqueous sodium hydroxide. The γ-lactone derivatives 11a34 and 11j were synthesized by coupling (S)-3-aminodihydrofuran-2-one hydrobromide with 6a and naphthalene-2-carbonyl chloride (6j), respectively. Amide 1223 was prepared by reacting 6a with cyclobutylamine. Cyclobutanone derivative 1523 was synthesized by the acid catalyzed reaction of 1,2-bis(trimethylsilanyloxy)cyclobutene and 3-phenylpropionamide (14)35, the latter obtained through alkaline hydrolysis of 4-phenylpropionitrile (13) by slight modifications of a literature procedure.36
Scheme 4.

a Reagents and conditions: (a) L-serine, NaOH, 0 °C, 4 h; (b,c) (S)-3-aminodihydrofuran-2-one.HBr or c-C4H7NH2, Et3N, CH2Cl2, 0 °C–room temperature, 3–3.5 h; (d) t-BuOH/H2O, NaOH, H2O2, room temperature, 3 h; (e) 1,2-bis(trimethylsilanyloxy)cyclobutene, THF, HCl/Et2O, reflux, 3 h.
The synthesis of threonine β-lactones 20a and 20b is reported in Scheme 5. The amino groups of L- and D-threonine were protected through treatment with di-tert-butyl dicarbonate in aqueous sodium bicarbonate to give 17a37 and 17b38, respectively, by means of slight modification of a literature procedure.39 Cyclization to β-lactones 18a40 and 18b was accomplished through treatment of 17a and 17b, respectively, with benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP reagent) by means of a literature procedure.26 Subsequent deprotection of the amino group with trifluoroacetic acid in presence of p-toluensulfonic acid, following the same synthetic protocol adopted for 4a,b, gave the tosylate salts (3S,4R)- and (3R,4S)-2-methyl-4-oxo-3-oxetanylammonium toluene-4-sulfonate (19a41 and 19b). Finally, the triethylamine catalyzed coupling of these tosylate salts with 6a afforded the desired compounds 20a and 20b.
Scheme 5.

a Reagents and conditions: (a) Boc2O, NaHCO3, H2O, MeOH, room temperature, 36 h; (b) BOP, Et3N, CH2Cl2, room temperature, 3 h; (c) CF3COOH, p-TsOH, 0 °C, 0.25 h; (d) 6a, Et3N, CH2Cl2, 0 °C, 0.5 h then room temperature, 3 h.
rac-3-(4-Phenylbutyl)oxetan-2-one 23 was prepared by means of slight modification of a literature procedure:42 alkanoic acid 22, obtained from unsaturated 2-methylene-6-phenylhexanoic acid (21)43, was cyclized in aqueous sodium hydroxide (Scheme 6).
Scheme 6.

a Reagents and conditions: (a) HBr/AcOH, CH2Cl2, 0 °C, 3 h then room temperature, 52 h; (b) NaOH, CHCl3, room temperature, 2 h.
The syntheses of the carboxylic acids 9g,44k,45m,46n are reported in Scheme 7. 9g was obtained via a Suzuki cross-coupling reaction between 3-bromobenzaldehyde (24) and phenylboronic acid (25),47 and an oxidation of the resulting biphenyl-3-carbaldehyde to carboxylic acid by using aqueous potassium permanganate. The monoester 9k was prepared by treating the symmetric dicarboxylic acid 26 with ethylbromide by means of a slight modification of a literature procedure.48 The reaction of phenol with terephthaloyl dichloride (27), followed by the selective hydrolysis of the resulting terephthalic acid diphenyl ester with sodium carbonate, furnished 9m. 9n49 was obtained by reaction between 4-hydroxybenzoic acid (28) and benzylbromide according to a literature procedure.50
Scheme 7.

a Reagents and conditions: (a) Bu4NBr, K2CO3, PdCl2, EtOH, room temperature, 3 h; (b) KMnO4, CH3C(O)CH3, H2O, room temperature, 5 h; (c) EtBr, Et3N, DMF, 80 °C, 4 h; (d) PhOH, Et3N, MeCN, room temperature, 4 h; (e) Na2CO3, room temperature, 15 h; (f) BnBr, KOH, EtOH/H2O, reflux, 24 h.
Results and Discussion
The new compounds were tested for their ability to inhibit heptadecenoylethanolamide hydrolysis by recombinant rat NAAA, heterologously expressed in HEK293 cells. IC50 values are reported in Tables 1 and 2.
Table 2.
Inhibitory Potencies (IC50) of N-(2-Oxo-3-oxetanyl)arylamides (7e-7n,8j,11j) on Rat NAAA Activity.
| Cpds. | Structure | IC50 (nM) ± S.E.M. |
|---|---|---|
| 7e | 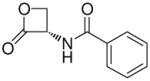 |
1,700 ± 400 |
| 7f | 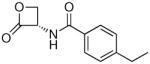 |
102 ± 21 |
| 7i | 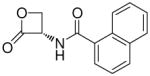 |
50,000 ± 19,000 |
| 7j | 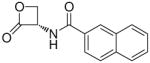 |
160 ± 40 |
| 8j | 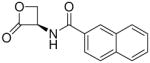 |
3,200 ± 800 |
| 11j | 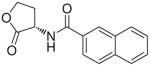 |
>100,000 |
| 7k | 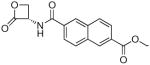 |
300 ± 20 |
| 7l | 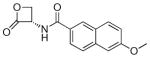 |
400 ± 20 |
| 7m | 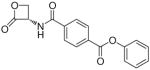 |
300 ± 100 |
| 7n | 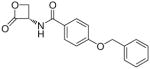 |
90 ± 10 |
| 7g | 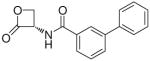 |
4,400 ± 1,200 |
| 7h | 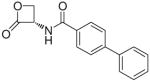 |
115 ± 13 |
Previous work with compounds 1, 7a and 8a23 indicated that (S)-2-oxo-3-oxetanylamides offer a promising scaffold for SAR analysis and potency optimization, and that an intact β-lactone ring is essential for NAAA inhibition. Confirming those results, we found that compounds lacking the β-lactone moiety were entirely devoid of inhibitory activity (10, 11a, 12, 15).
The β-lactone group may contribute to NAAA inhibition in two different ways. Assuming that the sulfhydryl group of the active cysteine (Cys131 of rodent NAAA) interacts covalently with the 2-oxo-3-oxetane moiety of 7a, the nucleophilic attack could occur either at the 2-carbonyl, giving a thioester as the result of enzyme acylation (route “a”, Figure 1), or at the 4-methylene group, resulting in enzyme alkylation (route “b”, Figure 1). Both mechanisms are documented in the literature. For example, compound 1 irreversibly inhibits hepatitis A virus 3C protease through a mechanism involving the covalent attack of the enzyme nucleophile on the lactone methylene, leading to alkylation of catalytic Cys172.26,51 On the other hand, the β-lactone hymeglusin inhibits human HMGS by covalent modification of the active Cys129,52 but this effect is reversed by incubation with hydroxylamine,53 and a thioester adduct resulting from the attack of the active Cys117 of Brassica juncea HMGS has been recently crystallized.54 A dialysis experiment on NAAA showed partial, but significant reversibility of inhibition by 7a (after inhibition by 10 μM of 7a, 44±2% of initial NAAA activity is recovered following 12-hour dialysis), suggesting that cysteine acylation (route “a”, Figure 1) is a likely mechanism.
Figure 1.

Possible mechanisms for covalent inactivation of NAAA by lactone derivatives.
To design new derivatives for further SAR exploration, we utilized a three-dimensional model of NAAA previously build by comparative modeling. This model had evidenced that essential features of the catalytic site of Ntn hydrolases20 are retained by NAAA, revealing critical roles for amino acid residues involved in the oxyanion hole arrangement (Asn292), the stabilization of Cys131 basic nitrogen (Asp150) or ligand recognition (Asn209 and Tyr151); these roles have been confirmed by mutational analysis.23
Using the NAAA model, we built the putative reaction intermediates resulting from nucleophilic attacks of the sulfhydryl group of Cys131 on the lactone carbonyls of 7a and 8a (see Figure 2A and 2B, respectively). These covalent intermediates, built from docking poses consistent with cysteine acylation, showed favorable polar interactions of the lactone ring and the amide fragment with critical amino acid residues, and accommodation of the phenethyl chain in the lipophilic pocket lined by the aromatic ring of Tyr151, where the acyl chain of PEA might also be located.23
Figure 2.

Representation of the putative tetrahedral intermediates resulting from the nucleophilic attack of catalytic cysteine 131 onto the lactone carbonyl of 7a (A, green carbons) or 8a (B, orange carbons). The backbone of the NAAA model, built by comparative modeling, is represented in grey. Hydrogen bonds between enzyme residues and the inhibitor are symbolized by yellow lines. Standard-atom color codes: black: carbon; red: oxygen; blue: nitrogen; white: hydrogen; yellow: sulfur.
Following bond formation between the sulfur of Cys131 and the lactone carbonyl, and oxyanion accommodation into the putative oxyanion hole, the β-methylene groups of the lactone rings of 7a and 8a appeared to be surrounded by different stereoelectronic environments: the syn hydrogen in the β-methylene of 7a was close to the carbonyl group of Asp150 (Figure 2A), while the corresponding hydrogen of 8a pointed toward a free region of the active site (Figure 2B). We exploited this difference to design two derivatives aimed at testing the acylation hypothesis, as implemented by the NAAA model: the enantiomeric syn-methyl derivatives of 7a and 8a, compounds 20a and 20b. The complete loss of activity observed for compound 20a and the tolerance for the syn-methyl group of 20b (Table 1) supported our interaction model, which was then considered as a working hypothesis for further SAR exploration. The recognition role of the amide fragment of 7a and 8a, illustrated in Figure 2, was also supported by the low potency of the racemic phenylbutyl lactone 23.
The model also implied that inhibitory potency could be improved by modulating the lipophilicity of the phenylpropionic chain of 7a [calculated LogP (LogPcalc): −0.43],55 which may occupy the presumed acyl chain-binding pocket.20 However, while a shorter phenylacetic chain (7b, LogPcalc: −0.91) gave an expected increase in IC50, a decrease was not observed for the phenylheptanoic derivative 7c (LogPcalc: 1.50), or for the heptanoic one 7d (LogPcalc: 0.10). Based on these contrasting results, and considering that flexible structures typically provide ambiguous information about the steric requirements of their binding pockets, we turned our attention toward the more rigid class of aromatic amides (Table 2).
Starting from the benzamide derivative 7e (LogPcalc: −0.64), addition of lipophilic substructures produced the expected increase in potency (7f, LogPcalc: 0.35). Steric factors also played a role, as evidenced by the comparison of the two naphthyl derivatives of similar lipophilicity 7i and 7j. The latter was modified to test whether major SARs observed in the series of aliphatic amides were retained in the aromatic amide series. This possibility was confirmed by the lower potency of the (R) enantiomer 8j and the inactivity of the γ-lactone 11j. Next, to explore the role of electronic effects, groups with electron-withdrawing or electron-donating properties were added at conjugated positions of the phenyl or naphthyl nuclei of 7e and 7j, respectively. No clear influence of electronic effects on inhibitory potency emerged from compounds 7k–7n. On the other hand, the role of steric effects on NAAA inhibition was confirmed by the different potencies of the two biphenyl derivatives 7g and 7h (Table 2 and Figure 3A). This suggests a preference of the inhibitor-binding site of NAAA for straight lipophilic structures, which appears to be a general feature of aromatic amides, as evidenced by the potency increases observed for all the derivatives of 7e having additional lipophilic groups in the para position (7f, 7j, 7m, 7n and 7h).
Figure 3.

Characterization of NAAA inhibition by compound 7h in vitro. (A) Concentration-response curves for inhibition of NAAA by 7g (meta-biphenyl derivative, closed triangles) and 7h (para-biphenyl derivative, closed squares). (B) Preincubation time-course of 7h. (C) Michaelis-Menten analysis of 7h (vehicle – closed squares; 100 nM 7h: closed upright triangles; 200 nM 7h: closed inverted triangles). (D) Overnight dialysis of 7h (pre-dialysis: filled bars; post-dialysis: shaded bars). *** P < 0.001 vs vehicle-preincubation (n = 3–5).
Pharmacological characterization of the potent inhibitor 7h revealed that the compound inhibited NAAA with a rapid, noncompetitive and reversible mechanism (Table 3, 7h Figure 3B-D), which is consistent with the proposed acylation mechanism. We also tested whether inhibits NAAA activity and attenuates inflammation in vivo. We induced a localized inflammatory response in mice by implanting subcutaneously polyethylene sponges instilled with the proinflammatory polysaccharide carrageenan and collected infiltrating cells three days after surgery. As previously reported,23 carrageenan lowered PEA levels in leukocytes, an effect that was prevented, in a dose-dependent manner, by addition of compounds 7h and 7a to the sponges (Figure 4A). The compounds also reduced carrageenan-induced leukocyte infiltration with potencies that paralleled their ability to inhibit NAAA in vitro and normalize PEA levels in vivo (Figure 4B).
Table 3.
Effects of Compound 7h on Michaelis–Menten Constant (Km) and Maximal Rate of Reaction (Vmax) of Recombinant NAAA expressed in HEK-293 Cells.
| Compound 7h (nM) | 0 | 100 | 200 |
|---|---|---|---|
| Km (μM) | 162.2 ± 9.0 | 128.7 ± 32.8 | 156.5 ± 23.8 |
| Vmax (nmol/min/mg) | 17.9 ± 1.6 | 10.8 ± 2.5* | 6.2 ± 0.2** |
See Experimental Section for details.
P < 0.01;
P < 0.05 vs vehicle
Figure 4.
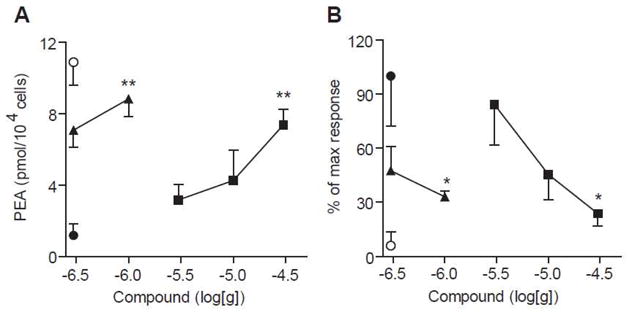
(A) Effects of compounds 7a and 7h on PEA levels in infiltrating leukocytes collected from carrageenan-instilled sponges implanted beneath the mouse skin. (B) Effects of compounds 7a and 7h on number of infiltrating leukocytes elicited by carrageenan. Open circles: vehicle/vehicle; closed circles: carrageenan/vehicle; closed triangles: carrageenan/7h; closed squares: carrageenan/7a. ** P < 0.01 vs carrageenan/vehicle; * P < 0.05 vs carrageenan/vehicle; (n = 3–5).
Conclusions
Figure 5 summarizes the SARs for the β-lactone NAAA inhibitors described in the present study. These SARs might be explained by hypothesizing a two-step mechanism of inhibition involving (i) inhibitor recognition at the substrate-binding site, and (ii) acylation of the active Cys131 by the lactone group. During the first step, the amide fragment attached to the lactone may undergo attractive polar interactions, favoring the correct positioning of the lactone warhead, as supported by the loss of potency of compounds lacking this fragment and by the stereoselectivity shown for positions 3 and 4 of the 2-oxo-3-oxetane portion. Moreover, the alkyl or aryl chain might assist inhibitor docking, by means of shape-dependent lipophilic interactions with the enzyme pocket responsible for the recognition of the fatty acyl chain of PEA. Modification of these fragments led us to discover a β-lactone-based NAAA inhibitor, compound 7h, which is potent at inhibiting NAAA in vitro and preventing carrageenan-induced inflammation after local administration in vivo. This compound may provide a chemical scaffold for the development of new tools to investigate the functional roles of NAAA and, possibly, of new anti-inflammatory and analgesic agents.
Figure 5.

Summary of SARs for β-lactone inhibitors of NAAA.
Experimental Section
(a) Chemicals, Materials, and Methods
All reagents were purchased from Sigma-Aldrich, Lancaster, NovaBiochem, or Acros in the highest quality commercially available. Solvents were RP grade unless otherwise indicated. Dry tetrahydrofuran was distilled over sodium and benzophenone. Dry dimethylformamide, dichloromethane and triethylamine were used as supplied. Petroleum ether refers to alkanes with boiling point 40–60 °C. Melting points were determined on a Büchi B-540 capillary melting point apparatus. The structures of the unknown compounds were unambiguously assessed by MS, 1H NMR and 13C NMR and IR. MS (EI) spectra were recorded with a Fisons Trio 1000 (70 eV) spectrometer; only molecular ions (M+) and base peaks are given. ESI-MS spectra were recorded with a Waters Micromass ZQ spectrometer in a positive mode using a nebulizing nitrogen gas at 400 L/min and a temperature of 250 °C, cone flow 40 mL/min, capillary 3.5 Kvolts and cone voltage 60 V; only molecular ions in positive or negative ion mode [M+H]+ or [M-H]− are given. 1H NMR and 13C NMR spectra were recorded on a Bruker AC 200 or 50, respectively, spectrometer and analyzed using the WIN-NMR software package. Chemical shifts were measured by using the central peak of the solvent. IR spectra were obtained on a Nicolet Atavar 360 FT spectrometer. Optical rotations were measured on a Perkin Elmer 241 digital polarimeter using a sodium lamp (589 nm) as the light source; concentrations are expressed in g/100 mL, and the cell length was 1 dm. Enantiomeric excess (ee) values were determined by direct HPLC analysis with a Shimadzu spectrometer: pump LC-10AS, UV detector SPD-10A, integrator C-R6A and the chiral column Chiralpak AD-H (0.46 × 25 cm) using a combination of n-hexane and isopropanol as eluent. Column chromatography purifications were performed under “flash” conditions using Merck 230–400 mesh silica gel. TLC was carried out on Merck silica gel 60 F254 plates which were visualized by exposure to ultraviolet light and by exposure to an aqueous solution of ceric ammonium molibdate. Reactions involving generation or consumption of β-lactone were conveniently followed by TLC using bromocresol green spray (0.04% in EtOH, made blue by NaOH) followed by heating of the plate to detect the β-lactone as a yellow spot against a blue background. Compound purity was assessed by elemental analysis, on a ThermoQuest FlashEA 1112 Elemental Analyzer, for C, H and N; measured percent values were within ±0.4 of theoretical ones. All the tested compounds possessed a purity >95%.
(S) -(2-Oxo-3-oxetanyl)carbamic Acid tert-Butyl Ester (3a)
White solid. Mp 120–122 °C dec (CHCl3/hexane) (lit. 119.5–120.5 dec).27 [α]20D −27 (c 0.1, MeCN) [lit. −26.7 (c 1, MeCN)].27 MS (EI) m/z: 188 (M+), 57 (100). 1H NMR and IR are according to the literature.27
(R)-(2-Oxo-3-oxetanyl)carbamic Acid tert-Butyl Ester (3b)
White solid. Mp 121–122 °C dec (CHCl3/hexane). [α]20D +26.7 (c 1, MeCN). MS (EI) m/z: 188 (M+), 57 (100). 1H NMR (CDCl3) δ: 1.47 (s, 9H), 4.41–4.51 (m, 2H), 4.85 (br s, 1H), 5.06–5.19 (m, 1H) ppm. IR (Nujol) νmax: 3357, 1844, 1717, 1683 cm−1.
(S)-2-Oxo-3-oxetanylammonium Toluene-4-sulfonate (4a).28
White solid. Mp and 1H NMR are according to the literature.28
(R)-2-Oxo-3-oxetanylammonium Toluene-4-sulfonate (4b).29
White solid. 1H NMR is according to the literature.29
General Procedure for the Synthesis of Amides 7a,b,d–f,i,j,8a,j
To a stirred mixture of 4a (0.260 g, 1 mmol) in dry CH2Cl2 (4 mL), at 0 °C and under N2 atmosphere, Et3N (0.304 g, 0.42 mL, 3 mmol) and the suitable acyl chloride 6a,b,d–f,i,j (1.5 mmol) were added. The mixture was stirred at 0 °C for 0.5 h and at room temperature for 2 h, then concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 1:1 for 7d–f,i,j,8j; 1:1 to 3:7 for 7a,8a; 4:6 for 7b) and recrystallization gave 7a,b,d–f,i,j,8a,j.
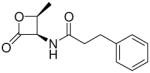
(S)-N-(2-Oxo-3-oxetanyl)-3-phenylpropionamide (7a).23
White solid. Yield: 60% (0.132 g). Mp 104–106 °C [CH3C(O)CH3/petroleum ether]. [α]20D −13 (c 0.5, MeOH). Ee > 98% [Chiral HPLC: AD-H; flow: 1 mL/min; λmax: 220 nm; eluent: n-hexane/isopropanol 85:15; tS: 8.9 min]. MS (EI) m/z: 219 (M+), 91 (100). 1H NMR (CDCl3) δ: 2.57 (t, 2H, J = 7.5 Hz), 2.99 (t, 2H, J = 7.5 Hz), 4.34 (t, 1H, J = 4.9 Hz), 4.43 (dd, 1H, J1 = 5.2 Hz, J2 = 6.5), 5.10–5.19 (m, 1H), 5.97 (br d, 1H), 7.18–7.36 (m, 5H) ppm. 13C NMR (CDCl3) δ: 31.2, 37.7, 58.4, 66.1, 126.5, 128.3, 128.7, 140.2, 168.4, 172.5 ppm. IR (Nujol) νmax: 3333, 1832, 1652 cm−1. Anal. (C12H13NO3) C, H, N.
(R)-N-(2-Oxo-3-oxetanyl)-3-phenylpropionamide (8a)
White solid. Yield: 33% (0.072 g). Mp 100–103 °C [CH3C(O)CH3/petroleum ether]. [α]20D +11 (c 1, MeOH). Ee > 98% (experimental conditions are identical with those for 7a; tR: 12.3 min). MS (EI) m/z: 219 (M+), 91 (100). 1H NMR (CDCl3) δ: 2.57 (t, 2H, J = 7.5 Hz), 2.99 (t, 2H, J = 7.5 Hz), 4.34 (t, 1H, J = 4.9 Hz), 4.43 (dd, 1H, J1 = 5.2 Hz, J2 = 6.5), 5.08–5.18 (m, 1H), 6.11 (br d, 1H), 7.17–7.34 (m, 5H) ppm. 13C NMR (CDCl3) δ: 31.2, 37.7, 58.4, 66.1, 126.5, 128.3, 128.7, 140.2, 168.4, 172.5 ppm. IR (Nujol) νmax: 3349, 1845, 1646 cm−1. Anal. (C12H13NO3) C, H, N.
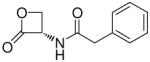
(S)-N-(2-Oxo-3-oxetanyl)-2-phenylacetamide (7b).31
White solid. Yield: 10% (0.021 g). Mp 112–114 °C dec (CHCl3/hexane) (lit. 122–123 °C).31 [α]20D −14.1 (c 0.34, MeOH). MS (EI) m/z: 205 (M+), 91 (100). 1H NMR (CDCl3) δ: 3.67 (s, 2H), 4.41–4.46 (m, 2H), 5.06–5.16 (m, 1H), 6.00 (br s, 1H), 7.25–7.43 (m, 5H) ppm. 13C NMR (CDCl3) δ: 43.1, 58.5, 65.9, 127.9, 129.3, 129.5, 133.6, 168.2, 171.3 ppm. IR (Nujol) νmax: 3321, 1845, 1814, 1651 cm−1. Anal. (C11H11NO3) C, H, N.
(S)-N-(2-Oxo-3-oxetanyl)heptanamide (7d)
White fluffy crystals. Yield: 49% (0.097 g). Mp 103–105 °C (CHCl3/n-hexane). [α]20D −16.0 (c 0.45, MeOH). MS (ESI) m/z: 200.5 [M+H]+. 1H NMR [CD3C(O)CD3] δ: 0.88 (t, 3H, J = 6.5 Hz), 1.25–1.36 (m, 6H), 1.52–1.67 (m, 2H), 2.24 (t, 2H, J = 7.4 Hz), 4.43 (d, 2H, J = 5.8 Hz), 5.21–5.31 (m, 1H), 7.86 (br s, 1H) ppm. 13C NMR [CD3C(O)CD3] δ: 13.4, 22.2, 25.1, 28.6, 31.4, 35.1, 58.5, 65.1, 169.3, 172.9 ppm. IR (Nujol) νmax: 3275, 1861, 1648, 1534 cm−1. Anal. (C10H17NO3) C, H, N.
(S)-N-(2-Oxo-3-oxetanyl)benzamide (7e)
White crystals. Yield: 41% (0.078 g). Mp 113–114 °C [CH3C(O)CH3/petroleum ether]. [α]20D −28.4 (c 0.5, MeCN). MS (EI) m/z: 191 (M+), 77 (100). 1H NMR [CD3C(O)CD3] δ: 4.52–4.63 (m, 2H), 5.47–5.57 (m, 1H), 7.46–7.60 (m, 3H), 7.91–7.97 (m, 2H), 8.63 (br s, 1H) ppm. 13C NMR [CD3C(O)CD3] δ: 58.8, 65.1, 127.3, 128.5, 131.9, 133.2, 166.7, 169.2 ppm. IR (Nujol) νmax: 3272, 1849, 1827, 1647 cm−1. Anal. (C10H9NO3) C, H, N.
(S)-4-Ethyl-N-(2-oxo-3-oxetanyl)benzamide (7f)
White fluffy crystals. Yield: 40% (0.088 g). Mp 118–119 °C [CH3C(O)CH3/petroleum ether]. [α]20D −9.5 (c 0.6, MeOH). MS (EI) m/z: 219 (M+), 133 (100). 1H NMR [CD3C(O)CD3] δ: 1.23 (t, 3H, J = 7.5 Hz), 2.70 (q, 2H, J = 7.5 Hz), 4.50–4.63 (m, 2H), 5.45–5.55 (m, 1H), 7.31–7.36 (m, 2H), 7.82–7.89 (m, 2H), 8.54 (br s, 1H) ppm. 13C NMR [CD3C(O)CD3] δ: 14.8, 28.4, 58.8, 65.1, 127.4, 128.0, 130.6, 148.6, 166.6, 169.3 ppm. IR (Nujol) νmax: 3298, 1832, 1642 cm−1. Anal. (C12H13NO3) C, H, N.
(S)-N-(2-Oxo-3-oxetanyl)-1-naphthamide (7i)
Off-white crystals. Yield: 67% (0.161 g). Mp 124–126 °C [CH3C(O)CH3/petroleum ether]. [α]20D −32.2 (c 0.45, MeCN). MS (EI) m/z: 241 (M+), 127 (100). 1H NMR [CD3C(O)CD3] δ: 4.63 (dd, 1H, J = 4.6, 6.7 Hz), 4.74 (t, 1H, J = 4.7 Hz), 5.56–5.65 (m, 1H), 7.51–7.62 (m, 3H), 7.78 (dd, 1H, J = 1.3, 7.1 Hz), 7.95–8.08 (m, 2H), 8.39–8.44 (m, 1H), 8.55 (br s, 1H) ppm. 13C NMR [CD3C(O)CD3] δ: 58.9, 65.2, 124.7, 125.5, 125.6, 126.4, 127.0, 128.3, 130.3, 130.9, 132.9, 133.8, 169.1, 169.2 ppm. IR (Nujol) νmax: 3291, 1842, 1637 cm−1. Anal. (C14H11NO3) C, H, N.
(S)-N-(2-Oxo-3-oxetanyl)-2-naphthamide (7j)
White solid. Yield: 47% (0.113 g). Mp 193–195 °C, decomposition and shrinkage starting from 180 °C, sealed capillary tube [CH3C(O)CH3/petroleum ether]. [α]20D −17 (c 0.1, MeCN). MS (ESI) m/z: 240.5 [M-H]+. 1H NMR [CD3C(O)CD3] δ: 4.56–4.68 (m, 2H), 5.54–5.64 (m, 1H), 7.56–7.69 (m, 2H), 7.96–8.06 (m, 4H), 8.52 (d, 1H, J = 1 Hz), 8.80 (br d, 1H) ppm. 13C NMR [CD3C(O)CD3] δ: 58.8, 65.1, 123.8, 126.9, 127.7, 127.9, 128.3, 129.0, 130.5, 132.6, 135.0, 166.8, 169.3 ppm. IR (Nujol) νmax: 3270, 1825, 1647 cm−1. Anal. (C14H11NO3) C, H, N.
(R)-N-(2-Oxo-3-oxetanyl)-2-naphthamide (8j)
White solid. Yield: 20% (0.048 g). Mp 195 °C, decomposition and shrinkage starting from 175 °C [CH3C(O)CH3/petroleum ether]. [α]20D +17 (c 0.1, MeCN). MS (EI) m/z: 241 (M+), 141 (100). 1H NMR [CD3C(O)CD3] δ: 4.56–4.68 (m, 2H), 5.54–5.64 (m, 1H), 7.57–7.68 (m, 2H), 7.96–8.06 (m, 4H), 8.52 (d, 1H, J = 1 Hz), 8.80 (br d, 1H) ppm. 13C NMR [CD3C(O)CD3] δ: 58.8, 65.1, 123.8, 126.9, 127.7, 127.9, 128.3, 129.0, 130.5, 132.6, 135.0, 166.8, 169.3 ppm. IR (Nujol) νmax: 3262, 1824, 1645 cm−1. Anal. (C14H11NO3) C, H, N.
General Procedure for the Synthesis of Amides 7c,g,h,k–n
To a stirred solution of the suitable acid 9c,g,h,k–n (2 mmol) in dry CH2Cl2 (5 mL), at 0 °C and under N2 atmosphere, (COCl)2 (0.381 g, 0.26 mL, 3 mmol) and a catalytic amount of DMF were added. After gas evolution the mixture was stirred at room temperature for 3 h (16 h, 7l; 6 h, 7m) and concentrated; the resulting crude acid chloride was used as such in the next step. A mixture of the acid chloride (2 mmol), 4a (0.337 g, 1.3 mmol), Et3N (6.7 mmol, 7c,g; 8 mmol, 7h,k; 5.5 mmol, 7n; 3.6 mmol, 7l,m) (a catalytic amount of 4-dimethylaminopyridine in the case of 7c,g is necessary) in dry CH2Cl2 (8 mL, 7c,g,k; 4 mL, 7l,m) or dry THF (20 mL, 7h; 8 mL, 7n) was stirred at 0 °C for 0.5 h and at room temperature for 3 h under N2 atmosphere, then concentrated. In the case of 7m further amounts of 4a (0.168 g, 0.65 mmol) and Et3N (0.101 g, 0.14 mL, 1 mmol) were added and the mixture was stirred for further 3 h, then concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 1:1 for 7c,g,h,n; CH2Cl2/EtOAc 9:1 for 7k-m) and recrystallization gave 7c,g,h,k–n.
(S)-N-(2-Oxo-3-oxetanyl)-7-phenylheptanamide (7c)
White fluffy solid. Yield: 39% (0.139 g). Mp 98–99 °C [CH3C(O)CH3/petroleum ether]. [α]20D −16.2 (c 0.5, MeOH). MS (ESI) m/z: 274.6 [M-H]+. 1H NMR (CDCl3) δ: 1.32–1.39 (m, 4H), 1.55–1.73 (m, 4H), 2.25 (t, 2H, J = 7.5 Hz), 2.61 (t, 2H, J = 7.6 Hz), 4.40–4.50 (m, 2H), 5.15–5.24 (m, 1H), 6.14 (br d, 1H, J = 7.5 Hz), 7.14–7.33 (m, 5H) ppm. 13C NMR (CDCl3) δ: 25.1, 28.8, 29.0, 31.2, 35.8, 58.4, 66.3, 125.6, 128.2, 128.4, 142.6, 168.5, 173.3 ppm. IR (Nujol) νmax: 3331, 1853, 1833, 1649 cm−1. Anal. (C16H21NO3) C, H, N.
(S)-N-(2-Oxo-3-oxetanyl)biphenyl-3-carboxamide (7g)
White fluffy solid. Yield: 68% (0.236 g). Mp 116–117 °C [CH3C(O)CH3/petroleum ether]. [α]20D −20 (c 0.55, MeCN). MS (ESI) m/z: 268.0 [M+H]+. 1H NMR [CD3C(O)CD3] δ: 4.54–4.66 (m, 2H), 5.51–5.61 (m, 1H), 7.37–7.74 (m, 6H), 7.86–7.96 (m, 2H), 8.20–8.22 (m, 1H), 8.75 (br s, 1H) ppm. 13C NMR [CD3C(O)CD3] δ: 58.9, 65.1, 125.7, 126.4, 126.9, 127.8, 129.0, 129.2, 130.3, 133.8, 140.0, 141.3, 166.6, 169.2 ppm. IR (Nujol) νmax: 3314, 1856, 1824, 1638 cm−1. Anal. (C16H13NO3) C, H, N.
(S)-N-(2-Oxo-3-oxetanyl)biphenyl-4-carboxamide (7h)
Off-white solid. Yield: 46% (0.160 g). Mp 218–220 °C, decomposition with colour change and shrinkage starting from 146 °C, sealed capillary tube [CH3C(O)CH3/petroleum ether]. [α]20D −20 (c 0.55, MeCN). MS (EI) m/z: 267 (M+), 167 (100). 1H NMR [CD3C(O)CD3] δ: 4.53–4.65 (m, 2H), 5.50–5.59 (m, 1H), 7.38–7.55 (m, 3H), 7.70–7.84 (m, 4H), 8.00–8.07 (m, 2H), 8.68 (br d, 1H, J = 7 Hz) ppm. 13C NMR [CD3C(O)CD3] δ: 58.8, 65.1, 126.9, 127.0, 128.0, 128.1, 129.0, 131.9, 139.7, 144.4, 166.4, 169.2 ppm. IR (Nujol) νmax: 3270, 1827, 1641 cm−1. Anal. (C16H13NO3) C, H, N.
(S)-6-(2-Oxo-3-oxetanylcarbamoyl)naphthalene-2-carboxylic Acid Ethyl Ester (7k)
White solid. Yield: 42% (0.171 g). Mp 205–207 °C [CH3C(O)CH3/petroleum ether]. [α]20D −19 [c 0.1, (CH3C(O)CH3)]. MS (ESI) m/z: 313.8 [M+H]+. 1H NMR (DMSO-d6) δ: 1.37 (t, 3H, J = 7.1 Hz), 4.39 (q, 2H, J = 7.1 Hz), 4.48–4.53 (m, 2H), 5.43–5.52 (m, 1H), 7.97–8.08 (m, 2H), 8.17 (d, 1H, J = 8.9 Hz), 8.27 (d, 1H, J = 8.8 Hz), 8.55 (s, 1H), 8.68 (s, 1H), 9.57 (d, 1H, J = 7.4 Hz) ppm. 13C NMR (DMSO-d6) δ: 14.7, 58.7, 61.5, 65.6, 125.2, 126.2, 128.3, 129.4, 130.1, 130.3, 130.6, 132.7, 134.0, 134.6, 166.0, 166.8, 170.4 ppm. IR (neat) νmax: 3408, 1831, 1799, 1710, 1654 cm−1. Anal. (C17H15NO5) C, H, N.
(S)-6-Methoxy-N-(2-oxo-3-oxetanyl)naphthalene-2-carboxamide (7l)
White solid. Yield: 10% (0.035 g). Mp 205 °C [CH3C(O)CH3/petroleum ether]. [α]20D −17 [c 0.08, CH3C(O)CH3]. MS (EI) m/z: 271 (M+), 171 (100). 1H NMR [CD3C(O)CD3] δ: 3.95 (s, 3H), 4.54–4.67 (m, 2H), 5.51–5.61 (m, 1H), 7.23 (dd, 1H, J = 9.0, 2.5 Hz), 7.38 (d, 1H, J = 2.5 Hz), 7.87–7.99 (m, 3H), 8.43 (s, 1H), 8.72 (br d, 1H) 13C NMR [CD3C(O)CD3] δ: 54.9, 58.9, 65.1, 105.7, 119.7, 124.4, 127.1, 127.8, 127.9, 128.1, 130.5, 136.7, 159.4, 169.4 ppm. IR (Nujol) νmax: 3419, 1825, 1720, 1627 cm−1. Anal. (C15H13NO4) C, H, N.
(S)-(2-Oxo-3-oxetanylcarbamoyl)terephthalamic Acid Phenyl Ester (7m)
White solid. Yield: 13% (0.053 g). Mp 222–226 °C dec from 150 °C (CHCl3/n-hexane). [α]20D −15.5 [c 0.09, CH3C(O)CH3]. MS (EI) m/z: 311 (M+), 65 (100). 1H NMR [CD3C(O)CD3] δ: 4.56–4.67 (m, 2H), 5.54–5.63 (m, 1H), 7.29–7.37 (m, 3H), 7.45–7.54 (m, 2H), 8.05–8.15 (m, 2H), 8.27–8.33 (m, 2H), 8.87 (br d, 1H) ppm. 13C NMR [CD3C(O)CD3] δ: 58.8, 65.0, 121.8, 126.0, 127.7, 129.5, 130.1, 132.6, 137.6, 151.1, 163.9, 165.8, 168.9 ppm. IR (Nujol) νmax: 3304, 1827, 1731, 1647 cm−1. Anal. (C17H13NO5) C, H, N.
(S)-4-(Benzyloxy)-N-(2-oxo-3-oxetanyl)benzamide (7n)
White solid. Yield: 61% (0.235 g). Mp 205–208 °C, decomposition with colour change and shrinkage starting from 170 °C, sealed capillary tube (CH2Cl2/hexane). [α]20D −17.6 (c 0.5, MeCN). MS (EI) m/z: 297 (M+), 91 (100). 1H NMR (DMSO-d6) δ: 4.41–4.46 (m, 2H), 5.17 (s, 2H), 5.29–5.38 (m, 1H), 7.09–7.14 (m, 2H), 7.32–7.48 (m, 5H), 7.80–7.87 (m, 2H), 9.15 (d, 1H, J = 7.5 Hz) ppm. 13C NMR (DMSO-d6) δ: 58.6, 65.7, 69.9, 115.0, 125.4, 128.3, 128.4, 128.9, 129.8, 137.0, 161.7, 166.4, 170.7 ppm. IR (Nujol) νmax: 3380, 1825, 1637 cm−1. Anal. (C17H15NO4) C, H, N.
Synthesis of (S)-3-Hydroxy-2-(3-Phenylpropionylamino)propionic Acid (10).33
To a stirred solution of L-serine (1.050 g, 10 mmol) in aqueous 2 N NaOH (10 mL), at 0 °C, 3-phenylpropionyl chloride (6a) (1.855 g, 1.67 mL, 11 mmol) and aqueous 2 N NaOH (10 mL) were alternatively added in 8 portions. The mixture was stirred at 0 °C for further 4 h, then acidified with 5 N HCl, filtered, and the filtrate extracted with EtOAc. The combined organic layers were dried (Na2SO4) and concentrated. Purification of the residue by recrystallization gave 10 as off-white crystals. Yield: 40% (0.950 g). Mp 127 °C (EtOAc). [α]20D +6.7 (c 0.36, MeOH). MS (EI) m/z: 237 (M+), 91 (100). 1H NMR [CD3C(O)CD3] δ: 2.54–2.62 (m, 2H), 2.88–2.97 (m, 2H), 3.77 (dd, 1H, J1 = 11.0 Hz, J2 = 4.2 Hz), 3.91 (dd, 1H, J1 = 11.0 Hz, J2 = 4.5 Hz), 4.51 (m, 1H), 7.16–7.31 (m, 6H) ppm. 13C NMR [CD3C(O)CD3] δ: 31.3, 37.3, 54.5, 62.2, 125.8, 128.2, 128.3, 141.6, 171.2, 171.7 ppm. IR (Nujol) νmax: 3303, 1730, 1715, 1647, 1625 cm−1. Anal. (C12H15NO4) C, H, N.
General Procedure for the Synthesis of (S)-N-(2-Oxotetrahydrofuran-3-yl)amides (11a,j)
To a stirred suspension of (S)-3-aminodihydrofuran-2-one hydrobromide (0.501 g, 2.75 mmol) in dry CH2Cl2 (7 mL), at 0 °C and under N2 atmosphere, Et3N (0.972 g, 1.35 mL, 9.68 mmol) was added; the salt was dissolved at first, then a white precipitate was produced. The opportune acid chloride 6a,j (5.38 mmol) was added dropwise to the mixture, which was stirred at 0 °C for 0.5 h and at room temperature for 3 h, added of CH3C(O)CH3 and filtered, then the filtrate was concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 7:3 to EtOAc) and recrystallization gave 11a,j.
(S)-N-(2-Oxotetrahydrofuran-3-yl)-3-phenylpropionamide (11a).34
White solid. Yield: 86% (0.550 g). Mp 149–150 °C [CH3C(O)CH3/petroleum ether] (lit. 150.0–150.8)56. [α]20D −23.6 (c 0.55, MeCN). MS (EI) m/z: 233 (M+), 91 (100). 1H NMR [CD3C(O)CD3] δ: 2.10–2.31 (m, 1H), 2.47–2.62 (m, 3H), 2.82–2.95 (m, 2H), 4.21–4.44 (m, 2H), 4.56–4.70 (m, 1H), 7.16–7.28 (m, 5H), 7.52 (br s, 1H) ppm. 13C NMR [CD3C(O)CD3] δ: 28.9, 31.2, 37.2, 48.3, 65.1, 125.9, 128.3, 141.4, 171.5, 174.7 ppm. IR (Nujol) νmax: 3294, 1776, 1644 cm−1. Anal. (C13H15NO3) C, H, N.
(S)-N-(2-Oxotetrahydrofuran-3-yl)-2-naphthamide (11j)
White fluffy needles. Yield: 87% (0.610 g). Mp 221–222 °C [CH3C(O)CH3/petroleum ether]. [α]20D −29.2 (c 0.4, MeCN). MS (EI) m/z: 255 (M+), 155 (100). 1H NMR [CD3C(O)CD3] δ: 2.40–2.78 (m, 2H), 4.32–4.56 (m, 2H), 4.89–5.03 (m, 1H), 7.56–7.66 (m, 2H), 7.96–8.04 (m, 4H), 8.37 (br s, 1H), 8.50 (s, 1H) ppm. 13C NMR [CD3C(O)CD3] δ: 28.7, 48.8, 65.2, 123.9, 126.7, 127.6, 127.7, 128.1, 128.9, 131.4, 132.7, 134.9, 166.4, 174.7 ppm. IR (Nujol) νmax: 3357, 1760, 1654 cm−1. Anal. (C15H13NO3) C, H, N.
Synthesis of N-Cyclobutyl-3-phenylpropionamide (12).23
To a stirred solution of c-C4H7NH2 (0.213 g, 0.26 mL, 3 mmol) and Et3N (0.404 g, 0.56 mL, 4 mmol) in dry CH2Cl2 (15 mL), at 0 °C and under N2 atmosphere, 6a (0.523 g, 0.46 mL, 3.1 mmol) was added dropwise. The solution was stirred at room temperature for 3 h and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 7:3) and recrystallization gave 12 as white needles. Yield: 82% (0.500 g). Mp 109–110 °C (Et2O/petroleum ether). MS (EI) m/z: 204 (M+), 174 (100). 1H NMR (CDCl3) δ: 1.68–1.85 (m, 4H), 2.18–2.36 (m, 2H), 2.42 (t, 2H, J = 8.0 Hz), 2.96 (t, 2H, J = 8.0 Hz), 4.28–4.48 (m, 1H), 5.50 (br s, 1H), 7.16–7.33 (m, 5H) ppm. 13C NMR (CDCl3) δ: 15.0, 31.2, 31.8, 38.5, 44.6, 126.2, 128.4, 128.5, 140.9, 171.2 ppm. IR (Nujol) νmax: 3297, 1642, 1544 cm−1. Anal. (C13H17NO) C, H, N.
Synthesis of 3-Phenylpropionamide (14).35
To a stirred solution of 3-phenylpropionitrile (13) (1.310 g, 1.3 mL, 10 mmol) in aqueous 60% tert-butanol (14.5 mL), aqueous 1 N NaOH (10.8 mL) and aqueous 30% H2O2 (1.8 mL) were added. The mixture was stirred at room temperature for 3 h, concentrated, acidified with 1 N HCl and extracted with CH2Cl2. The combined organic layers were washed with brine, dried (Na2SO4) and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 1:1 to EtOAc) and trituration with hexane gave 14 as a white solid. Yield: 50% (0.750 g). Mp, MS, 1H NMR and IR are according to the literature.57
Synthesis of N-(2-Oxocyclobutyl)-3-phenylpropionamide (15).23
To a stirred solution of 14 (0.067 g, 0.45 mmol) in dry THF (5 mL) and ethereal 2 N HCl (5 mL), at 0 °C and under N2 atmosphere, 1,2-bis(trimethylsilanyloxy)cyclobutene (0.099 g, 0.11 mL, 0.43 mmol) was added. The solution was stirred at reflux for 3 h, then concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 6:4 to 3:7) and trituration of the colorless oil previously obtained gave 15 as an off-white solid. Yield: 70% (0.065 g). Mp 70–73 °C (hexane). MS (EI) m/z: 217 (M+), 91 (100). 1H NMR (CDCl3) δ: 1.87–2.06 (m, 1H), 2.32–2.58 (m, 3H), 2.90–3.00 (m, 4H), 4.86–4.99 (m, 1H), 5.91 (br d, 1H, J = 6.4 Hz), 7.18–7.35 (m, 5H) ppm. 13C NMR (CDCl3) δ: 19.7, 31.4, 37.8, 42.1, 64.1, 126.3, 128.3, 128.6, 140.5, 171.8, 205.3 ppm. IR (Nujol) νmax: 3287, 1784, 1646 cm−1. Anal. (C13H15NO2) C, H, N.
Synthesis of (2S,3R)-2-tert-Butoxycarbonylamino-3-hydroxybutyric Acid (17a).37
To a stirred solution of L-threonine (16a) (1.001 g, 8.4 mmol) and NaHCO3 (1.092 g, 13 mmol) in H2O (17 mL), MeOH (17 mL) and Boc2O (2.684 g, 12.3 mmol) were added. The mixture was stirred at room temperature for 36 h, concentrated, acidified with 1% HCl until pH 2 and extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4) and concentrated to give 17a as a colorless oil. Yield: 87% (1.600 g). MS (EI) m/z: 220 (M+), 101 (100). 1H NMR (CDCl3) δ: 1.25 (d, 3H, J = 6.1 Hz), 1.44 (s, 9H), 4.25–4.41 (m, 2H), 5.75 (d, 1H, J = 9.2 Hz), 7.35 (br s, 2H) ppm.
Synthesis of (2R,3S)-2-tert-Butoxycarbonylamino-3-hydroxybutyric Acid (17b).38
The same protocol applied to D-threonine (16b) gave 17b as a colorless oil. Yield: 95% (1.748 g). MS (EI) and 1H NMR data are identical to those of 17a.
Synthesis of (2R,3S)-2-Methyl-4-oxo-3-oxetanylcarbamic Acid tert-Butyl Ester (18a).40
To a stirred suspension of N-Boc-L-threonine (17a) (1.600 g, 7.3 mmol) in dry CH2Cl2 (150 mL), at 0 °C and under N2 atmosphere, Et3N (2.21 g, 3.05 mL, 21.9 mmol) and BOP reagent (3.892 g, 8.8 mmol) were added. The mixture was stirred at room temperature for 3 h, then concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 1:1) and recrystallization gave 18a as an off-white solid. Yield: 63% (0.930 g). Mp 141–143 °C (CHCl3/hexane) (lit. 141–142 °C)58. [α]D20 +25.4 (c 0.7, MeOH) [lit. +20.4 (c 1.16, CHCl3)].58 MS (EI) m/z: 202 (M+1), 57 (100). 1H NMR (CDCl3) δ: 1.45–1.48 (m, 12H), 4.80–4.93 (m, 1H), 5.27 (br d, 1H), 5.40–5.47 (m, 1H) ppm. IR (nujol) νmax: 3353, 1830, 1701 cm−1.
Synthesis of (2S,3R)-2-Methyl-4-oxo-3-oxetanylcarbamic Acid tert-Butyl Ester (18b)
The same protocol applied to 17b gave 18b as a white solid. Yield 70% (0.510 g). Mp 140–141 °C (CHCl3/hexane). [α]20D −26.8 (c 0.6, MeOH). MS (EI) m/z: 202 (M+1), 57 (100). 1H NMR (CDCl3) δ: 1.45–1.48 (m, 12H), 4.80–4.93 (m, 1H), 5.27 (br d, 1H), 5.40–5.47 (m, 1H) ppm. IR (nujol) νmax: 3352, 1829, 1713 cm−1.
Synthesis of (3S,4R)-2-Methyl-4-oxo-3-oxetanylammonium Toluene-4-sulfonate (19a).41
To a stirred mixture of 18a (0.402 g, 2 mmol) and dry p-TsOH (0.406 g, 2.13 mmol), at 0 °C and under N2 atmosphere, CF3COOH (4.5 mL) was added dropwise in the course of 10 min. The solution was stirred at 0 °C for 0.25 h and concentrated at a temperature below 30 °C. The oily residue was kept under vacuum for 1 h and added of dry Et2O. Purification of the resulting solid by repeated trituration with dry Et2O gave 19a as an off-white solid, which starts to decompose from 120 °C [lit. 120 °C dec (EtOAc/hexane)].41 Yield: 92% (0.500 g). 1H NMR and IR are according to the literature.41
Synthesis of (3R,4S)-2-Methyl-4-oxo-3-oxetanylammonium Toluene-4-sulfonate (19b)
The same protocol applied to 18b afforded 19b as an off-white solid which starts to decompose from 120 °C. Yield: 77% (0.420 g). 1H NMR (DMSO-d6) δ: 1.51 (d, 3H, J = 6.4 Hz), 2.28 (s, 3H), 4.97 (m, 1H), 5.16 (d, 1H, J = 6.2 Hz), 7.12 (d, 2H, J = 7.9 Hz), 7.47 (d, 2H, J = 8.2 Hz), 8.90 (br s, 3H) ppm. IR (Nujol) νmax: 3392, 3162, 1838, 1717 cm−1.
Synthesis of (2R,3S)-N-(2-Methyl-4-oxo-3-oxetanyl)-3-phenylpropionamide (20a)
To a stirred mixture of 19a (0.402 g, 1.47 mmol) in dry CH2Cl2 (8 mL), at 0 °C and under N2 atmosphere, Et3N (0.591 g, 0.81 mL, 5.84 mmol) was added dropwise and, successively, 3-phenylpropionyl chloride (6a) (0.372 g, 0.33 mL, 2.21 mmol). The mixture was reacted at 0 °C for 0.5 h and at room temperature for 3 h, then concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 7:3 to EtOAc) and recrystallization gave 20a as a white solid. Yield: 53% (0.180 g). Mp 115–116 °C (CHCl3/n-hexane). [α]20D +32.8 (c 0.5, MeOH). MS (EI) m/z: 233 (M+, 100). 1H NMR (CDCl3) δ: 1.23 (d, 3H, J = 6.3 Hz), 2.55–2.64 (m, 2H), 2.99 (t, 2H, J = 7.5 Hz), 4.78–4.90 (m, 1H), 5.59 (dd, 1H, J = 7.9, 6.0 Hz), 6.36 (br d, 1H, J = 7.3 Hz), 7.18–7.30 (m, 5H) ppm. 13C NMR (CDCl3) δ: 14.8, 31.3, 37.6, 58.7, 74.9, 126.6, 128.3, 128.7, 140.0, 169.3, 172.1 ppm. IR (Nujol) νmax: 3282, 1827, 1656 cm−1. Anal. (C13H15NO3) C, H, N.
Synthesis of (2S,3R)-N-(2-Methyl-4-oxo-3-oxetanyl)-3-phenylpropionamide (20b)
The same protocol procedure applied to 19b gave 20b as a white fluffy solid. Yield: 58% (0.199 g). Mp 117–119 °C (CHCl3/n-hexane). [α]20D −33.1 (c 0.55, MeOH). MS (EI) m/z: 233 (M+), 91 (100). 1H NMR (CDCl3) δ: 1.23 (d, 3H, J = 6.3 Hz), 2.55–2.64 (m, 2H), 2.99 (t, 2H, J = 7.5 Hz), 4.78–4.90 (m, 1H), 5.59 (dd, 1H, J = 7.9, 6.0 Hz), 6.16 (br d, 1H, J = 7.3 Hz), 7.19–7.27 (m, 5H) ppm. 13C NMR (CDCl3) δ: 14.8, 31.3, 37.6, 58.7, 74.8, 126.5, 128.3, 128.7, 140.0, 169.1, 172.0 ppm. IR (Nujol) νmax: 3282, 1827, 1655 cm−1. Anal. (C13H15NO3) C, H, N.
Synthesis of 2-(Bromomethyl)-6-phenylhexanoic Acid (22)
To a stirred solution of 2-methylene-6-phenylhexanoic acid (21) (0.239 g, 1.17 mmol) in CH2Cl2 (3 mL), at 0 °C, 5.7 M HBr in CH3COOH (0.6 mL, 3.42 mmol) was added dropwise. The mixture was stirred at 0 °C for 3 h and at room temperature for 36 h. A further amount of 5.7 M HBr in CH3COOH (0.2 mL, 1.14 mmol) was added and the mixture stirred for further 16 h. The mixture was poured onto ice H2O (250 mL) and extracted with Et2O. The combined organic layers were washed with brine, dried (Na2SO4) and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 8:2 to 1:1 to EtOAc) gave 22 as a colorless oil. Yield: 48% (0.160 g). MS (EI) m/z: 285/287 (M+), 91 (100). 1H NMR (CDCl3) δ: 1.38–1.49 (m, 2H), 1.64–1.76 (m, 4H), 2.64 (t, 2H, J = 7.8 Hz), 2.78–2.92 (m, 1H), 3.45–3.63 (m, 2H), 7.16–7.32 (m, 5H) ppm. IR (neat) νmax: 3026, 2931, 2858, 1709 cm−1.
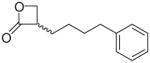
Synthesis of rac-3-(4-Phenylbutyl)oxetan-2-one (23)
To a stirred suspension of 22 (0.148 g, 0.52 mmol) in aqueous 1 N NaOH (0.60 mL, 0.6 mmol), CHCl3 (2 mL) was added. The mixture was stirred vigorously at room temperature for 10 min. H2O (1 mL) and 1 N NaOH (0.50 mL) were added, the organic layer was separated and CHCl3 (5 mL) was added to the aqueous layer. The mixture was stirred for 1 h and the organic layer separated. Further amount of CHCl3 (5 mL) was added to the aqueous layer, the mixture was stirred for 1 h and organic layer separated. The combined organic layers were dried (Na2SO4) and concentrated. Purification of the residue by column chromatography (cyclohexane/EtOAc 8:2) gave 23 as a light brown oil. Yield: 29% (0.030 g). MS (EI) m/z: 204 (M+), 91(100). 1H NMR (CDCl3) δ: 1.38–2.01 (m, 6H), 2.64 (t, 2H, J = 7.8 Hz), 3.64–3.77 (m, 1H), 4.00 (t, 1H, J = 5.0 Hz), 4.36 (dd, 1H, J1 = 5.1 Hz, J2 = 6.3 Hz), 7.15–7.29 (m, 5H) ppm. 13C NMR (CDCl3) δ: 26.4, 28.0, 31.0, 35.6, 52.0, 65.0, 125.9, 128.3, 128.4, 142.0, 171.7 ppm. IR (neat) νmax: 3026, 2931, 2859, 1821 cm−1. Anal. (C13H16O2) C, H.
Synthesis of Biphenyl-3-carboxylic Acid (9g).44
To a stirred solution of biphenyl-3-carbaldehyde (0.601 g, 3.3 mmol), the 1H NMR of which is according to the literature,59 in CH3C(O)CH3 (46 mL), KMnO4 (1.012 g, 6.4 mmol) in H2O (32 mL) was added. The mixture was stirred at room temperature for 5 h, concentrated, added of CH2Cl2/MeOH 9:1, filtered, and the filtrate concentrated. Purification of the residue by column chromatography (CH2Cl2/MeOH 9:1) gave 9g as a white solid. Yield: 60% (0.380 g). Mp, MS and 1H NMR data are according to the literature.60
Synthesis of Naphthalene-2,6-dicarboxylic Acid Monoethyl Ester (9k).45
A suspension of naphthalene-2,6-dicarboxylic acid (26) (1.500 g, 6.94 mmol), Et3N (1.404 g, 1.93 mL, 13.88 mmol) and EtBr (0.907 g, 0.62 mL, 8.33 mmol) in dry DMF (10 mL), under N2 atmosphere, was stirred at 80 °C for 4 h. The mixture was cooled to room temperature, diluted with H2O, then acidified with 2 N HCl; the precipitate was filtered and the solid was washed with H2O, and dried. Purification of the solid by column chromatography (CH2Cl2/MeOH 9:1) gave 9k as a white solid. Yield: 32% (0.540 g). Mp 233–237 °C. MS (EI) m/z: 244 (M+), 115 (100). 1H NMR [DMSO-d6] δ: 1.37 (t, 3H, J = 7.1 Hz), 4.39 (q, 2H, J = 7.1 Hz), 8.01–8.07 (m, 2H), 8.23 (d, 2H, J = 8.5 Hz), 8.66 (d, 2H, J = 4.7 Hz), 13.11 (br s, 1H) ppm. IR (Nujol) νmax: 3389, 1705, 1680 cm−1.
Synthesis of Terephthalic Acid Monophenyl Ester (9m).46
To a stirred solution of terephthaloyl dichloride (27) (3.490 g, 17.2 mmol) in MeCN (60 mL), phenol (1.619 g, 17.2 mmol) and Et3N (3.491 g, 4.81 mL, 34.5 mmol) were added. The mixture was stirred at room temperature for 4 h, then an aqueous solution of 2 N Na2CO3 (80 mL) was added and the mixture was stirred again for 15 h, then acidified with 2 N HCl and filtered. Purification of the solid by column chromatography (CH2Cl2/MeOH 9:1 to CH2Cl2/MeOH/CH3COOH 9:1.9:0.1) and recrystallization gave 9m as a white solid. Yield: 16% (0.666 g). Mp 230–235 °C (EtOH) [the sample was heated so as to raise its temperature of 20 °C/min; when heated more slowly (3 °C/min), it tends to decompose with colour and volume change, instead of melting] [lit. 238–240 (H2O/EtOH)]61. MS (EI) m/z: 242 (M+), 65 (100). IR (Nujol) νmax: 1739, 1686 cm−1. 1H NMR is according to the literature.62
Synthesis of 4-Benzyloxybenzoic acid (9n).49
MS (EI) m/z: 228 (M+, 100). Pf, 1H NMR and IR are according to the literature.50
(b) Pharmacology
NAAA assay
Recombinant NAAA, expressed as described below, was incubated at 37 °C for 30 min in 0.2 mL of sodium hydrogen phosphate buffer (50 mM, pH 5.0) containing 0.1% Triton X-100, 3 mM dithiothreitol (DTT) and 50 μM heptadecenoylethanolamide as substrate. The reaction was terminated by the addition of 0.2 mL cold methanol containing 1 nmol of heptadecanoic acid (NuChek Prep, Elysian, MN). Samples were analyzed by liquid chromatography/mass spectrometry (LC/MS). Heptadecenoic and heptadecanoic acids were eluted on an XDB Eclipse C18 column isocratically at 2.2 mL/min for 1 min with a solvent mixture of 95% methanol and 5% water, both containing 0.25% acetic acid and 5 mM ammonium acetate. The column temperature was 50 °C. ESI was in the negative mode, capillary voltage was 4 kV, and fragmentor voltage was 100 V. N2 was used as drying gas at a flow rate of 13 L/min and a temperature of 350 °C. Nebulizer pressure was set at 60 psi. We monitored [M-H]− in the selected-ion monitoring mode. Calibration curves were generated using commercial heptadecenoic acid (Nu-Chek Prep, m/z = 267).
Lipid extractions
Lipids were extracted using a chloroform/methanol mixture (2:1, v/v, 3 mL) containing internal standards. The organic phases were collected, dried under N2 and dissolved in methanol/chloroform (3:1, v/v) for LC/MS analyses.
LC/MS analysis
We used an Agilent 1100-LC system coupled to a 1946A-MS detector equipped with an ESI interface (Agilent Technologies, Inc., Palo Alto, CA). PEA was separated on an XDB Eclipse C18 column (50′4.6 mm i.d., 1.8 mm, Zorbax, Agilent Technologies) with a gradient of methanol in water (from 85% to 90% methanol in 2.0 min and 90% to 100% in 3.0 min) at a flow rate of 1.5 ml/min. Column temperature was kept at 40 °C. MS detection was in the positive ionization mode, capillary voltage was 3 kV and fragmentor voltage was 120 V. N2 was used as drying gas at a flow rate of 13 L/min and a temperature of 350 °C. Nebulizer pressure was set at 60 psi.
Expression of recombinant NAAA
We amplified the full-length coding sequence of rat NAAA by polymerase chain reaction (PCR) using High Fidelity PCR Master (Roche, Indianapolis, IN) and rat brain cDNAs as templates. We designed two primers using sequences obtained from the NCBI database: 5′rNAAA (5′-ATGGGGACCCCAGCCATCCGG-3′) and 3′rNAAA (5′-TCAGCTTGGGTTTCTGATCATGGT-3′). The PCR product was subcloned into a pCMV-Flag vector (Stratagene, La Jolla, CA) by HindIII and XhoI (Roche, Indianapolis, IN) sites to construct a mammalian expression vector encoding Flag-tagged rNAAA. We transfected HEK293 cells with pCMV-Flag-rNAAA using SuperFect reagent (Qiagen, Valencia, CA) and screened with G418 (0.3 mg/mL). We harvested and washed cells stably expressing rNAAA, sonicated them in 20 mM Tris-HCl (pH 7.5) with 0.32 M sucrose, and centrifuged them at 800′g for 15 min at 4 °C. The supernatant was ultra-centrifuged at 12,000′g for 30 min at 4 °C. The pellet was suspended in phosphate-buffered saline (PBS) and subjected to 2 freeze-thaw cycles at −80 °C. The suspension was centrifuged at 105,000′g for 1 h at 4 °C, and the supernatant containing rNAAA was kept at −80 °C until use.
Carrageenan-induced inflammation
Sterile polyethylene sponges (1 cm3) were implanted under the dorsal skin of mice. Carrageenan (1.0% in 90 μL sterile saline/sponge), and drugs or vehicle (DMSO, 10 μL/sponge) were instilled into the sponges, wounds were sutured and mice were allowed to recover. Seventy two hours following sponge implantation (2/mouse), mice were euthanized by cervical dislocation following isoflurane (Sigma) anesthesia and the sponges were collected. Volume of exudate and number of leukocytes were quantified using a hemocytometer.
(c) Molecular modelling
Molecular modelling studies were carried out employing a previously developed three-dimensional model of rat NAAA–PEA tetrahedral adduct (residues 131–362),23 built using MODELLER 7.0 program,63 starting from the crystallographic coordinates of conjugated bile acid hydrolase (CBAH),20 a related member of the Ntn family. For the present work, in order to perform docking studies, PEA atoms were deleted from the active site, and the hydrogen atoms of the catalytic Cys131 were reassigned to form a hydrogen bond between the thiol hydrogen atom and its neutral nitrogen one. The resulting NAAA structure was submitted to a geometry optimization of hydrogen atoms, minimizing its energy to a gradient of 0.01 kcal/(mol·Å) with the MMFF94s force field64 (as implemented in Macromodel).65 Inhibitor models were built using Maestro 8.566 and their geometries were optimised to an energy gradient of 0.01 kcal/(mol·Å) with the MMFF94s force field. Docking experiments were performed using Glide 5.067 starting from minimum-energy conformations of the ligands, placed in an arbitrary starting position within a region centered on residues Cys131, Asp150 and Asn292, using enclosing and bounding boxes of 20 and 10 Å on each side, respectively. Van der Waals radii of the protein atoms were not scaled, while Van der Waals radii of the ligand atoms, having partial atomic charges between −0.15 and 0.15, were scaled by 0.8. Standard precision mode was applied and docking solutions were ranked according to their Emodel values. An alternative conformation of the NAAA model was generated by rearranging the side chains of Asn209 and Thr258 as shown in Figure 2B. The geometry of the resulting structure was optimized to an energy gradient of 0.01 kcal/(mol·Å) and employed for docking, using the same computational settings as described above.
Docking complexes with compounds 7a and 8a, selected among the five poses having highest E-model scores, and having the lactone ring sufficiently close to Cys131 to allow a reactive event, were employed to build tetrahedral intermediates, by imposing a covalent bond between the β-lactone carbonyl carbon and the sulfur atom of Cys131. After model building of the covalent complexes, atom types and protonation states were opportunely modified and the resulting structures were minimized (with MMFF94s force field) to an energy-gradient of 0.05 kcal/(mol·Å), keeping the backbone atoms of the protein frozen.
Acknowledgments
This work was supported by Italian MiUR (Ministero dell’Università e della Ricerca), Universities of Parma and Urbino “Carlo Bo”, the Sandler Foundation (to D.P.) and the National Institute on Drug Abuse (to D.P.).
Abbreviations
- CBAH
conjugated bile acid hydrolase
- LogPcalc
calculated LogP
- FAE
fatty acid ethanolamide
- NAAA
N-acylethanolamine-hydrolyzing acid amidase
- PEA
palmitoylethanolamide
- PPAR
peroxisome proliferator-activated receptor
- SAR
structure-activity relationship
References
- 1.Russo R, LoVerme J, La Rana G, Compton TR, Parrott J, Duranti A, Tontini A, Mor M, Tarzia G, Calignano A, Piomelli D. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther. 2007;322:236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- 2.McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 3.Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 4.Mazzari S, Canella R, Petrelli L, Marcolongo G, Leon A. N-(2-Hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur J Pharmacol. 1996;300:227–236. doi: 10.1016/0014-2999(96)00015-5. [DOI] [PubMed] [Google Scholar]
- 5.Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- 6.Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- 7.LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-α. J Pharmacol Exp Ther. 2006;319:1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- 8.Petrosino S, Iuvone T, Di Marzo V. N-palmitoyl-ethanolamine: biochemistry and new therapeutic opportunities. Biochimie. 2010;92:724–727. doi: 10.1016/j.biochi.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: involvement of CB1, TRPV1 and PPARγ receptors and neurotrophic factors. Pain. 2008;139:541–550. doi: 10.1016/j.pain.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim H-Y, Kunos G. A biosynthetic pathway for anandamide. Proc Natl Acad Sci USA. 2006;103:13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ueda N, Yamanaka K, Yamamoto S. Purification and characterization of an acid amidase selective for N-palmitoylethanolamine, a putative endogenous anti-inflammatory substance. J Biol Chem. 2001;276:35552–35557. doi: 10.1074/jbc.M106261200. [DOI] [PubMed] [Google Scholar]
- 13.Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J Biol Chem. 2005;280:11082–11092. doi: 10.1074/jbc.M413473200. [DOI] [PubMed] [Google Scholar]
- 14.Tsuboi K, Takezaki N, Ueda N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA) Chem Biodivers. 2007;4:1914–1925. doi: 10.1002/cbdv.200790159. [DOI] [PubMed] [Google Scholar]
- 15.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. Cyclohexylcarbamic acid 3′- or 4′-substituted biphenyl-3-yl esters as fatty acid amide hydrolase inhibitors: synthesis, quantitative structure–activity relationships, and molecular modeling studies. J Med Chem. 2004;47:4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 16.Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 17.Tsuboi K, Hilligsmann C, Vandevoorde S, Lambert DM, Ueda N. N-cyclohexanecarbonylpentadecylamine: A selective inhibitor of the acid amidase hydrolysing N-acylethanolamines, as a tool to distinguish acid amidase from fatty acid amide hydrolase. Biochem J. 2004;379:99–106. doi: 10.1042/BJ20031695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vandevoorde S, Tsuboi K, Ueda N, Jonsson K-O, Fowler CJ, Lambert DM. Esters, retroesters, and a retroamide of palmitic acid: pool for the first selective inhibitors of N-palmitoylethanolamine-selective acid amidase. J Med Chem. 2003;46:4373–4376. doi: 10.1021/jm0340795. [DOI] [PubMed] [Google Scholar]
- 19.Saturnino C, Petrosino S, Ligresti A, Palladino C, De Martino G, Bisogno T, Di Marzo V. Synthesis and biological evaluation of new potential inhibitors of N-acylethanolamine hydrolyzing acid amidase. Bioorg Med Chem Lett. 2010;20:1210–1213. doi: 10.1016/j.bmcl.2009.11.134. [DOI] [PubMed] [Google Scholar]
- 20.Rossocha M, Schultz-Heienbrok R, von Moeller H, Coleman JP, Saenger W. Conjugated bile acid hydrolase is a tetrameric N-terminal thiol hydrolase with specific recognition of its cholyl but not of its tauryl product. Biochemistry. 2005;44:5739–5748. doi: 10.1021/bi0473206. [DOI] [PubMed] [Google Scholar]
- 21.Zhao L-Y, Tsuboi K, Okamoto Y, Nagahata S, Ueda N. Proteolytic activation and glycosylation of N-acylethanolamine-hydrolyzing acid amidase, a lysosomal enzyme involved in the endocannabinoid metabolism. Biochim Biophys Acta. 2007;1771:1397–1405. doi: 10.1016/j.bbalip.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Zhao L-Y, Uyama T, Tsuboi K, Tonai T, Ueda N. Amino acid residues crucial in pH regulation and proteolytic activation of N-acylethanolamine-hydrolyzing acid amidase. Biochim Biophys Acta. 2008;1781:710–717. doi: 10.1016/j.bbalip.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S, Mor M, Russo R, Maccarrone M, Antonietti F, Duranti A, Tontini A, Cuzzocrea S, Tarzia G, Piomelli D. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci US A. 2009;106:20966–20971. doi: 10.1073/pnas.0907417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer RJ, Louis-Flamberg P, Elliott JD, Fisher M, Leber J. Inhibition of 3-hydroxy-3-methylglutaryl coenzyme A synthase by antibiotic 1233A and other β-lactones. Biochem Biophys Res Commun. 1990;169:610–616. doi: 10.1016/0006-291x(90)90374-v. [DOI] [PubMed] [Google Scholar]
- 25.Chauhan D, Hideshima T, Anderson KC. A novel proteasome inhibitor NPI-0052 as an anticancer therapy. Br J Cancer. 2006;95:961–965. doi: 10.1038/sj.bjc.6603406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lall MS, Ramtohul YK, James MNG, Vederas JC. Serine and threonine β-lactones: A new class of hepatitis A virus 3C cysteine proteinase inhibitors. J Org Chem. 2002;67:1536–1547. doi: 10.1021/jo0109016. [DOI] [PubMed] [Google Scholar]
- 27.Arnold LD, Kalantar TH, Vederas JC. Conversion of serine to stereochemically pure β-substituted α-amino acids via β-lactones. J Am Chem Soc. 1985;107:7105–7109. [Google Scholar]
- 28.Arnold LD, May RG, Vederas JC. Synthesis of optically pure α-amino acids via salts of α-amino-β-propiolactone. J Am Chem Soc. 1988;110:2237–2241. [Google Scholar]
- 29.Marinez ER, Salmassian EK, Lau TT, Gutierrez CG. Enterobactin and enantioenterobactin. J Org Chem. 1996;61:3548–3550. [Google Scholar]
- 30.Sliedregt KM, Schouten A, Kroon J, Liskamp RMJ. Reaction of N-trityl amino acids with BOP: Efficient synthesis of t-butyl esters as well as N-trytil serine- and threonine-β-lactones. Tetrahedron Lett. 1996;37:4237–4240. [Google Scholar]
- 31.Parker WL, Rathnum ML, Liu WC. SQ 26,517 – A β-lactone produced by a Bacillus species. J Antibiot. 1982;35:900–902. doi: 10.7164/antibiotics.35.900. [DOI] [PubMed] [Google Scholar]
- 32.Montalbetti CAGN, Falque V. Amide bond formation and peptide coupling. Tetrahedron. 2005;61:10827–10852. [Google Scholar]
- 33.White AJ, Wharton CW. Hydrogen-bonding in enzyme catalysis. Fourier-trasform infrared detection of ground-state electronic strain in acyl-chymotrypsins and analysis of the kinetic consequences. Biochem J. 1990;270:627–637. doi: 10.1042/bj2700627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geske GD, O’Neill JC, Miller DM, Mattmann ME, Blackwell HE. Modulation of bacterial quorum sensing with synthetic ligands: Systematic evaluation of N-acylated homoserine lactones in multiple species and new insights into their mechanism of action. J Am Chem Soc. 2007;129:13613–13625. doi: 10.1021/ja074135h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hutchinson A. Note on the reduction of aromatic amides. J Chem Soc Trans. 1890;57:957. [Google Scholar]
- 36.Bessodes M, Saiah M, Antonakis K. A new, versatile and stereospecific route to unusual amino acids: the enantiospecific total synthesis of statine amide and its three stereoisomers. J Org Chem. 1992;57:4441–4444. [Google Scholar]
- 37.Hofmann K, Schmiechen R, Wells RD, Wolman Y, Yanaihara N. Studies on Polypeptides. XXIX. Synthetic peptides related to the N-terminus of bovine pancreatic ribonuclease A (Position 1–7) J Am Chem Soc. 1965;87:611–619. doi: 10.1021/ja01081a039. [DOI] [PubMed] [Google Scholar]
- 38.Miller MJ, Mattingly PG, Morrison MA, Kerwin JF., Jr Synthesis of β-lactams from substituted hydroxamic acids. J Am Chem Soc. 1980;102:7026–7032. [Google Scholar]
- 39.Cohen SB, Halcomb RL. Application of serine- and threonine-derived cyclic sulfamidates for the preparation of S-linked glycosyl amino acids in solution- and solid-phase peptide synthesis. J Am Chem Soc. 2002;124:2534–2543. doi: 10.1021/ja011932l. [DOI] [PubMed] [Google Scholar]
- 40.Higashibayashi S, Kohno M, Goto T, Suzuki K, Mori T, Hashimoto K, Nakata M. Synthetic studies on thiostrepton family of peptide antibiotics: synthesis of the pentapeptide segment containing dihydroxyisoleucine, thiazoline and dehydroamino acid. Tetrahedron Lett. 2004;45:3707–3712. [Google Scholar]
- 41.Pu Y, Martin FM, Vederas JC. Synthesis and acylation of salts of L-threonine β-lactone: A route to β-lactone antibiotics. J Org Chem. 1991;56:1280–1283. [Google Scholar]
- 42.Sakai N, Ageishi S, Isobe H, Hayashi Y, Yamamoto Y. Lipase promoted asymmetric trans-esterification of 4-alkyl-, 3-alkyl- and 3,4-dialkyloxetan-2-ones with ring-opening. J Chem Soc, Perkin Trans. 2000;1:71–77. [Google Scholar]
- 43.Park DJ, Kim DH. Cysteine derivatives as inhibitors for carboxypeptidase A: Synthesis and structure–activity relationships. J Med Chem. 2002;45:911–918. doi: 10.1021/jm010272s. [DOI] [PubMed] [Google Scholar]
- 44.Gomberg M, Pernert JC. Methylbiphenyls. J Am Chem Soc. 1926;48:1372–1384. [Google Scholar]
- 45.Xiao Z-Y, Zhao X, Jiang X-K, Li Z-T. Self-assembly of porphyrin–azulene–porpfyrin and porphyrin–azulene–conjugates. Org Biomol Chem. 2009;7:2540–2547. doi: 10.1039/b904009a. [DOI] [PubMed] [Google Scholar]
- 46.Fraser RTM, Taube H. Activation effects and rates of electron transfer. J Am Chem Soc. 1961;83:2242–2246. [Google Scholar]
- 47.Deng Y, Gong L, Mi A, Liu H, Jiang Y. Suzuki coupling catalyzed by ligand-free palladium(II) species at room temperature and by exposure to air. Synthesis. 2003:337–339. [Google Scholar]
- 48.Müller CE, Schobert U, Hipp J, Geis U, Frobenius W, Pawlowski M. Configurationally stable analogs of styrylxanthines as A2A adenosine receptor antagonists. Eur J Med Chem. 1997;32:709–719. [Google Scholar]
- 49.Orton KJP, Soper FG, Wiliams G. The chlorination of anilides. Part III. N-Chlorination and C-chlorination as simultaneous side reactions. J Chem Soc. 1928:998–1005. [Google Scholar]
- 50.Cativiela C, Serrano JL, Zurbano MM. Synthesis of 3-substituted pentane-2,4-diones: Valuable intermediates for liquid crystals. J Org Chem. 1995;60:3074–3083. [Google Scholar]
- 51.Yin J, Bergmann EM, Cherney MM, Lall MS, Jain RP, Vederas JC, James MNG. Dual modes of modification of hepatitis A virus 3C protease by a serine-derived β-lactone: Selective crystallization and formation of a functional catalytic triad in the active site. J Mol Biol. 2005;354:854–871. doi: 10.1016/j.jmb.2005.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tomoda H, Ohbayashi N, Morikawa Y, Kumagai H, Omura S. Binding site for fungal β-lactone hymeglusin on cytosolic 3-hydroxy-3-methylglutaryl coenzyme A synthase. Biochim Biophys Acta. 2004;1636:22–28. doi: 10.1016/j.bbalip.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 53.Greenspan MD, Bull HG, Yudkovitz JB, Hanf DP, Alberts AW. Inhibition of 3-hydroxy-3-methylglutaryl-CoA synthase and cholesterol biosynthesis by β-lactone inhibitors and binding of these inhibitors to the enzyme. Biochem J. 1993;289:889–895. doi: 10.1042/bj2890889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pojer F, Ferrer JL, Richard SB, Nagegowda DA, Chye M-L, Bach TJ, Noel JP. Structural basis for the design of potent and species-specific inhibitors of 3-hydroxy-3-methylglutaryl CoA synthases. Proc Natl Acad Sci USA. 2006;103:11491–11496. doi: 10.1073/pnas.0604935103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.ACD/LogP, version 800. Advanced Chemistry Development, Inc; Toronto ON, Canada: 2004. www.acdlabs.com. [Google Scholar]
- 56.Oliver CM, Schaefer AM, Greenberg EP, Sufrin JR. Microvawe synthesis and evaluation of phenacylhomoserine lactones as anticancer compounds that minimally activate quorum sensing pathways in Pseudomonas aeruginosa. J Med Chem. 2009;52:1569–1575. doi: 10.1021/jm8015377. [DOI] [PubMed] [Google Scholar]
- 57.Lu X, Lin S. Pd(II)-Bipyridine catalyzed conjugate addition of arylboronic acid to α,.β-unsaturated carbonyl compounds. J Org Chem. 2005;70:9651–9653. doi: 10.1021/jo051561h. [DOI] [PubMed] [Google Scholar]
- 58.Wang Z, Gu C, Colby T, Shindo T, Balamurugan R, Waldmann H, Kaiser M, van der Hoorn RAL. β-Lactone probes identify a papain-like peptide ligase in Arabidopsis thaliana. Nat Chem Biol. 2008;4:557–563. doi: 10.1038/nchembio.104. [DOI] [PubMed] [Google Scholar]
- 59.Tao B, Boykin DW. Simple amine/Pd(OAc)2-catalyzed Suzuki coupling reactions of aryl bromides under mild aerobic conditions. J Org Chem. 2004;69:4330–4335. doi: 10.1021/jo040147z. [DOI] [PubMed] [Google Scholar]
- 60.Tzschucke CC, Bannwart W. Fluorous-silica-supported perfluoro-tagged palladium complexes catalyze Suzuki couplings in water. Helv Chim Acta. 2004;87:2882–2889. [Google Scholar]
- 61.Chase BH. Ester exchange in the hydrolysis of diphenyl terephthalate. J Chem Soc. 1963:1334–1335. [Google Scholar]
- 62.Wietzerbin K, Bernadou J, Meunier B. Mechanism of the catalytic oxidation of tertiary alcohols by the water-soluble Mn-TMPyP/KHSO5 system: β-Fragmentation versus O-neophyl rearrangement. Eur J Inorg Chem. 1999;9:1467–1477. [Google Scholar]
- 63.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 64.Halgren TA. MMFF VI. MMFF94s option for energy minimization studies. J Comput Chem. 1999;20:720–729. doi: 10.1002/(SICI)1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 65.Macromodel version 85. Schrödinger L.L.C; New York, NY: 2008. [Google Scholar]
- 66.Maestro, version 85. Schrödinger, L.L.C; New York, NY: 2008. [Google Scholar]
- 67.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]