

Abstract
(±)-Citalopram (1, 1-(3-(dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile), and its eutomer, escitalopram (S(+)-1) are selective serotonin reuptake inhibitors (SSRIs) that are used clinically to treat anxiety and depression. To further explore structure-activity relationships at the serotonin transporter (SERT), a series of (±)-4- and 5-substituted citalopram analogues were designed, synthesized and evaluated for binding at the SERT, dopamine transporter (DAT) and norepinephrine transporter (NET) in native rodent tissue. Many of these analogues showed high SERT binding affinities (Ki = 1–40 nM) and selectivities over both NET and DAT. Selected enantiomeric pairs of analogues were synthesized and both retained enantioselectivity as with S- and R-1, wherein S > R at the SERT. In addition, the enantiomeric pairs of 1 and 5 were tested for binding at the homologous bacterial Leucine transporter (LeuT), wherein low affinities and the absence of enantioselectivity suggested distinctive binding sites for these compounds at SERT as compared to LeuT. These novel ligands will provide molecular tools to elucidate drug-protein interactions at the SERT and to relate those to behavioral actions, in vivo.
Introduction
The serotonin transporter (SERT) is a monoamine transporter that along with the norepinephrine transporter (NET) and the dopamine transporter (DAT) belongs to the neurotransmitter: sodium symporter (NSS) family. The SERT acts as a serotonergic signal terminator in the brain, by transporting excess serotonin from the synapse into the presynaptic cell terminal.1 Selective serotonin reuptake inhibitors (SSRIs) are used clinically to treat anxiety and depression. Citalopram ((±)-1) is a clinically used antidepressant that binds with high affinity (Ki = 1.94 nM) and selectivity to the SERT relative to other monoamine transporters.2 Despite the clinical success of the SSRIs, elucidation of the specific drug-protein interactions that lead to clinical efficacy remain unknown.
The 3-dimensional (3-D) structure of proteins in the NSS family (also called SLC6) was unknown until 2005 when the leucine transporter (LeuT) was cocrystallized along with the substrate leucine and two sodium ions.3 LeuT is a bacterial NSS homolog with 12 transmembrane segments with both N and C- termini located intracellularly.3 Using the crystal structure of LeuT as a model, drug-protein interactions between substrates and inhibitors at the monoamine transporters have been studied.4–20 Homology models have predicted critical residues for binding the SSRIs and tricyclic antidepressants, guiding site-directed mutagenesis and pharmacological studies to elucidate further drug-protein interactions and relate these to the behavioral actions of these drugs.5–8, 11, 13, 15, 17, 18, 21–25
Several reports have been published describing citalopram (1) and its eutomer, S(+)-1, which has ∼30 fold higher binding affinity at SERT than its R(−)-enantiomer.26, 27 Site-directed mutagenesis studies on the SERT have also revealed considerable differences in binding and uptake profiles between S- and R-1.28 Additional studies suggest that R-1 may attenuate the effects of S-1, which has been hypothesized to involve an allosteric binding domain 22, 29, 30 and is supported by a recent clinical study.31 It has been suggested that differing conformational changes induced by S- and R-1 at the SERT may effect downstream signaling proteins differently and these in turn may be related to antidepressant actions, in vivo.28
Only limited structure-activity relationships (SAR) studies have been reported on the citalopram pharmacophore, especially with enantiomeric pairs.32, 33 In the first report published in 1977 by Bigler, et al., compound 1 and a large series of analogues with substitutions in the isobenzofuranyl and pendant phenyl rings were reported. Substitutions including halogen, nitrile, methoxy and trifluoromethyl groups resulted in a narrow range of high potencies for inhibition of [14C]5-HT uptake.32 More recently emphasis on the dimethylamino group as playing a principal role for the selectivity at SERT over NET was reported.33 However, enantioselectivity of 1 and several analogues was only recently reported wherein site-specific mutagenesis was used to guide computational protein-ligand docking studies, which predicted detailed binding domain interactions at the SERT.24 In the present study the synthesis of (±)-4- and 5-substituted analogues of 1 and their evaluation for binding at SERT, NET, and DAT is reported. In addition, the S- and R-enantiomers of two of these analogues (5 and 9) were also prepared and evaluated for enantioselective binding at all three monoamine transporters. The enantioselective binding of R- and S-fluoxetine to the homologous bacterial transporter LeuT recently appeared.8 In this report, S- and R-1 as well as the enantiomers of 5 were evaluated for competition of [3H]Leucine in LeuT.
Chemistry
All analogues were synthesized by the strategy shown in Scheme 1, starting from either the commercially available 5-bromophthalide or 6-bromophthalide (3), which was synthesized from commercially available 3-bromo-2-methylbenzoic acid using a two-step procedure.34 The second step of this procedure used chromyl chloride to oxidize the methyl group of 3-bromo-2-methylbenzoate (2) and form the isobenzofuran ring in a one-pot reaction. The reaction yielded the expected compound 3 (57% yield) with another side product 4 as reported.
Scheme 1.

Synthesis of (±)4- and 5-substituted citalopram analoguesa
a Reagents and conditions: (a) MeOH, conc. H2SO4, reflux, 12 h; (b) chromyl chloride, CCl4, 0°C ∼ reflux, 20 h; (c) THF, 0°C ∼ rt, 3 h; (d) THF, 0°C ∼ rt, overnight; (e) HCl/EtOH (1:1), 10 min; (f) Boronic acids, Na2CO3, Pd(PPh3)4, DME, H2O, 70–80°C, overnight; (g) CuI, KI, HMPA, 150°C, 3h.
The fluorophenyl group and the dimethylamino moiety were introduced by a double Grignard reaction using a modification of a previously described procedure,35 to give diol intermediates (not shown). In this double Grignard reaction, a commercially available THF solution of 4-fluorophenylmagnesium bromide was used as the first Grignard reagent, while (3-(dimethylamino)propyl)magnesium chloride was freshly made and used as the second Grignard reagent. To make the magnesium chloride reagent, dibromoethane was used as an initiator (See details in Experimental Methods section). By treating with HCl in ethanol, the ring closed compounds 5 and 6 were obtained. Suzuki coupling of the two benzofurans 5 and 6 gave a set of 4- and 5- substituted analogues 7–20 shown in Scheme 1. In addition, compound 21 was synthesized by halogen exchange using CuI and KI, at 150 °C.
These novel isobenzofuran analogues 5–21 were assessed for SERT, NET and DAT binding affinities, which will be discussed in detail in the SAR section. The resulting SAR and our interest in further investigating SERT tolerance of analogues with extended steric bulk lead us to choose the Br-analogue 5 and the vinyl compound 9, for further synthesis of enantiomeric pairs, as shown in Scheme 2. Compounds 1, S-1 and R-1 were also synthesized for comparison by a similar procedure also shown in Scheme 2.
Scheme 2.

Synthesis of Chiral Analoguesa
a Reagents and conditions: (a) (4-Fluorophenyl)magnesium bromide, THF, 0°C ∼ rt, 3h; (b) (3-(Dimethylamino)propyl)magnesium chloride, THF, 0 °C ∼ rt, overnight; (c) Triethylamine, MsCl, 0 °C, 3h; (d) Resolution with (+)-di-p-toluoyl-D-tartaric acid or (−)-di-p-toluoyl-L-tartaric acid monohydrate; (e) trans-phenylvinylboronic acid, Na2CO3, Pd(PPh3)4, DME, H2O, 70–80 °C, overnight.
Chiral resolution of the diols 22 and 23 was successfully performed by(+)-di-p-toluoyl-D-tartaric acid or (−)-di-p-toluoyl-L-tartaric acid. The ring closure reactions under the condition of triethylamine and methanethiosulfonyl chloride gave compounds S(+)-1, R(−)-1, S(+)-5, and R(−)-5. Compounds S(+)-9, and R(−)-9 were obtained by Suzuki coupling of S(+)-5, and R(−)-5, with trans-phenylvinylboronic acid, respectively.
All the compounds were purified by flash column chromatography, analytically characterized as the free bases, and then converted to the oxalate salts for biological testing, unless otherwise described in the experimental methods.
Biological Results
All the compounds were tested in radioligand competition binding assays for SERT, NET, and DAT, using [3H]citalopram, [3H]nisoxetine and [3H]WIN 35,428 in rat brain stem, frontal cortex, and caudate-putamen, respectively. The Ki values are displayed in Table 1 and Table 2. Experimental details of these assays have been previously published.36
Table 1.
In Vitro Data for (±)1 and its (±)analoguesa
 | |||||||
|---|---|---|---|---|---|---|---|
| compound | R | R’ | SERT Ki ± SEM (nM) |
NET Ki ± SEM (nM) |
DAT Ki ± SEM (nM) |
Ki(NET)/ Ki (SERT) |
Ki(DAT)/ Ki (SERT) |
| 1 | CN | H | 1.94 ± 0.198 | 5950 ± 77.4 | 9270 ± 872 | 3070 | 4780 |
| 5b | Br | H | 1.04 ± 0.126 | 28400 (global fit no SEM) |
1650 ± 112 | >10,000 | 1590 |
| 6 | H | Br | 3.87 ± 0.575 | 6170 ± 521 | 216 ± 18.1 | 1590 | 56 |
| 7 | Ph | H | 40.0 ± 4.01 | 16900 ± 1380 | 1800 ± 167 | 423 | 45 |
| 8 | 3-NH2-Ph | H | 10.4 ± 1.34 | 3820 ± 318 | 1690 ± 231 | 367 | 163 |
| 9 | Ph-CH=CH | H | 9.32 ± 1.36 | 11400 ± 1090 | 836 ± 14.9 | 1220 | 90 |
| 10 | 3-CN-Ph | H | 6.70 ± 0.983 | 4980 ± 115 | 737 ± 68.5 | 743 | 110 |
| 11 | 3-OCH3-Ph | H | 9.20 ± 1.32 | 2610 ± 301 | NT | 284 | |
| 12 | 3,4-diCl-Ph | H | 143 ± 21.2 | 12700 ± 1290 | 2510 ± 71.3 | 89 | 18 |
| 13 | 3,5-diF-Ph | H | 108 ± 16.2 | 2000 ± 260 | NT | 19 | |
| 14 | Ph-CH2CH2 | H | 38.1 ± 1.58 | 4630 ± 255 | NT | 122 | |
| 15 | 3-F-Ph | H | 21.8 ± 1.09 | 4200 ± 489 | NT | 193 | |
| 16 | 3-Cl-Ph | H | 61.2 ± 6.11 | 5710 ± 640 | 1390 ± 172 | 93 | 23 |
| 17 | 4-Cl-Ph- CH=CH |
H | 33.6 ± 4.48 | 25100 ± 3220 | 2000 ± 132 | 747 | 60 |
| 18 | H | Ph | 5.07 ± 0.350 | 7160 ± 791 | 542 ± 39.1 | 1410 | 107 |
| 19 | H | Ph-CH=CH | 46.4 ± 4.91 | 11900 ± 534 | 1140 ± 52.5 | 256 | 25 |
| 20 | H | 3-F-Ph | 8.22 ± 1.12 | 7670 ± 1100 | 490 ± 26.2 | 933 | 60 |
| 21c | I | H | 1.42 ± 0.155 | 32500 ± 4630 | 1350 ± 72.9 | >10,000 | 950 |
Table 2.
In Vitro Data for (±)1 and its chiral analoguesa
 | ||||||
|---|---|---|---|---|---|---|
| compound | R’ | SERT Ki ± SEM (nM) |
NET Ki ± SEM (nM) |
DAT Ki ± SEM (nM) |
Ki(NET)/Ki (SERT) |
Ki(DAT)/Ki (SERT) |
| (±)-1 | CN | 1.94 ± 0.198 | 5950 ± 77.4 | 9270 ± 872 | 3070 | 4780 |
| S(+)-1 | CN | 0.89 ± 0.132 | 10500 ± 893 | 8150 ± 314 | 11800 | 9160 |
| R(−)-1 | CN | 28.3 ± 3.62 | 4980 ± 515 | 7980±646 | 178 | 282 |
| (±)-5b | Br | 1.04 ± 0.126 | 28400 (global fit no SEM) |
1650 ± 112 | >10,000 | 1590 |
| S(+)-5 | Br | 0.92 ± 0.056 | 8410 ± 202 | 2250 ± 115 | 9140 | 2450 |
| R(−)-5 | Br | 23.6 ± 1.76 | 21600 ± 3210 | 1350 ± 44 | 915 | 57 |
| (±)-9 | Ph-CH=CH | 9.32 ± 1.36 | 11400 ± 1090 | 836 ± 14.9 | 1220 | 90 |
| S(+)-9 | Ph-CH=CH | 10.6 ± 1.09 | 16800 ± 1360 | 1810 ± 46.8 | 1590 | 171 |
| R(−)-9 | Ph-CH=CH | 113 ± 14.7 | 5190 ± 427 | 541 ± 40.7 | 46 | 4.79 |
Structure-Activity Relationships
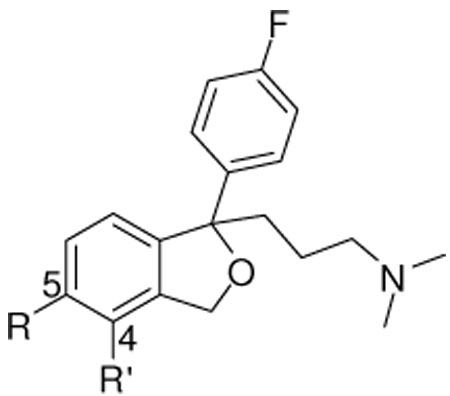
SAR results showed most modifications at the 4- and 5-positions of the dihydroisobenzofuran ring were well tolerated at the SERT and none of the compounds demonstrated high binding affinity at NET or DAT (Table 1). The 5-Br analogue 5, was as active (Ki = 1.04 nM) and at least as selective (Ki ratio > 10,000) for SERT over NET as 1 (Ki = 1.94 nM, ratio = 3070), and was chosen for chiral resolution (Scheme 2) to give the enantiomerically pure compounds S(+)-5 and R(−)-5. The 5-I-analogue 21 also demonstrated high binding affinity (Ki = 1.42 nM) and selectivity (ratio >10,000 over NET) for SERT. A ∼1000-fold selectivity for SERT over DAT was also observed for these racemic compounds, in which the 5-Br compound 5 was more selective than its iodo analogue, 21. Likewise the 4-Br-analogue 6 showed high binding affinity (Ki = 3.87 nM) with selectivity (Ki) ratios for SERT of 1590 over NET and 56 over DAT.
The additional extended aryl substitutions at the 4- and 5-positions were typically well tolerated (e.g., 7 to 20) at SERT. All the 5-(3’-substituted-phenyl) analogues showed moderate to high binding affinities at SERT (Ki range 1–60 nM), while di-substitution of the phenyl ring (e.g., 12 (Ki = 143 nM) v. 16 (Ki = 61.2 nM) and 13 (Ki = 108 nM) v. 15 (Ki = 21.8)) showed lower affinities, suggesting steric limitations at the 5-position. The same trend was observed for NET and DAT affinities with 12 and 16.
Compound 9, which has the phenylvinyl substituent, showed higher binding affinity at SERT (Ki = 9.32 nM) compared to its saturated analogue 14 (Ki = 38.1 nM). This suggested that the more rigid structure of the trans-styryl moiety was favored. Further modification, as in compound 17, which introduced a chloro group in the vinylphenyl ring, retained SERT affinity (Ki = 33.6 nM) as well as selectivity (ratio = 747 over NET, 60 over DAT). As we are interested in identifying positions on the citalopram pharmacophore where sterically bulky groups might be extended, the results with compound 9 suggested it was the best candidate for chiral resolution, in order to potentially optimize SERT binding affinity with an enantiopure compound. Thus synthesis of the enantiomers S(+)-9 and R (−)-9 was undertaken. (Scheme 2).
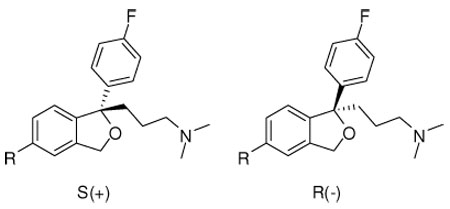
The enantiomers of 5 and 9 (Table 2), showed the expected chiral selectivity at SERT, with the S(+)-enantiomers being more active than the R(−)-enantiomers in all cases. Specifically, S(+)-5 (Ki = 0.92) showed ∼ 26 fold higher binding affinity at SERT than its enantiomer R(−)-5 (Ki = 23.6). Compound S(+)-9 (Ki = 10.6) showed ∼ 11 fold higher binding affinity at SERT than its enantiomer R(−)-9 (Ki = 113).
The enantiomers of 1 and 9, showed the opposite enantioselectivity at NET compared to SERT, in which the R(−)-compounds showed higher binding affinities over the S(+)-enantiomers; while the enantiomers of 5 showed the same enantioselectivity at SERT and NET, although in all cases the enantioselectivity was low (< 2–3-fold).
When the 3D structures of S- and R-5 were superimposed, an interesting overlap in structures was observed (Fig. 2.) We reasoned that the enantioselectivity at SERT for the citalopram analogues might be due to the subtle differences in the binding of the two aryl ring systems. Moreover, this overlap corresponds to a recently described study using a different experimental approach in which Kolodso et al,24 studied several sets of enantiomers of citalopram and close analogues in 15 SERT mutants to confirm that the 180° rotation of the dihydroisobenzofuran ring at the chiral center explains different binding modes of the citalopram enantiomers at SERT. This study and complimentary investigations19, 20, 25 provide 3-D models of S-citalopram binding in the S1 substrate site of SERT.
Figure 2.

3D-superimposition of S- and R-538
3D–superimposition of S- and R-5 suggests the interchangeability of the halogen groups in compound 5 (Br v. F).
As the crystal structures of SERT and DAT have yet to be elucidated, 3D molecular models are based on high-resolution crystal structures of LeuT.3, 14, 23 In addition, LeuT has been used directly for binding and functional studies using selected SSRI’s. Selected SSRIs and tricyclic antidepressants have also been shown to bind in the extracellular vestibule of LeuT, although these compounds have very low affinity for LeuT relative to SERT.3, 8, 14, 18, 23 The extracellular vestibule also appears to comprise a second substrate site (S2) in LeuT that is essential for transport.6, 11, 39 It has recently been shown that cocaine analogs bind to the primary substrate site (S1) in DAT,16 and it seems likely that the high affinity site for inhibition in SERT that is responsible for the behavioral actions of the SSRIs is also the S1 site.5, 24, 25, 28 However, it has been argued that the S2 site in SERT, by analogy with LeuT, is also a target for inhibitor binding.8
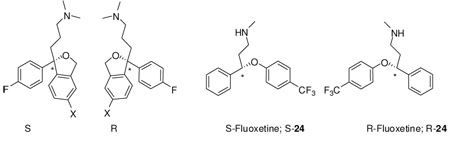
The potential halogen interchangeability in compound 5 as compared to the parent compound 1 (Br v. F, see Fig. 3) coupled with the recent report on the R- and S-enantiomers of the SSRI fluoxetine at LeuT8 led us to investigate the binding affinities of S- and R-1 and the Br-analogues S-5 and R-5 for competition with [3H]Leucine in LeuT. As fluoxetine and 1 share significant structural overlap, we reasoned that the pendant F-phenyl substituent on our analogues might access the described halogen binding pocket in an enantioselective manner and further, that by substituting the 5-CN group of 1 with a Br group, the interchangeability of the halogens might render this compound less enantioselective. Interestingly, both sets of enantiomers showed low binding affinities for LeuT similar to R-fluoxetine, but in contrast, the analogues of 1 showed no enantioselectivity (Table 3). It is possible that 1) 1 and fluoxetine might bind differently to LeuT such that the analogues of 1 do not access the halogen binding pocket, although it seems more likely that 2) the 5-CN group of 1 can interact at this binding site equivalently to the halogenated CF3 group of fluoxetine and therefore the interchangeability of the F with the CN would in essence, cancel enantioselectivity (Fig. 3). This interpretation would broaden characterization of the halogen binding pocket in LeuT to be an electron withdrawing group pocket and not simply limited to halogens. Hence, our findings support previous studies on SERT24, 25 and also differentiate this binding mode at SERT from that of LeuT, for these compounds.
Figure 3.

3D-superimposition of S-1 and R- and S-fluoxetine38
3D-superimposition of R-, S-24 and S-1 suggests the interchangeability of the F with the CN of 1 could be an explanation for the canceling effect on enantioselectivity. Note: The carbon chain of S-1 is shown in turquoise blue, S-24 in green and R-24 in orange for clarity.
Table 3.
LeuT Binding data
This competition was done at 4 nM Protein, 20 nM 3H-Leucine.
Published data.8
In summary, a series of 1-(3-(dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile analogues was synthesized many of which showed moderate to high SERT binding affinities (Ki range 1–40 nM) and selectivities over NET and DAT comparable to the parent ligand, 1. In addition, the novel chiral compounds of 5 and 9 showed the same enantioselectivity at SERT with S > R. The S(+)- and R(−)-enantiomers of 1 and 5 demonstrated low affinity binding (IC50 = 0.4–1.7 mM) to LeuT, comparable to the values recently reported for R-fluoxetine (R-24.)8 However, in contrast to the fluoxetine enantiomers, no enantioselectivity was demonstrated with the enantiomers of 1 and 5, at LeuT, despite their having halogenated aryl ring(s) that might be expected to interact with the halogenated binding pocket described.8 Further investigation into the SAR of the S2 v. S1 site in SERT will likely show further divergence between these two sites and ultimately provide another set of molecular tools with which to study the SERT-drug interactions that lead to the behavioral actions of the SSRIs.
Experimental Methods
Reaction conditions and yields were not optimized, and spectroscopic data refer to the free base unless otherwise described for each compound. Flash chromatography was performed using silica gel (EMD Chemicals, Inc.; 230–400 mess, 60 Å). 1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer. Chemical shifts are reported in parts-per-million (ppm) and referenced according to deuterated solvent for 1H spectra (CDCl3, 7.26; (CD3)2SO, 2.50; CD3OD, 3.31), 13C spectra (CDCl3, 77.2; (CD3)2SO, 39.5; CD3OD, 49.0), 19F spectra (CFCl3, 0). Infrared spectra were recorded as a KBr thin film using a Perkin-Elmer Spectrum RZ I FT-IR spectrometer or recorded as powder using an Avatar 370 FT-IR thermo Nicolet spectrometer. Gas chromatography-mass spectrometry (GC/MS) data were acquired using an Agilent Technologies (Santa Clara, CA) 6890N GC equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 µM film thickness) and a 5973 mass-selective ion detector in electron-impact mode. Ultrapure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (100°C) was held for 3 min and then increased to 295 at 15 °C/min over 13 min, and finally maintained at 295 °C for 10 min. Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) and agrees within 0.4% of calculated values. Melting point determinations were conducted using a Thomas-Hoover melting point apparatus and are uncorrected. Anhydrous solvents were purchased from Aldrich or J. T. Baker and were used without further purification, except for tetrahydrofuran, which was freshly distilled from sodium-benzophenone ketyl. All other chemicals and reagents were purchased from Aldrich Chemical Co., Combi-Blocks, TCI, America., Matrix Scientific; Lancaster Synthesis, Inc. (Alfa Aesar) and AK Scientific, Inc. The final products were converted into oxalate salts, typically by treating the free base with 1:1 molar ratio of oxalic acid in acetone followed by precipitation from a combination of organic solvents. On the basis of NMR, GC-MS, and combustion data, all final compounds are > 95% pure; Chiral purity was determined either by HPLC or by NMR with a chemical shift reagent as described.
General Procedure A for double Grignard reaction
To a cooled (0°C) suspension of substituted phthalide (40 mmol) in dry THF (60 mL) was added a solution of 4-fluorophenylmagnesium bromide in THF (1.0 M, 44 mL, 44 mmol) dropwise over 30 min. The reaction was allowed to warm to room temperature and stirred for 3 hr, then cooled to 0 °C. The second freshly made Grignard reagent (3-(Dimethylamino)propyl)magnesium chloride (∼50 mmol, 0.8 M, 60 mL) was added dropwise over 30 min. The reaction mixture was warmed to room temperature and stirred overnight. The reaction mixture was treated with aq NH4Cl solution (sat.), and extracted with ether several times. The organic layer was washed with sat. NaHCO3, brine, dried over MgSO4, and concentrated to give the crude diol product.
General Procedure B for coupling of heteroaryl bromides with boronic acid
To a suspension of boronic acid (1–1.5 eq), heteroaryl bromide (1 eq), and Na2CO3 in a mixture of solvents DME/H2O (3/1, 4 mL for 1 mmol scale reaction) was added Pd(PPh3)4 (5 mol%) under Argon. The mixture was warmed to 80 °C overnight. The solvent was then removed under reduced pressure, and the residue was extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated to a crude product, which was then purified by flash column chromatography to give the pure product.
General Procedure C for resolution
To a solution of the purified racemic product in 2-propanol was added a solution of (+)-di-p-toluoyl-D-tartaric acid or (−)-di-p-toluoyl-L-tartaric acid monohydrate (4:1) in a small volume of 2-propanol. The salt precipitated and was collected by filtration, dried and converted to the free base in aq NaOH (2 M). The mixture was extracted with chloroform, dried over MgSO4, and concentrated. The resolution was repeated three times to give the enantiomerically pure free base.
General Procedure D for ring closure reaction
To a solution of diol (1 eq) in dichloromethane at 0 °C, was added mesyl chloride (1.5 eq) and triethylamine (4 eq) dropwise. The reaction mixture was stirred on ice for 3 hr, and then extracted from dichloromethane. The organic layer was washed with aq NaHCO3 (sat.) and brine, dried over MgSO4, concentrated and purified by flash column chromatography to give pure product.
3-(5-Cyano-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine, citalopram (1)
Compound 1 was prepared by following the general procedure D using 22 (10 g, 30 mmol), eluting with chloroform/MeOH (10:1, 5:1)to give the product (5.4 g) in 55% yield; GC-MS (EI) m/z 324 (M+). The oxalate salt was precipitated from 2-propanol; mp 156–157 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.79 (s, 1H), 7.77 (d, J = 7.6 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.57-7.54 (m, 2H), 7.14 (dd, J = 9.2, 8.4 Hz, 2H), 5.18 (dd, J = 13.2, 14.0 Hz, 1H), 5.14 (dd, J = 13.2, 14.0 Hz, 1H), 2.91 (t, J = 8.0 Hz, 2H), 2.58 (s, 6H), 2.18 (t, J = 8.0 Hz, 2H), 1.45 (m, 1H), 1.38 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 165.3, 149.5, 140.7, 140.6, 132.8, 127.7, 127.6, 126.5, 123.8, 119.5, 116.0, 115.8, 111.3, 91.0, 71.8, 57.5, 43.2, 37.7, 20.2; IR (powder) 1158, 1234, 2226 cm−1; Anal. (C20H21FN2O·C2H2O4) C, H, N.
(S)-(+)-3-(5-Cyano-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine, escitalopram (S-1)
Compound S-1 was prepared by following the general procedure D using S-22 (1.63 g, 4.77 mmol), eluting with chloroform/MeOH (10:1, 5:1) to give product (0.5 g) in 46% yield. The oxalate salt was precipitated from acetone/EtOAc; mp 144–146 °C; Anal. (C20H21FN2O•C2H2O4) C, H, N; HPLC (Shim-pack HRC-CN 4.6 × 250 mm, 5.0 µM, with 12 mM β-CD in aqueous buffer (10% CAN, 1% TEA, with AcOH to adjust pH4.0))40 area ratio of the peaks with retention time 44.38 : 46.04 = 100 : 0 (ee > 99%); [α]D27 = 11.69 ± 0.06 (c = 2, MeOH).
(R)-(−)-3-(5-Cyano-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (R-1)
Compound R-1 was prepared by following the general procedure D using R-22 (1 g, 2.91 mmol), eluting with chloroform/MeOH (10:1, 5:1) to give product (0.48 g) in 51% yield. GC-MS (EI) m/z 324 (M+). The oxalate salt was precipitated from acetone/EtOAc; mp 148–149 °C; Anal. (C20H21FN2O·C2H2O4·1/4 H2O) C, H, N; HPLC (Shim-pack HRC-CN 4.6 × 250 mm, 5.0 µM, with 12 mM β-CD in aqueous buffer (10% CAN, 1% TEA, with AcOH to adjust pH4.0))40 area ratio of the peaks with retention time 44.38 : 46.04 = 0 : 100 (ee > 99%); [α]D24 = −11.69 ± 0.05 (c = 2, MeOH).
Methyl 3-bromo-2-methylbenzoate (2).34
Concentrated H2SO4 (0.5 mL) was added to a stirred solution of 3-bromo-2-methylbenzoic acid (2.15 g, 10 mmol) in MeOH (20 mL) at room temperature. The resulting mixture was stirred at reflux for 12 h. Methanol was partially removed under reduced pressure, and ether (150 mL) was added to dilute the residue. The solution was washed with aq NaHCO3, water, brine and dried over Na2SO4. The resulting organic layer was concentrated to give an oil that later solidified as a brown solid. Yield: 2.22 g (97%); mp 30–31 °C; 1H NMR (400 MHz, CDCl3) δ 7.74-7.69 (m, 2H), 7.09 (t, J = 8 Hz, 1H), 3.90 (s, 3H), 2.63 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.9, 138.6, 135.8, 132.6, 129.1, 127.0, 126.7, 52.3, 20.6; GC-MS (EI) m/z 228, 230 (M+).
6-Bromo-phthalide (3)
To a solution of Methyl 3-bromo-2-methylbenzoate (2)34 (1.02 g, 4.4 mmol) in CCl4 (6 mL) was added a solution of chromyl chloride (0.72 mL, 8.9 mmol) in CCl4 (6 mL) dropwise over a period of 1 h, at 0 °C. The reaction mixture was stirred at room temperature for 2 h and slowly heated to reflux for 20 h. The reaction mixture was cooled to room temperature and then quenched by pouring into an ice-cold aq Na2SO3 solution (sat., 6 mL). The mixture was extracted with EtOAc. The organic layer was washed with water and brine, dried over MgSO4, and concentrated to give the crude product, which was purified by flash column chromatography eluting with hexane/EtOAc (6:1) to give 3 (0.54 g) in 57% yield. 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 7.6 Hz, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.46 (t, J = 8.0 Hz, 1H), 5.24 (s, 2H); GC-MS (EI) m/z 212, 214 (M+). Side product 4: 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 4.0 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 5.20 (s, 2H), 3.96 (s, 3H); GC-MS (EI) m/z 264 (M+).
3-(5-bromo-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (5)
Compound 5 was prepared by following the general procedure A using 5-bromophthalidite to give the crude diol (6 g, 15 mmol), followed by treating with aq HCl (sat. 37%, 10 mL) in EtOH (15 mL) for 10–15 min. The reaction mixture was concentrated under vacuum and purified by flash column chromatography eluting with chloroform/MeOH (30:1). 1H NMR (400 MHz, CDCl3) δ 7.44-7.39 (m, 3H), 7.34 (s, 1H), 7.13 (d, J = 8 Hz, 1H), 6.98 (dd, J = 8.8, 8.8 Hz, 2H), 5.11 (d, J = 4.8 Hz, 2H), 2.22 (dd, J = 7.6, 6.8 Hz, 2H), 2.13 (s, 6H), 2.12 (m, 2H), 1.46 (m, 1H), 1.33 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 165.0, 143.8, 142.0, 141.4, 131.2, 127.6, 125.3, 124.6, 121.5, 115.9, 115.7, 90.6, 71.7, 57.4, 37.8, 20.0; 19F NMR (376 MHz, DMSO-d6) δ −116.5. The oxalate precipitated from acetone; mp 142–144 °C; IR (powder) 1164, 1216 cm−1; GC-MS (EI) m/z 377, 379 (M+); Anal. (C19H21BrFNO· C2H2O4) C, H, N.
(S)-(+)-3-(5-bromo-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (S-5)
Compound S-5 was prepared by following the general procedure D using S-23 (2.65 g, 6.6 mmol) to give product (1.14 g) in 45% yield. GC-MS (EI) m/z 377, 379 (M+). The oxalate salt precipitated from acetone; mp 141–143 °C; Anal. (C19H21BrFNO· C2H2O4·H2O) C, H, N. [α]D25 = 2.73 ± 0.17 (c = 1, MeOH) (lit. 2.2)35
(R)-(−)-3-(5-bromo-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (R-5)
Compound R-5 was prepared by following the general procedure D using R-23 (4.77 g, 12 mmol) to give product (3.15 g) in 70% yield; GC-MS (EI) m/z 377, 379 (M+). The oxalate salt precipitated from acetone; mp 154–155 °C; Anal. (C19H21BrFNO· C2H2O4) C, H, N; [α]D26 = −2.80 ± 0.40 (c = 1, MeOH). (lit. −2)35
3-(4-Bromo-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (6)
Compound 6 was prepared by following the general procedure A using 6-bromophthalidite 3 (0.54 g, 2.5 mmol) to give the crude diol. The crude product (GC-MS (EI) m/z 377, 379 (M+)) was treated with aq HCl (sat. 37%, 10 mL) in EtOH (15 mL) for 10–15 min. The reaction mixture was concentrated under vacuum and purified by flash column chromatography eluting with chloroform/MeOH (10:1, 5:1) to give the product as a syrup (0.88 g) in 90% yield; GC-MS (EI) m/z 377, 379 (M+). The oxalate precipitated from acetone; mp 172–173 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.57-7.46 (m, 4H), 7.26 (t, J = 8.0 Hz, 1H), 7.12 (dd, J = 8.0, 8.8 Hz, 2H), 5.15 (dd, J = 13.2, 10 Hz, 1H), 5.11 (dd, J = 12.8, 9.6 Hz, 1H), 2.96 (t, J = 8.0 Hz, 2H), 2.62 (s, 6H), 2.17 (t, J = 7.6 Hz, 2H), 1.48 (m, 1H), 1.41 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 164.7, 146.5, 141.3, 139.2, 131.3, 131.0, 127.6, 127.5, 121.2, 115.9, 115.7, 92.4, 73.1, 57.2, 42.9, 37.9, 19.9; 19F NMR (376 MHz, DMSO-d6) δ −116.2; IR (powder) 1174, 1225 cm−1; Anal. (C19H21BrFNO· C2H2O4) C, H, N.
3-(1-(4-Fluorophenyl)-5-phenyl-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (7)
Compound 7 was prepared by following the general procedure B using 5 (0.3 g, 0.8 mmol) and phenyl boronic acid (0.1 g, 0.82 mmol), eluting with chloroform/MeOH (10:1) to give the product (0.22 g) in 75% yield; GC-MS (EI) m/z 375 (M+). The oxalate was precipitated from Acetone/EtOAc; mp 112–115 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.60-7.52 (m, 7H), 7.41 (dd, J = 7.2, 8.0 Hz, 2H), 7.32 (t, J = 7.6 Hz, 1H), 7.12 (dd, J = 8.8, 8.8 Hz, 2H), 5.16 (dd, J = 12.8, 12.0 Hz, 1H), 5.13 (dd, J = 12.8, 12.0 Hz, 1H), 2.54 (m, 2H), 2.31 (s, 6H), 2.14 (m, 2H), 1.35 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 165.1, 141.3, 129.6, 127.4, 121.6, 119.7, 117.2, 112.9, 110.6, 105.8, 105.4, 90.52, 71.7, 58.0, 44.4, 21.4; IR (powder) 1159, 1220 cm−1; Anal. (C25H26FNO· 1/2C2H2O4·3/8 H2O) C, H, N.
3-(5-(3-Amino-phenyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (8)
Compound 8 was prepared by following the general procedure B using 5 (0.3 g, 0.8 mmol) and 3-amino-phenyl boronic acid (0.12 g, 0.79 mmol), eluting with chloroform/MeOH (30:1, 10:1) to give product (0.29 g) in 95% yield; GC-MS (EI) m/z 390 (M+). The oxalate was precipitated from Acetone; mp 115–117 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.23 (br, 1H), 8.04 (br, 1H), 7.61-7.43 (m, 5H), 7.15 (dd, J = 8.4, 7.6 Hz, 2H), 7.06 (t, J = 8.0 Hz, 1H), 6.76 (s, 1H), 6.71 (d, J = 6.4 Hz, 1H), 6.52 (d, J = 7.6 Hz, 1H), 5.19 (dd, J = 12.8, 12.8 Hz, 1H), 5.15 (dd, J = 12.8, 12.8 Hz, 1H), 2.96 (m, 2H), 2.63 (s, 6H), 2.18 (m, 2H), 1.47 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 164.8, 160.6, 155.5, 149.8, 141.5, 141.3, 130.1, 127.6, 127.5, 126.9, 122.7, 120.0, 115.8, 115.6, 115.1, 112.8, 104.9, 90.6, 72.2, 57.7, 43.0, 20.2; 19F NMR (376 MHz, DMSO-d6) δ −117.2; IR (powder) 1158, 1228 cm−1; Anal. (C25H27FN2O· C2H2O4·1/2 H2O) C, H, N.
(E)-3-(1-(4-Fluorophenyl)-5-styryl-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (9)
Compound 9 was prepared by following the general procedure B using 5 (0.2 g, 0.53 mmol) and 2-phenylvinyl boronic acid (0.08 g, 0.79 mmol), eluting with chloroform/MeOH (30:1, 10:1) to give the product (0.2 g) in 95% yield; GC-MS (EI) m/z 401(M+); 1H NMR (400 MHz, CDCl3) δ 7.44 (m, 10H), 6.97 (dd, J = 8.8 Hz, 2H), 6.35 (m, 2H), 5.11 (d, J = 9.2 Hz, 2H), 2.30 (m, 4H), 2.18 (s, 6H), 1.49 (m, 1H), 1.38 (m, 1H). The oxalate was precipitated from Acetone; mp 118–120 °C; 1H NMR (400 MHz, MeOD) δ 7.86-7.48 (m, 6H), 7.40 (d, J = 8 Hz, 1H), 7.33 (dd, J = 7.6, 8 Hz, 2H), 7.23 (m, 1H), 7.18 (s, 2H), 7.06, (dd, J = 9.2, 8.4 Hz, 2H), 5.19 (dd, J = 12, 11.2 Hz, 1H), 5.17 (dd, J = 11.2, 12.4 Hz, 1H), 3.09 (t, J = 8 Hz, 2H), 2.75 (s, 6H), 2.27 (m, 2H), 1.71 (m, 1H), 1.69 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 164.9, 143.8, 141.8, 139.9, 137.6, 136.5, 131.8, 129.6, 129.4, 128.6, 127.6, 127.5, 127.2, 119.6, 117.2, 115.8, 115.6, 110.0, 90.6, 77.9, 57.5, 43.1, 36.0, 20.2; 19F NMR (376 MHz, DMSO-d6) δ −117.0; IR (powder) 1169, 1230, 1718 cm−1; Anal. (C27H28FNO· C2H2O4·1/2 H2O) C, H, N.
(S)-(+)-(E)-3-(1-(4-Fluorophenyl)-5-styryl-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (S-9)
Compound S-9 was prepared by following the general procedure B using S-5 (0.4 g, 1.1 mmol) and trans-phenylvinylboronic acid (0.15 g, 1 mmol) to give the product (0.2 g) in 49% yield; GC-MS (EI) m/z 401 (M+). The oxalate salt precipitated from acetone; mp 138–139 °C; Anal. (C27H28FNO·C2H2O4·H2O) C, H, N; [α]D25 = 11.0 ± 0.00 (c = 0.83, MeOH).
(R)-(+)-(E)-3-(1-(4-Fluorophenyl)-5-styryl-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (R-9)
Compound R-9 was prepared by following the general procedure B using R-5 (0.4 g, 1.1 mmol) and trans-phenylvinylboronic acid (0.15 g, 1 mmol) to give product (0.13 g) in 30% yield; GC-MS (EI) m/z 401 (M+). The oxalate salt precipitated from acetone; mp 114–117 °C; Anal. (C27H28FNO·C2H2O4·H2O) C, H, N; [α]D25 = −11.2 ± 0.32 (c = 0.82, MeOH).
3-(5-(3’-Cyano-phenyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (10)
Compound 10 was prepared by following the general procedure B using 5 (0.6 g, 1.6 mmol) and 3-cyanophenyl boronic acid (0.23 g, 1.6 mmol), eluting with chloroform/MeOH (30:1, 10:1) to give the product (0.54 g) in 84% yield; GC-MS (EI) m/z 400 (M+). The oxalate salt was precipitated from 2-propanol. mp 157–159 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.11 (s, 1H), 7.98 (m, 1H), 7.80 (m, 1H), 7.65-7.59 (m, 6 H), 7.15 (m, 2H), 5.19 (dd, J = 6.6, 6.8 Hz, 1H), 5.18 (dd, J = 6.6, 6.8 Hz, 1H), 2.96 (m, 2H), 2.61 (s, 6H), 2.20 (m, 2H), 1.49 (m, 1H), 1.47 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 165.1, 163.1, 160.7, 144.6, 141.7, 140.3, 138.6, 132.3, 131.8, 131.0, 130.9, 127.6, 127.5, 123.1, 120.8, 119.5, 115.9, 115.6, 112.8, 90.7, 72.1, 57.4, 42.9, 38.0, 20.1; 19F NMR (376 MHz, DMSO-d6) δ −116.7; IR (powder) 1164, 1225, 2226 cm−1; Anal. (C26H25FN2O· C2H2O4·1/4 H2O) C, H, N.
3-(1-(4-Fluorophenyl)-5-(3’-methoxyphenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (11)
Compound 11 was prepared by following the general procedure B using 5 (0.6 g, 1.6 mmol) and 3-methoxyphenyl boronic acid (0.27 g, 1.8 mmol), eluting with chloroform/MeOH (30:1, 10:1) to give product (0.52 g) in 86% yield. The oxalate salt precipitated from acetone. GC-MS (EI) m/z 405(M+). Recrystallization from methanol gave purer compound. mp 147–149 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.57 (m, 5H), 7.34 (t, J = 8 Hz, 1H), 7.18-7.13 (m, 4H), 6.90 (d, J = 7.2 Hz, 1H), 5.20 (dd, J = 12.8, 12.4 Hz, 1H), 5.17 (dd, J = 12.8, 12.8 Hz, 1H), 3.79 (s, 3H), 2.97 (t, J = 8.0 Hz, 2H), 2.63 (s, 6H), 2.20 (t, J = 7.6 Hz, 2H), 1.46 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 160.4, 143.8, 142.1, 140.7, 140.1, 130.7, 127.6, 127.5, 127.3, 122.9, 120.5, 119.8, 115.8, 115.6, 113.8, 113.0, 90.6, 72.2, 57.5, 55.8, 43.0, 38.0, 20.1; IR (powder) 1181, 1222 cm−1; Anal. (C26H28FNO2· C2H2O4·1/2 H2O) C, H, N.
3-(5-(3,4-Dichlorophenyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (12)
Compound 12 was prepared by following the general procedure B using 5 (0.5 g, 1.3 mmol) and 3, 4-dichlorophenyl boronic acid (0.29 g, 1.5 mmol), eluting with chloroform/MeOH (30:1, 10:1) to give the product (0.52 g) in 90% yield. The oxalate salt was precipitated from acetone. mp 151–152 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.89 (s, 1H), 7.69-7.56 (m, 7H), 7.14 (dd, J = 9.2, 8.4 Hz, 2 H), 5.17 (ddd, J = 12.4, 11.6, 12 Hz, 2H), 2.93 (m, 2H), 2.60 (s, 6H), 2.25 (m, 2H), 1.47 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 163.6, 141.1, 140.3, 138.0, 131.7, 129.3, 127.7, 127.6, 127.5, 123.1, 120.7, 115.9, 115.6, 90.7, 72.1, 57.6, 43.1, 20.2; IR (powder) 1125, 1154 cm−1; GC-MS (EI) m/z 443 (M+). Recrystallization from methanol gave pure 12. Anal. (C25H24Cl2FNO· C2H2O4) C, H, N.
3-(5-(3,5-Difluorophenyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine oxalate (13)
Compound 13 was prepared by following the general procedure B using 5 (0.66 g, 1.75 mmol) and 3, 5-difluorophenyl boronic acid (0.32 g, 2 mmol), eluting with chloroform/MeOH (30:1, 10:1) to give product (0.7 g) in 97% yield. The oxalate salt precipitated from acetone; mp 145–147 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.67-7.57 (m, 5H), 7.40 (d, J = 7.6 Hz, 2H), 7.19 (m, 3 H), 5.20 (dd, J = 12.8, 12.4 Hz, 1H), 5.17 (dd, J =12.8, 13.6 Hz, 1H), 2.96 (t, J = 8 Hz, 2H), 2.62 (s, 6H), 2.20 (t, J = 7.2 Hz, 2H), 1.50 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 164.9, 162.3, 160.7, 144.9, 144.2, 140.2, 138.1, 127.6, 127.4, 123.1, 120.8, 115.9, 115.7, 113.0, 110.7, 110.5, 90.7, 57.4, 46.8, 43.0, 20.1; 19F NMR (376 MHz, DMSO-d6) δ −110.3, −116.9; IR (powder) 1158, 1222, 1345 cm−1; GC-MS (EI) m/z 411 (M+). Recrystallization from methanol gave pure 13. Anal. (C25H24F3NO· C2H2O4·1/4 H2O) C, H, N.
3-(1-(4-Fluorophenyl)-5-phenethyl-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (14)
A suspension of 9 (140 mg, 0.35 mmol) and a catalytic amount of Pd/C in MeOH was hydrogenated under 45 psi at RT for 5 hrs. The reaction mixture was filtered and the filtrate was concentrated to give the oxalate salt (80 mg) in 55% yield; mp 109–111 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.51 (m, 2H), 7.35 (d, J = 8 Hz, 1H), 7.25-7.08 (m, 9H), 5.08 (dd, J = 12.4, 12.4 Hz, 1H), 5.05 (dd, J = 12.4, 12.8 Hz, 1H), 3.97 (t, J = 7.6 Hz, 2H), 2.81 (m, 2H), 2.58 (s, 6H), 2.45 (m, 2H), 2.11 (m, 2H), 1.13 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 165.1, 142.1, 142.0, 139.3, 129.0, 128.9, 128.5, 127.5, 127.4, 126.5, 122.2, 121.8, 115.7, 115.5, 90.5, 72.1, 57.4, 42.9, 38.2, 37.8, 37.6, 20.0; 19F NMR (376 MHz, DMSO-d6) δ −116.99, IR (powder) 1159, 1210 cm−1; GC-MS (EI) m/z 403 (M+) Anal. (C27H30FNO· C2H2O4·1/3 H2O) C, H, N.
3-(5-(3-Fluorophenyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (15)
Compound 15 was prepared by following the general procedure B using 5 (0.85 g, 2.2 mmol) and 3-fluorophenylboronic acid (0.49 g, 2.3 mmol), eluting with chloroform/MeOH (30:1) to give product (0.67 g) in 67% yield. GC-MS (EI) m/z 393 (M+). The oxalate salt precipitated from EtOAc; mp 139–141 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.61-7.48 (m, 8H), 7.16 (m, 3H), 5.20 (dd, J = 12.8, 12.8 Hz, 1H), 5.17 (dd, J = 12.8, 12.8 Hz, 1H), 2.96 (m, 2H), 2.62 (s, 6H), 2.20 (m, 2H), 1.47 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 164.6, 143.7, 141.9, 140.2, 133.6, 127.6, 127.5, 123.6, 123.0, 120.6, 115.9, 115.6, 90.6, 72.2, 57.5, 43.0, 38.0, 20.2; 19F NMR (376 MHz, DMSO-d6) δ −113.5, 117.0; IR (powder) 1159, 1199, 1222 cm−1; Anal. (C25H25F2NO· C2H2O4·1/4 H2O) C, H, N.
3-(5-(3-Chlorophenyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (16)
Compound 16 was prepared by following the general procedure B using 5 (0.39, 1 mmol) and 3-chlorophenylboronic acid (0.19 g, 1.1 mmol), eluting with chloroform/MeOH (30:1) to give product (0.39 g) in 95% yield. GC-MS (EI) m/z 409 (M+). The oxalate salt was precipitated from acetone; mp 110–111 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.68 (s, 1H), 7.58 (m, 6H), 7.47 (t, J = 6.8 Hz, 1 H), 7.40 (d, J = 8.4 Hz, 1H), 7.16 (dd, J = 7.2 Hz, 2H), 5.20 (dd, J = 12.4, 12.8Hz, 1H), 5.17 (dd, J = 11.6, 12.0 Hz, 1H), 2.96 (t, J = 7.2 Hz, 2H), 2.62 (s, 6H), 2.20 (m, 2H), 1.50 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 164.9, 144.4, 142.8, 141.7, 140.3, 139.2, 134.6, 131.6, 128.2, 127.9, 127.7, 127.3, 126.2, 126.3, 123.2, 120.4, 115.8, 115.5, 90.9, 72.2, 57.3, 43.0, 20.3; 19F NMR (376 MHz, DMSO-d6) δ −116.7; IR (powder) 1170, 1222 cm−1; Anal. (C25H25ClFNO· C2H2O4·1/4 H2O) C, H, N.
(E)-3-(5-(4-Chlorostyryl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (17)
Compound 17 was prepared by following the general procedure B using 5 (0.38, 1 mmol) and trans-4-chlorophenylvinylboronic acid (0.18 g, 1 mmol), eluting with chloroform/MeOH (30:1, 10:1) to give product (0.4 g) in 90% yield; GC-MS (EI) m/z 435 (M+). The oxalate salt was precipitated from acetone; mp 113–115 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.42-7.57 (m, 7H), 7.40 (d, J = 7.6 Hz, 2 H), 7.24 (m, 2H), 7.13 (dd, J = 8.4, 8.4 Hz, 2H), 5.13 (dd, J = 12.8, 13.6Hz, 1H), 5.12 (dd, J =12.4, 13.6 Hz, 1H), 2.96 (t, J = 7.2 Hz, 2H), 2.61 (s, 6H), 2.16 (t, J = 7.2 Hz, 2H), 1.49 (m, 1H), 1.45 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 165.2, 163.1, 160.6, 144.1, 141.8, 140.0, 137.4, 136.6, 132.7, 129.5, 129.4, 128.8, 128.0, 127.6, 127.5, 127.3, 122.8, 120.0, 115.8, 115.6, 90.6, 72.0, 57.3, 42.8, 38.0, 31.4, 20.0; 19F NMR (376 MHz, DMSO-d6) δ −117.5; IR (powder) 1159, 1222, 1718 cm−1; Anal. (C27H27ClFNO· C2H2O4·1/3 H2O) C, H, N.
3-(1-(4-Fluorophenyl)-4-phenyl-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (18)
Compound 18 was prepared by following the general procedure B using 6 (0.2 g, 0.53 mmol) and phenyl boronic acid (0.065 g, 0.53 mmol), eluting with chloroform/MeOH (10:1) to give product (0.2 g) in 95% yield. GC-MS (EI) m/z 375 (M+). The oxalate salt was precipitated from 2-propanol; mp 152–154 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.59 (m, 2H), 7.50-7.35 (m, 8H), 7.14 (dd, J = 8.8, 9.2 Hz, 2H), 5.32 (d, J = 12 Hz, 1H), 5.16 (d, J = 13.6 Hz, 1H), 2.89 (m, 2H), 2.57 (s, 6H), 2.29 (m, 2H), 1.41 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 165.0, 139.9, 128.7, 128.6, 127.9, 127.7, 127.6, 127.1, 127.0, 120.8, 115.0, 114.8, 90.3, 71.5, 57.9, 42.2, 37.6, 19.9; 19F NMR (376 MHz, CD3OD) δ −118.4; IR (powder) 1170, 1234 cm−1; GC-MS (EI) m/z 375 (M+). Anal. (C25H26FNO· C2H2O4·1/4 H2O) C, H, N.
(E)-3-(1-(4-Fluorophenyl)-4-styryl-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (19)
Compound 19 was prepared by following the general procedure B using 6 (0.28 g, 0.74 mmol) and 2-phenylvinyl boronic acid (0.12 g, 0.75 mmol), eluting with chloroform/ MeOH (30:1, 10:1) to give the product (0.27 g) in 91% yield. GC-MS (EI) m/z 401(M+). The oxalate salt was made from Acetone. Recrystallization from methanol gave pure 19; mp 175–176 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.59-7.16 (m, 14H), 5.38 (dd, J = 13.6, 11.6 Hz, 1H), 5.36 (dd, J =11.2, 12.0 Hz, 1H), 2.97 (t, J = 8.0 Hz, 2H), 2.62 (s, 6H), 2.19 (t, J = 7.6 Hz, 2H), 1.51 (m, 1H), 1.45 (m, 1H);13C NMR (100 MHz, DMSO-d6) δ 164.0, 145.0, 137.5, 136.9, 131.9, 129.4, 129.0, 127.6, 127.5, 127.4, 126.3, 126.0, 115.8, 115.6, 90.8, 72.1, 57.5, 46.8, 43.1, 20.2; 19F NMR (376 MHz, DMSO-d6) δ −116.7; IR (powder) 1158, 1228, 1718 cm−1; GC-MS (EI) m/z 401 (M+). Anal. (C27H28FNO· C2H2O4·1/4 H2O) C, H, N.
3-(5-(3-Fluorophenyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (20)
Compound 20 was prepared by following the general procedure B using 6 (0.2, 0.53 mmol) and 3-fluorophenylvinylboronic acid (0.09 g, 0.64 mmol), eluting with chloroform/MeOH (30:1, 10:1) to give the product (0.2 g) in 95% yield. GC-MS (EI) m/z 393 (M+). The oxalate salt was precipitated from acetone; mp 1441-146 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.59 (dd, J = 4.8, 5.2 Hz, 2H), 7.53-7.38 (m, 4H), 7.30 (dd, J = 10.4 Hz, 7.6 Hz, 2H), 7.17 (m, 3H), 5.34 (d, J = 12.8 Hz, 1H), 5.18 (d, J = 13.2 Hz, 1H), 2.93 (t, J = 8 Hz, 2H), 2.60 (s, 6H), 2.18 (t, J = 7.6 Hz, 2H), 1.45 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 164.9, 145.5, 141.7, 136.9, 134.7, 131.5, 131.4, 129.4, 128.4, 127.7, 127.6, 124.7, 122.2, 115.8, 115.6, 115.4, 115.1, 90.8, 71.9, 57.5, 43.0, 38.1, 20.1; 19F NMR (376 MHz, DMSO-d6) δ −113.6, −117.2; IR (powder) 1169, 1220 cm−1; Anal. (C25H25F2NO· C2H2O4) C, H, N.
3-(1-(4-Fluorophenyl)-5-iodo-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (21)
A suspension of 5 (0.38, 1 mmol), KI (2.46 g, 15 mmol) and CuI (0.95 g, 5 mmol) in HMPA (3 mL) was heated at 150 °C for 3 h. The reaction mixture was purified by flash column chromatography eluting with chloroform/MeOH (30:1, 5:1) to give the product (0.2 g) in 47% yield; GC-MS (EI) m/z 425 (M+). The oxalate salt was precipitated from EtOAc; mp 140–142 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.57 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.42 (dd, J = 5.6 Hz, 7.6 Hz, 2H), 7.21 (d, J = 8 Hz, 1H), 7.02 (dd, J = 8.8 Hz, 8.8 Hz, 2H), 5.00 (dd, J = 13.2 Hz, 13.6 Hz, 1H), 4.96 (dd, J = 13.2 Hz, 13.6 Hz, 1H), 2.82 (t, J = 7.2 Hz, 2H), 2.37 (s, 6H), 2.03 (t, J = 7.6 Hz, 2H), 1.35 (m, 1H), 1.29 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 164.6, 143.9, 141.8, 136.6, 130.8, 127.2, 124.5, 123.4, 115.5, 115.3, 90.4, 71.2, 57.1, 42.7, 37.5, 19.7; 19F NMR (376 MHz, DMSO-d6) δ −112.8; IR (powder) 1152, 1222 cm−1; GC-MS (EI) m/z 425 (M+). Anal. (C19H21FINO· C2H2O4) C, H, N.
4-(4-(Dimethylamino)-1-(4-fluorophenyl)-1-hydroxybutyl)-3-(hydroxymethyl)benzonitrile (22)
Compound 22 was prepared by following the general procedure A using 5-cyanophthalidite (6.36 g, 40 mmol) to give the crude diol, which was purified by chromatography eluting with chloroform /MeOH (10:1) to give pure product (11 g) in 79% yield. 1H NMR (400 MHz, CDCl3) δ 7.61-7.54 (m, 3H), 7.31-7.26 (m, 2H), 6.97 (dd, J = 8.4, 8.8 Hz, 2H), 4.42 (d, J = 12 Hz, 1H), 4.16 (d, J = 12 Hz, 1H), 2.37 (m, 4H), 2.22 (s, 6H), 1.66 (m, 1H), 1.57 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ 161.4, 149.6, 142.9, 130.7, 130.3, 128.4, 127.6, 119.8, 115.0, 110.3, 77.4, 60.5, 59.8, 45.6, 41.1, 22.1; GC-MS (EI) m/z 342 (M+); 1H NMR (400 MHz, CDCl3) with the chemical shift reagent (R)-(−)-1-(9-Anthryl)-2,2,2-trifluoroethanol (TFAE, 1/1 weight ratio), area ratio of the peaks with chemical shift 2.17 : 2.15 = 48 : 52 (ee = 4%).
(S)-(−)-4-(4-(Dimethylamino)-1-(4-fluorophenyl)-1-hydroxybutyl)-3-(hydroxymethyl)benzonitrile (S-22)
Compound S-22 was prepared by following the general procedure C using 22 (3.5 g, 10 mmol) and (+)-di-p-toluoyl-D-tartaric acid monohydrate (1 g, 2.5 mmol) to give the enantiomerically pure free base (0.5 g) in 14 % yield. 1H NMR (400 MHz, CDCl3) with the chemical shift reagent (R)-(−)-1-(9-Anthryl)-2,2,2-trifluoroethanol (TFAE, 1/2 weight ratio), area ratio of the peaks with chemical shift 2.19 : 2.17 = 99/1 (ee = 98%). [α]D26 = −61.09 ± 0.075 (c = 2.01, MeOH).
(R)-(+)-4-(4-(Dimethylamino)-1-(4-fluorophenyl)-1-hydroxybutyl)-3-(hydroxymethyl)benzonitrile (R-22)
Compound R-22 was prepared by following the general procedure C using 22 (7.8 g, 23 mmol) and (−)-di-p-toluoyl-L-tartaric acid monohydrate (2.3 g, 5.75 mmol) to give the enantiomerically pure free base (1.27 g) in 15% yield. 1H NMR (400 MHz, CDCl3) with the chemical shift reagent (R)-(−)-1-(9-Anthryl)-2,2,2-trifluoroethanol (TFAE, 1/2 weight ratio), area ratio of the peaks with chemical shift 2.19 : 2.17 = 1/99 (ee = 98%). [α]D24 = +59.02 ± 0.074 (c =3.98, MeOH).
4-(4-(Dimethylamino)-1-(4-fluorophenyl)-1-hydroxybutyl)-3-(hydroxymethyl)benzobromide (23)
Compound 23 was prepared by following the general procedure A using 5-bromophthalide (8.5 g, 40 mmol). The reaction mixture became a green cloudy suspension after the addition of two Grignard reagents. The pure di-magnesium salt was filtered and treated with aq NH4Cl (sat.) to give the pure diol (2.38 g) in 15% yield. More diol was obtained from the filtrate left by following the same work-up procedure in general procedure A. 1H NMR (400 MHz, CDCl3) δ 7.41, 7.32 (2 m, 5H), 6.94 (dd, J = 8.8, 8.8 Hz, 2H), 4.33 (d, J = 12 Hz, 1H), 4.07 (d, J = 12 Hz, 1H), 2.45 (m, 2H), 2.32 (m, 2H), 2.22 (s, 6H), 1.68 (m, 1H), 1.54 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 162.9, 160.5, 145.2, 143.9, 143.8, 143.3, 135.7, 130.1, 128.4, 127.9, 127.8, 121.5, 114.9, 114.7, 64.6, 60.2, 45.0, 44.3, 22.5; 19F NMR (376 MHz, DMSO-d6) δ −115.7; GC-MS (EI) m/z 394 (M+); 1H NMR (400 MHz, CDCl3) with the chemical shift reagent (R)-(−)-1-(9-Anthryl)-2,2,2-trifluoroethanol (TFAE, 1/2 weight ratio), area ratio of the peaks with chemical shift 2.22 : 2.20 = 49 : 51(ee = 2%).
(S)-(−)-4-(4-(Dimethylamino)-1-(4-fluorophenyl)-1-hydroxybutyl)-3-(hydroxymethyl)benzobromide (S-23)
Compound S-23 was prepared by following the general procedure C using 23 (9.6 g, 24 mmol) and (+)-di-p-toluoyl-D-tartaric acid monohydrate (2.44 g, 6 mmol) to give the pure salt (2.65 g) in 28% yield. [α]D25 = 11.0 ± 0.74 (c = 1.00, MeOH); Free base was checked by 1H NMR (400 MHz, CDCl3) with the chemical shift reagent (R)-(−)-1-(9-Anthryl)-2,2,2-trifluoroethanol (TFAE, 1/1 weight ratio), only one peak with chemical shift 2.22 (ee > 99%). GC-MS (EI) m/z 394 (M+).
(R)-(+)-4-(4-(Dimethylamino)-1-(4-fluorophenyl)-1-hydroxybutyl)-3-(hydroxymethyl)benzobromide (R-23)
Compound R-23 was prepared by following the general procedure C using 23 (9 g, 23 mmol) and (−)-di-p-toluoyl-L-tartaric acid monohydrate (2.18 g, 5.8 mmol) to give the pure salt (2.4 g) in 27% yield. [α]D25 = −11.2 ± 0.62 (c = 0.65, MeOH); Free base was checked by 1H NMR (400 MHz, CDCl3) with the chemical shift reagent (R)-(−)-1-(9-Anthryl)-2,2,2-trifluoroethanol (TFAE, 1/1 weight ratio), only one peak with chemical shift 2.21 (ee > 99%).
Biology
Scintillation Proximity-Based Binding Studies
Binding of 3H-leucine (140 Ci/mmol; Moravek) to purified LeuT was performed by means of the scintillation proximity assay (SPA) as described6, 11 with 4 nmol of purified protein and 20 nM 3H-leucine per assay in buffer composed of 50 mM Tris, Mes (pH 8.0), 100 mM NaCl, 1 mM TCEP, 20% glycerol, 1 mM DDM and concentration (10−7 - 10−2 M) of the indicated compounds. Note that the 3H–Leu and test compounds were added simultaneously.
Supplementary Material
Figure 1.

Citalopram and its enantiomers S-1 and R-1.
Acknowledgements
This work was funded by the NIDA-Intramural Research Program and by NIH grants DA022413 and DA17293 (JAJ). GC was supported by a NIH Post-Baccalaureate Intramural Research Training Award. A special thanks to Dr. Yuri Belov for the chiral HPLC analyses of S(+)- and R(−)-1.
Abbreviations
- SERT
serotonin transporter
- NET
norepinephrine transporter
- DAT
dopamine transporter
- LeuT
leucine transporter
- NSS
neurotransmitter:sodium (Na+/Cl−) symporter
- SSRI
selective serotonin reuptake inhibitor
Footnotes
Supporting Information Available
Elemental analysis results are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Butler SG, Meegan MJ. Recent developments in the design of anti-depressive therapies: targeting the serotonin transporter. Curr Med Chem. 2008;15(17):1737–1761. doi: 10.2174/092986708784872357. [DOI] [PubMed] [Google Scholar]
- 2.Waugh J, Goa KL. Escitalopram: a review of its use in the management of major depressive and anxiety disorders. CNS Drugs. 2003;17(5):343–362. doi: 10.2165/00023210-200317050-00004. [DOI] [PubMed] [Google Scholar]
- 3.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437(7056):215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 4.Xhaard H, Backstrom V, Denessiouk K, Johnson MS. Coordination of Na(+) by monoamine ligands in dopamine, norepinephrine, and serotonin transporters. J Chem Inf Model. 2008;48(7):1423–1437. doi: 10.1021/ci700255d. [DOI] [PubMed] [Google Scholar]
- 5.Sinning S, Musgaard M, Jensen M, Severinsen K, Celik L, Koldso H, Meyer T, Bols M, Jensen HH, Schiott B, Wiborg O. Binding and orientation of tricyclic antidepressants within the central substrate site of the human serotonin transporter. J Biol Chem. 2010;285(11):8363–8374. doi: 10.1074/jbc.M109.045401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30(6):667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Indarte M, Madura JD, Surratt CK. Dopamine transporter comparative molecular modeling and binding site prediction using the LeuT(Aa) leucine transporter as a template. Proteins. 2008;70(3):1033–1046. doi: 10.1002/prot.21598. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith ME, Wang DN. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat Struct Mol Biol. 2009;16(6):652–657. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang CI, Lewis RJ. Emerging structure-function relationships defining monoamine NSS transporter substrate and ligand affinity. Biochem Pharmacol. 2009;79(8):1083–1091. doi: 10.1016/j.bcp.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 10.Ravna AW, Sylte I, Dahl SG. Structure and localisation of drug binding sites on neurotransmitter transporters. J Mol Model. 2009;15(10):1155–1164. doi: 10.1007/s00894-009-0478-1. [DOI] [PubMed] [Google Scholar]
- 11.Quick M, Winther AM, Shi L, Nissen P, Weinstein H, Javitch JA. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc Natl Acad Sci U S A. 2009;106(14):5563–5568. doi: 10.1073/pnas.0811322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufmann KW, Dawson ES, Henry LK, Field JR, Blakely RD, Meiler J. Structural determinants of species-selective substrate recognition in human and Drosophila serotonin transporters revealed through computational docking studies. Proteins. 2009;74(3):630–642. doi: 10.1002/prot.22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andersen J, Taboureau O, Hansen KB, Olsen L, Egebjerg J, Stromgaard K, Kristensen AS. Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants. J Biol Chem. 2009;284(15):10276–10284. doi: 10.1074/jbc.M806907200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh SK. LeuT: A prokaryotic stepping stone on the way to a eukaryotic neurotransmitter transporter structure. Channels (Austin) 2008;2(5):380–389. doi: 10.4161/chan.2.5.6904. [DOI] [PubMed] [Google Scholar]
- 15.Celik L, Sinning S, Severinsen K, Hansen CG, Moller MS, Bols M, Wiborg O, Schiott B. Binding of serotonin to the human serotonin transporter. Molecular modeling and experimental validation. J Am Chem Soc. 2008;130(12):3853–3865. doi: 10.1021/ja076403h. [DOI] [PubMed] [Google Scholar]
- 16.Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci. 2008;11(7):780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317(5843):1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448(7156):952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- 19.Jorgensen AM, Tagmose L, Jorgensen AM, Bogeso KP, Peters GH. Molecular dynamics simulations of Na+/Cl(−)-dependent neurotransmitter transporters in a membrane-aqueous system. ChemMedChem. 2007;2(6):827–840. doi: 10.1002/cmdc.200600243. [DOI] [PubMed] [Google Scholar]
- 20.Jorgensen AM, Tagmose L, Jorgensen AM, Topiol S, Sabio M, Gundertofte K, Bogeso KP, Peters GH. Homology modeling of the serotonin transporter: insights into the primary escitalopram-binding site. ChemMedChem. 2007;2(6):815–826. doi: 10.1002/cmdc.200600242. [DOI] [PubMed] [Google Scholar]
- 21.Celik L, Schiott B, Tajkhorshid E. Substrate binding and formation of an occluded state in the leucine transporter. Biophys J. 2008;94(5):1600–1612. doi: 10.1529/biophysj.107.117580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plenge P, Gether U, Rasmussen SG. Allosteric effects of R- and S-citalopram on the human 5-HT transporter: evidence for distinct high- and low-affinity binding sites. Eur J Pharmacol. 2007;567(1–2):1–9. doi: 10.1016/j.ejphar.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 23.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322(5908):1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koldso H, Severinsen K, Tran TT, Celik L, Jensen HH, Wiborg O, Schiott B, Sinning S. The two enantiomers of citalopram bind to the human serotonin transporter in reversed orientations. J Am Chem Soc. 2010;132(4):1311–1322. doi: 10.1021/ja906923j. [DOI] [PubMed] [Google Scholar]
- 25.Andersen J, Olsen L, Hansen KB, Taboureau O, Jorgensen FS, Jorgensen AM, Bang-Andersen B, Egebjerg J, Stromgaard K, Kristensen AS. Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter. J Biol Chem. 2010;285(3):2051–2063. doi: 10.1074/jbc.M109.072587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez C, Bogeso KP, Ebert B, Reines EH, Braestrup C. Escitalopram versus citalopram: the surprising role of the R-enantiomer. Psychopharmacology (Berl) 2004;174(2):163–176. doi: 10.1007/s00213-004-1865-z. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez C, Bergqvist PB, Brennum LT, Gupta S, Hogg S, Larsen A, Wiborg O. Escitalopram, the S-(+)-enantiomer of citalopram, is a selective serotonin reuptake inhibitor with potent effects in animal models predictive of antidepressant and anxiolytic activities. Psychopharmacology (Berl) 2003;167(4):353–362. doi: 10.1007/s00213-002-1364-z. [DOI] [PubMed] [Google Scholar]
- 28.Henry LK, Field JR, Adkins EM, Parnas ML, Vaughan RA, Zou MF, Newman AH, Blakely RD. Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. J Biol Chem. 2006;281(4):2012–2023. doi: 10.1074/jbc.M505055200. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez C, Kreilgaard M. R-citalopram inhibits functional and 5-HTP-evoked behavioural responses to the, SSRI, escitalopram. Pharmacol Biochem Behav. 2004;77(2):391–398. doi: 10.1016/j.pbb.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Storustovu S, Sanchez C, Porzgen P, Brennum LT, Larsen AK, Pulis M, Ebert B. R-citalopram functionally antagonises escitalopram in vivo and in vitro: evidence for kinetic interaction at the serotonin transporter. Br J Pharmacol. 2004;142(1):172–180. doi: 10.1038/sj.bjp.0705738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasper S, Sacher J, Klein N, Mossaheb N, Attarbaschi-Steiner T, Lanzenberger R, Spindelegger C, Asenbaum S, Holik A, Dudczak R. Differences in the dynamics of serotonin reuptake transporter occupancy may explain superior clinical efficacy of escitalopram versus citalopram. Int Clin Psychopharmacol. 2009;24(3):119–125. doi: 10.1097/YIC.0b013e32832a8ec8. [DOI] [PubMed] [Google Scholar]
- 32.Bigler AJ, Bogeso KP, Toft A, Hansed V. Quantitative structure-activity relationships in a series of selective 5-HT uptake inhibitors. Eur. J. Med. Chem. - Chimaca Therapeutica. 1977;12(3):289–295. [Google Scholar]
- 33.Eildal JN, Andersen J, Kristensen AS, Jorgensen AM, Bang-Andersen B, Jorgensen M, Stromgaard K. From the selective serotonin transporter inhibitor citalopram to the selective norepinephrine transporter inhibitor talopram: synthesis and structure-activity relationship studies. J Med Chem. 2008;51(10):3045–3048. doi: 10.1021/jm701602g. [DOI] [PubMed] [Google Scholar]
- 34.Xiao X, Antony S, Pommier Y, Cushman M. Total synthesis and biological evaluation of 22-hydroxyacuminatine. J Med Chem. 2006;49(4):1408–1412. doi: 10.1021/jm051116e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nannapaneni V. chowdary Processes for the preparation of escitalopram and its precursor. 2004 WO 2004/065375 A1. [Google Scholar]
- 36.Xu L, Izenwasser S, Katz JL, Kopajtic T, Klein-Stevens C, Zhu N, Lomenzo SA, Winfield L, Trudell ML. Synthesis and biological evaluation of 2-substituted 3beta-tolyltropane derivatives at dopamine, serotonin, and norepinephrine transporters. J Med Chem. 2002;45(6):1203–1210. doi: 10.1021/jm010453u. [DOI] [PubMed] [Google Scholar]
- 37.Kumar N, Nayyar S, Gupata M. Processes for the preparation of citalopram and its intermediate from 5-aminophthalide. 2006 WO 2006/103550 A1. [Google Scholar]
- 38.MacroModel. Maestro using the default setting of commond "flexible ligand alignment". New York, NY: Schrödinger, LLC; 2008. 9.6; 8.5. [Google Scholar]
- 39.Jorgensen AM, Topiol S. Driving Forces for Ligand Migration in the Leucine Transporter. Chem. Biol. Drug Des. 2008;72:265–277. doi: 10.1111/j.1747-0285.2008.00707.x. [DOI] [PubMed] [Google Scholar]
- 40.El-Gindy A, Emara S, Mesbah MK, Hadad GM. Liquid chromatography determination of citalopram enantiomers using beta-cyclodextrin as a chiral mobile phase additive. J AOAC Int. 2006;89(1):65–70. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.