

Abstract
The mammalian 26S proteasome is a 2500 kDa multi-catalytic complex involved in intracellular protein degradation. We describe the synthesis and properties of a novel series of non-covalent di-peptide inhibitors of the proteasome used on a capped tri-peptide that was first identified by high-throughput screening of a library of approx. 350000 compounds for inhibitors of the ubiquitin–proteasome system in cells. We show that these compounds are entirely selective for the β5 (chymotrypsin-like) site over the β1 (caspase-like) and β2 (trypsin-like) sites of the 20S core particle of the proteasome, and over a panel of less closely related proteases. Compound optimization, guided by X-ray crystallography of the liganded 20S core particle, confirmed their non-covalent binding mode and provided a structural basis for their enhanced in vitro and cellular potencies. We demonstrate that such compounds show low nanomolar IC50 values for the human 20S β5 site in vitro, and that pharmacological inhibition of this site in cells is sufficient to potently inhibit the degradation of a tetra-ubiquitin–luciferase reporter, activation of NFκB (nuclear factor κB) in response to TNF-α (tumour necrosis factor-α) and the proliferation of cancer cells. Finally, we identified capped di-peptides that show differential selectivity for the β5 site of the constitutively expressed proteasome and immunoproteasome in vitro and in B-cell lymphomas. Collectively, these studies describe the synthesis, activity and binding mode of a new series of non-covalent proteasome inhibitors with unprecedented potency and selectivity for the β5 site, and which can discriminate between the constitutive proteasome and immunoproteasome in vitro and in cells.
Keywords: chymotrypsin-like, immunoproteasome, 26S proteasome, proteasome inhibitor, β5-subunit, ubiquitin–proteasome system (UPS)
Abbreviations: Ac, acetyl; AMC, 7-amino-4-methylcoumarin; Boc, t-butoxycarbonyl; HBTU, O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate; HEK, human embryonic kidney; LC50, half-maximal lethal concentration; MPD, 2-methyl-2,4-pentanediol; NFκB, nuclear factor κB; IκB, inhibitory protein of NFκB; NFκB-Luc, NFκB–luciferase; PA, proteasomal activator; PDL, poly-D-lysine; RNAi, RNA interference; siRNA, small interfering RNA; Suc, succinyl; TEV, tobacco etch virus; TNF-α, tumour necrosis factor-α; 4xUb-Luc, tetra-ubiquitin–luciferase; UPS, ubiquitin–proteasome system; Z, benzyloxycarbonyl
INTRODUCTION
The 26S proteasome is a very large (2500 kDa) multi-subunit proteolytic complex found in high abundance in all eukaryotic cells that is responsible for the regulated degradation of the majority of intracellular proteins, including those involved in signal transduction, cell-cycle control, apoptosis and inflammatory responses [1–6]. It is composed of the 20S catalytic core particle that is capped at one or both ends by the 19S regulatory complex, also called PA700 [1–6]. The 20S core particle is comprises four stacked heteroheptameric rings that form a cylindrical structure with 2-fold axial symmetry. The two outer α rings gate substrate entry through their interaction with the 19S regulatory complex, which itself serves to bind, unfold, de-ubiquitinate and translocate into the catalytic core substrate proteins that have been conjugated to poly-ubiquitin [1–6]. The two inner β rings form the central proteolytic chamber and each contain three active sites with distinct substrate specificities. These are located on the β1, β2 and β5 subunits and are referred to as having caspase-like, trypsin-like and chymotrypsin-like activities on the basis of their preference for cleaving peptides after acidic, basic or hydrophobic amino acid residues respectively [1–6]. A second form of the 20S proteasome termed the immunoproteasome exists in cells of lymphoid origin and can be induced in most, if not all, cells in response to the pro-inflammatory cytokine interferon-γ [5–7]. This enzyme contains distinct catalytic subunits, designated β1i, β2i and β5i, and is primarily involved in the generation of antigenic peptides for MHC class I presentation, although a function for the immunoproteasome in cytokine production has also been described [8].
For each of the 20S catalytic β-subunits, the hydroxy group of the N-terminal threonine residue (Thr1Oγ) serves as the nucleophile that initiates cleavage of the peptide bond [4,5,9]. Various natural and synthetic compounds that contain electrophilic centres or ‘warheads’ inhibit the proteasome by forming covalent adducts with these active-site threonine residues, including peptide aldehydes, vinyl sulfones, boronic acids, α′β′-epoxyketones, 2-keto-1,3,4-oxadiazoles and β-lactones (Figure 1A) [4–6,9–11]. The di-peptide boronic acid bortezomib (PS-341 or VELCADE®, Millennium Pharmaceuticals, Inc.) is among the most potent, stable and selective of these inhibitors [12–15], and shows nanomolar potency with respect to cytotoxicity across a broad range of human tumour cell types in vitro [13,14]. It is in clinical use for the treatment of multiple myeloma [16–19] and refractory mantle cell lymphoma [20], and is being evaluated for the treatment of other malignancies [21–23]. Bortezomib induces cell death through a variety of transcriptional, translational and post-translational mechanisms, and may be preferentially cytotoxic to cancer cells by enhancing endoplasmic reticulum stress, increasing the expression of pro-apoptotic factors and/or inhibiting pro-survival or DNA-damage repair pathways [4–6,21–23]. More recently, two further closely related di-peptide boronic acids, CEP-18870 and MLN9708, have been described that inhibit cancer cell proliferation in vitro and show anti-tumour activity in solid and haematological preclinical tumour models [24,25].
Figure 1. Examples of covalent (A) and non-covalent (B) proteasome inhibitors.

Bortezomib binds with very high affinity to the β5 site of the proteasome, and to a lesser extent the β1 and β2 sites [15], and behaves as a slowly reversible inhibitor (t½~110 min for dissociation from the β5 site [25]). Analysis of the crystal structure of the yeast 20S in complex with bortezomib indicates that the high affinity of the drug is partly mediated by the covalent interaction of the boron atom with the nucleophilic oxygen of Thr1Oγ, and by the hydrogen bonding between the two acidic boronate hydroxy groups and the amine groups of Gly47N and Thr1N within each active site [5,9,26]. These properties are likely to contribute to the effectiveness of bortezomib as an anti-cancer agent for the treatment of haematological malignancies, but may limit its distribution and therefore broader utility in solid tumours due to the high tissue abundance of the target (the proteasome has been estimated to comprise 2% of total cellular protein [6]). More recently, the β-lactone salinosporamide A (NPI-0052) [27] and the epoxyketone carfilzomib (PR-171) (Figure 1A) [28], which are analogues of lactacystin and epoxomicin respectively, have demonstrated activity in preclinical models of multiple myeloma and chronic lymphocytic leukaemia, and have entered clinical trials for advanced solid and haematological malignancies [23]. These covalent inhibitors have comparable potency to bortezomib, but differ in that they form essentially irreversible adducts with the active site Thr1Oγ residues of the proteasome [4,5,9,10,15,27–30].
Although the reactive ‘warheads’ of covalent proteasome inhibitors can contribute to enzyme potency, they can also lack specificity and be excessively reactive and unstable [10,11,15], properties which may limit their efficacy in vivo. Non-covalent inhibitors that are readily reversible in their interactions with the catalytic 20S β-subunits provide an alternative mechanism for proteasome inhibition. Such inhibitors may offer therapeutic advantages by enabling more widespread tissue distribution through their more rapid binding and dissociation kinetics. TMC-95A is a complex natural cyclic tri-peptide (Figure 1B) that competitively and non-covalently inhibits the chymotrypsin-like activity of the proteasome with nanomolar potency, and also inhibits the caspase-like and trypsin-like activities at higher concentrations [4,5,9,10,31]. The high affinity of TMC-95A is due to its rigid heterocyclic ring system, which provides specific hydrogen-bonding interactions within each 20S active site without covalent modification of the catalytic threonine residues [4,5,9,32]. In addition, synthetic analogues of TMC-95A have been described, including endocyclic biphenyl-ether macrocyles and non-constrained linear tri-peptides that bind non-covalently with altered affinities for the trypsin- and caspase-like sites relative to the chymotrypsin-like site [5,9,33,34]. TMC-95A shows cytotoxicity in the low micromolar range in cancer cells, although the mechanism-based effects of TMC-95A or its derivatives have not been demonstrated [4]. Other non-covalent proteasome inhibitors include a series of 5-methoxy-1-indanone di-peptide benzamides (e.g. CVT-659) [35,36] and 2-aminobenzylstatine derivatives (Figure 1B) [37] that selectively inhibit the chymotrypsin-like site with submicromolar potencies, although these compounds also have poor cellular activity. However, homology modelling of a 2-aminobenzylstatine compound in the chymotrypsin-like binding pocket of the proteasome has led to the design a non-covalent tri-methoxy-L-phenylalanine-containing tri-peptide with maintained selectivity and greatly increased cell potency (Figure 1B) [38]. This compound is the most potent non-covalent proteasome inhibitor described to date, inhibiting the chymotrypsin-like activity in MDA-MB-435 cells and their proliferation with IC50 values of 20 nM and 60 nM respectively [38].
In an effort to identify cell-active inhibitors of the UPS (ubiquitin–proteasome system) with novel chemical scaffolds, we have screened a library of approx. 350000 compounds using a cell-based assay that monitors accumulation of a 4xUb-Luc (tetra-ubiquitin–luciferase) reporter in response to proteasome inhibition [15,39]. Using this approach, we report the identification of a C- and N-terminally capped tri-peptide derived from the unnatural amino acid S-homo-phenylalanine that potently and selectively inhibits the chymotrypsin-like activity of the mammalian and yeast 20S proteasomes. Further optimization, guided by X-ray crystallography of compounds in complex with purified yeast 20S, has yielded a series of non-covalent di-peptide inhibitors of the proteasome with unprecedented in vitro and cellular potencies. The synthesis, binding mode and cellular activity of these compounds are described in the present study.
EXPERIMENTAL
Cell culture
Cells were from the A.T.C.C. (Manassas, VA, U.S.A.), with the exception of the diffuse large B-cell lymphoma lines which were obtained from the following sources: Karpas-1106P, Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ; Braunschweig, Germany); WSU-DLCL2, Asterand (Detroit, MI, U.S.A.); and OCI-Ly10, provided by Dr Louis M. Staudt (National Cancer Institute, National Institutes of Health, Bethesda, MD, U.S.A.). Cells were cultured at 37 °C in a humidified air/6% CO2 atmosphere in medium supplemented with 10% fetal bovine serum, except for the medium for Karpas-1106P and OCI-Ly10 cells which contained 20% fetal bovine serum, and 100 units/ml penicillin/100 μg/ml streptomycin (all from Invitrogen), as specified: Calu6 cells, minimum essential medium; H460, WSU-DLCL2 and Karpas-1106P cells, RPMI 1640 medium; HCT116 and HT29 cells, McCoy’s 5a medium; and OCI-Ly-10, Iscove’s modified Dulbecco’s medium. Clonally-derived stable MDA-MB-231 cells expressing four tandem copies of ubiquitin fused to firefly luciferase (4xUb-Luc) and HEK (human embryonic kidney)-293 cells expressing NFκB-Luc [NFκB (nuclear factor κB)–luciferase] were generated and maintained as described previously [15].
Reporter assays
Cells were seeded at 10000 cells per well in white BioCoat™ PDL (poly-D-lysine)-coated 384-well plates (BD Biosciences) at 16–24 h prior to compound treatment. For the 4xUb-Luc assays, MDA-MB-231 cells were incubated with compound for 8 h. For NFκB-Luc assays, HEK-293 cells were pre-treated for 1 h with proteasome inhibitor and then stimulated with 10 ng/ml recombinant human TNF-α (tumour necrosis factor-α) (R&D Systems) for a further 3 h in the continued presence of the compound. Firefly luciferase activity was measured using Bright-Glo™ reagents according to the manufacturer’s instructions (Promega) in a LEADseeker™ plate reader (GE Healthcare Life Sciences). Inhibition of NFκB-Luc activity was calculated relative to a no-compound (DMSO) control, whereas 4xUb-Luc reporter accumulation was expressed as a fold increase in luciferase activity over the DMSO control.
Cell viability assay
Calu6, HT29, MDA-MB-231 cells (each at 2000 cells/well), H460 cells (1000 cells/well) and HCT116 cells (1500 cells/well) were plated in black clear-bottomed BioCoat™ PDL-coated 384-well plates (BD Biosciences). Cells were incubated with compound for 72 h, after which the medium was removed to leave 25 μl per well. An equal volume of ATPlite™ reagent (PerkinElmer) was then added and luminescence was measured using a LEADseeker™ instrument.
siRNA (small interfering RNA) transfection and assay
MDA-MB-231 4xUb-Luc cells were transfected in a 384-well format with 10 nM siRNAs (siGENOME SMARTpool, Dharmacon) using DharmaFECT 1 (DH1) reagent (Dharmacon) as follows. For the preparation of the RNAi (RNA interference) transfection mixture for each time point, 40 μl of OptiMEM I (Invitrogen) was dispensed into duplicate wells of a 384-well plate each containing 9 μl/well of 0.5 μM siRNA in siRNA buffer (Dharmacon). OptiMEM (50 μl) containing 0.53 μl of DH2 transfection reagent (Dharmacon) was then added to each well and the plates were incubated at room temperature (22 °C) for 20 min. The transfection mixture (14 μl) from duplicate wells was then transferred into six replicate wells of two separate BioCoat™ PDL-coated 384-well cell plates (BD Biosciences), one for 4xUb-Luc assay and one for ATPlite assay (white and black clear-bottomed respectively). Reverse transfection was performed by the addition of 50 μl of MDA-MB-231 4xUb-Luc cells to each well to give 1200 cells/well. At 48, 72 or 96 h after transfection, one set of duplicate plates was assayed for 4xUb-Luc reporter activity and cell viability. For each time point, six replicate wells of cells transfected with buffer only and the non-targeting GL2 luciferase control duplex were each treated with either DMSO or 1 μM bortezomib for 8 h prior to performing the 4xUb-Luc assay.
Cell-based Proteasome-Glo™ β5 assays
Calu6 cells were plated as for the reporter assays and incubated with compound for 1 h at 37 °C. B-cell lymphoma subtypes Karpas-1106P, WSU-DLCL2 and OCI-Ly10 were plated at 20000 cells per well in 384-well plates and treated with compounds identically. The activity of the 26S proteasome was measured in situ after compound removal by monitoring hydrolysis of the β5 (chymotrypsin-like) substrate Suc-LLVY-aminoluciferin (where Suc is succinyl) in the presence of luciferase using the Proteasome-Glo™ assay reagents according to the manufacturer's instructions (Promega). Luminescence was measured using a LEADseeker™ instrument.
In vitro assays of purified 20S
The peptidase activities of purified human erythrocyte and peripheral blood monocyte 20S proteasomes (Boston Biochem™) were assayed using fluorogenic tri-and tetra-peptide substrates coupled to AMC (7-amino-4-methylcoumarin) [40] (AnaSpec) in the presence of recombinant PA28α (proteasomal activator 28α; Boston Biochem™). The following selective substrates were obtained from AnaSpec, with the exception of Z-LLE-AMC (where Z is benzyloxycarbonyl), which was from Boston Biochem™, and were each used at a final concentration of 15 μM to assay the constitutive (c) and immunoproteasome (i) sub-sites: β1c, Z-LLE-AMC; β2c, Ac-KQL-AMC (where Ac is acetyl); β5c, Ac-WLA-AMC; β1i, Ac-PAL-AMC; β2i, Ac-KQL-AMC; and β5i, Ac-ANW-AMC. Reactions were performed at 37 °C in 384-well black microtitre plates (Corning) using 0.25 nM 20S and 12 nM PA28α in a final volume of 50 μl of buffer containing 20 mM Hepes, pH 7.4, 0.5 mM EDTA and 0.01% BSA. Peptidase activity was measured by monitoring the AMC liberation over time with a Polarstar Galaxy fluorimeter (BMG Labtechnologies) using excitation and emission wavelengths of 340 nm and 460 nm respectively. Percentage inhibition of 20S activity was calculated relative to the controls with DMSO and 10 μM ML599698, an analogue of bortezomib that contains a phenyl group in place of the N-terminal pyrazine cap.
In vitro assays of 20S proteasomes in cell extracts
B-cell lymphoma extracts were prepared by hypotonic lysis and assayed for proteasome activity as described previously [15] using 10 μg of protein extract per reaction in the presence of 12 nM PA28α and the following substrates, each at 100 μM, to monitor the catalytic activity of the c or i 20S sub-sites, as indicated: β1i, Ac-PAL-AMC; β5i, Ac-ANW-AMC; β5c, Ac-WLA-AMC; and β5 Suc-LLVY-AMC.
Kinetic analysis of purified 20S inhibition
Inactivation of purified human erythrocyte 20S was determined by monitoring the hydrolysis of the β5-specific fluorigenic peptide substrate Suc-LLVY-AMC in the presence of different concentrations of inhibitor. Substrate cleavage was continuously monitored as a change in the fluorescence emission at 460 nm (excitation wavelength, 360 nm) and plotted as a function of time. Assays were performed at 37 °C in cuvettes in a final volume of 2 ml in a reaction buffer containing 20 mM Hepes, pH 7.5, 0.5 mM EDTA, 0.01% SDS, 0.25 nM 20S and 10 μM Suc-LLVY-AMC, with continuous stirring. The apparent dissociation constant, Kiapp, was determined by non-linear least-fit of the fractional velocity, vs/v0, as a function of [I], where vs is the steady-state residual activity of the enzyme in the presence of inhibitor (I) and v0 is the initial velocity in the absence of inhibitor:
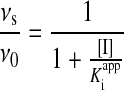 |
The dissociation constant, Ki, was calculated from the apparent dissociation constant, Kiapp, using the following expression, where [S] is the substrate concentration and Km is the substrate binding constant (for 20 μM Suc-LLVY-AMC):
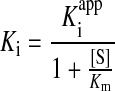 |
Protein turnover assay
Bulk protein turnover was measured in HCT116 cells according to previously published procedures [41], as described in the Supplementary material at http://www.BiochemJ.org/bj/430/bj4300461add.htm.
Synthesis of compound 1, the capped tri-peptide 5-methyl-N-((S)-1-((S)-1-((S)-1-(4-methylbenzylamino)-1-oxo-4-phenylbutan-2-ylamino)-1-oxo-4-phenylbutan-2-ylamino)-1-oxo-4-phenylbutan-2-yl)pyrazine-2-carboxamide
The trimeric compound 1 was identified by LC-MS as an impurity in an active well identified during the high-throughput screen [42] and its structure was confirmed by full characterization of material prepared according to Scheme 1, as detailed in the Supplementary Experimental section at http://www.BiochemJ.org/bj/430/bj4300461add.htm.
Scheme 1. Synthesis of compound 1.

(i) ArCH2-NH2, HBTU, NMM (N-methylmorpholine), DMF (N,N-dimethylformamide), 25 °C, 24 h; (ii) 4 M HCl, dioxane; (iii) N-Boc-homo-phenylalanine, TBTU [O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium-tetrafluoroborate], collidine, DMF, 25 °C, 3 h; (iv) 4-methylpyrazinecarboxylic acid, TBTU, collidine, DMF, 25 °C, 3 h.
Synthesis of capped di-peptide proteasome inhibitors
The synthesis of di-peptide analogues was carried out using standard peptide-coupling conditions, typically using HBTU (O-benzotriazole-N,N,N′,N′-tetramethyluronium hexafluorophosphate) to activate the carboxylic acids according to Scheme 2. The P1 and P2 residues (R1 and R2 respectively) were installed as described above for the preparation of compound 1 by coupling the appropriate amine to a Boc (t-butoxycarbonyl)-protected α-amino acid, followed by de-protection to give intermediates II as hydrochloride salts. The three-step elaboration of II to the di-peptide analogues of compound 1 (IV) was carried out using automated solution-phase parallel synthesis techniques in deep-well plates without purification of intermediates, but with rigorous purification and full characterization of final products. A representative procedure for the parallel synthesis of compound arrays of IV can be found in the Supplementary Experimental section.
Scheme 2. Synthesis of compound IV.

(i) R1-NH2, HBTU, NMM (N-methylmorpholine), DMF (N,N-dimethylformamide), 25 °C, 24 h; (ii) 4 M HCl, dioxane; (iii) P3 Boc-amino acid, HBTU, NMM, DMF, 25 °C, 24 h; (iv) R4COOH, HBTU, NMM, DMF, 25 °C, 24 h.
Purification of yeast 20S
Yeast 20S wild-type and open-gate (an N-terminal tail deletion from the α3 and α7 subunits, designated α3/7ΔN) mutant containing a TEV (tobacco etch virus)-protease-cleavable Protein A-derived tag in the core particle subunit Pre1 (β4) were affinity-purified from the SDL135 and yMS159 strains respectively (gifts from Dr M. Schmidt and Dr D. Finley, Harvard University Medical School, Boston, MA, U.S.A.) [43,44] on IgG–Sepharose 6 Fast Flow (GE Healthcare), according to the procedure of Leggett et al. [45], except that ATP was omitted from the buffers and the enzyme was eluted from the IgG resin by incubation with the His6–TEV protease (Invitrogen) overnight at 4 °C. The resultant yeast 20S was purified further by gel filtration on a 24 ml Superose 6 10/30 column (GE Healthcare) in 10 mM Tris/HCl, pH 7.5, containing 1 mM EDTA, concentrated to 30 mg/ml using an Amicon Centriplus YM-100 centrifugal filter device (Millipore) and stored at −80 °C.
X-ray crystallography
Crystals of purified wild-type and the open-gate mutant yeast 20S proteasome were grown in hanging drops at room temperature, as described previously [46], using a drop volume of 1.5 μl of 20S (20 mg/ml in 10 mM Tris/HCl, pH 7.5, and 1 mM EDTA) and 0.5 μl of reservoir solution containing 100 mM Mes, pH 7.0, 40 mM Mg(CH3COO)2, 15% MPD (2-methyl-2,4-pentanediol) and 10 mM EDTA. Proteasome–inhibitor complexes were generated by soaking crystals overnight in reservoir buffer containing 1 mM compound, 10% DMSO and 20% MPD followed by an additional 5 h in reservoir buffer containing 1 mM compound, 10% DMSO and 25% MPD before being flash-cooled in liquid nitrogen. Crystal data and refinement statistics are given in Supplementary Table S1 at http://www.BiochemJ.org/bj/430/bj4300461add.htm. Data were collected using the Structural GenomiX (SGX)-CAT beamline at the Advanced Photon Source (APS) synchrotron of the Argonne National Laboratory (U.S. Department of Energy Chicago, IL, U.S.A.) and processed using the programs iMOSFLM [47] and SCALA [48]. Starting co-ordinates for each of the proteasome–inhibitor structures were taken from PDB entry 1G0U [43]. SigmaA-weighted Fo−Fc difference electron density for the β5 active site was used to model inhibitor co-ordinates, starting with a conformation generated using the small-molecule topology generator PRODRG [49]. Model building was performed using the program Coot [50] and refinement was carried out using the CCP4i graphical user interface [51] to the REFMAC program [52].
RESULTS
Cell-based screen for inhibitors of the UPS
To identify novel cell-active small-molecule inhibitors of the UPS, a high-throughput screen of the Millennium Pharmaceuticals, Inc., library was performed for compounds that stabilized 4xUb-Luc in MDA-MB-231 cells. In this system, bortezomib and other catalytic 20S inhibitors cause a concentration-dependent accumulation of the constitutively expressed reporter by blocking its intracellular degradation, as reported previously [15]. To further validate the dependence of this reporter on the 26S proteasome, siRNAs were used to deplete selected components of the 20S core particle and 19S regulatory complex in MDA-MB-231-4xUb-Luc cells. As shown in Figure 2, depletion of subunits of either the 20S (α1 and β6) or 19S (rpn8 and rpn11) complex resulted in a time-dependent increase in the 4xUb-Luc signal comparable with that observed by an 8 h treatment of the cells with a near-saturating concentration of bortezomib of 1 μM.
Figure 2. Effects of bortezomib and depletion of 26S subunits by RNAi on accumulation of 4xUb-Luc in cells.

MDA-MB-231 cells stably expressing a 4xUb-Luc reporter were reverse-transfected with 10 nM siRNA SMARTPools to deplete components of the 19S regulatory cap or 20S catalytic core particle of the 26S proteasome, as indicated. At 48, 72 or 96 h after transfection, the accumulation of the 4xUb-Luc was measured as luminescence using firefly luciferase reagents, as described in the Experimental section. Mock transfections with buffer or with the GL2 luciferase control duplex were also performed to monitor basal expression of the 4xUb-Luc reporter and its stabilization by 1 μM bortezomib for the final 8 h of transfection. Results are the means±S.E.M. for six assay points and are representative of four independent experiments. PSMA, proteasome subunit α-type; PSMB, proteasome subunit β-type; PSMD, proteasome 26S subunit non-ATPase; rpn, regulatory particle non-ATPase.
High-throughput screening of approx. 352500 compounds at 50 μM gave 3015 hits that stabilized 4xUb-Luc by 2-fold or greater, representing a primary hit rate of 0.86%, and 611 of these positives were confirmed when re-tested in triplicate at the same concentration and cut-off, representing a re-test rate of 20.3%. The majority of these compounds were either peptide boronic acid inhibitors of the proteasome or ubiquitin-activating enzyme (E1) inhibitors that were part of the library generated by proprietary medicinal chemistry and served as screening controls. There were 191 compounds which were previously uncharacterized as inhibitors of the UPS, for which cell-based potency determinations were performed. Of these, 52 gave modest 2–3-fold stabilization, whereas 102 compounds gave a 3–10-fold stabilization and 37 compounds gave >10-fold stabilization of the reporter at concentrations up to 50 μM.
Identification of compound 1 as a potent selective 20S β5 inhibitor
The cell-active compounds were assessed for inhibition of purified rat brain 20S in vitro by measuring hydrolysis of the β5 (chymotrypsin-like) substrate Ac-WLA-AMC in the presence of PA28α. Only a small minority of compounds inhibited 20S, showing low and sub-micromolar potencies (results not shown). Among the most potent of these were four closely related peptidic benzyl amides synthesized by combinatorial chemistry, of which one was subsequently identified by MS and re-synthesis as a C- and N-terminally capped tri-peptide derived from the unnatural amino acid S-homo-phenylalanine, and designated as compound 1 (Figure 3) In vitro, this compound inhibited the β5 activity of the human constitutive and immunoproteasome with low nanomolar potencies, comparable with those of bortezomib, but was essentially inactive against the β1 (caspase-like) and β2 (trypsin-like) sites of either proteasome isoform (Table 1). β5 selectivity is in contrast with that of bortezomib, which also inhibits the β1 and, to a lesser extent, the β2 sites of the constitutive and immunoproteasome (Table 1). Assays against a diverse panel of ten proteases indicated that compound 1 was highly selective for the proteasome, showing only partial (<50%) inhibitory activity against cathepsin L, coagulation Factor β-XIIa, thrombin, tissue plaminogen activator and trypsin at concentrations approx. 10000-fold higher than its IC50 value for the 20S β5 site (Supplementary Table S2 at http://www.BiochemJ.org/bj/430/bj4300461add.htm). Kinetic analysis of human 20S β5 inhibition by compound 1 showed that it behaved as a rapid-equilibrium inhibitor with a Ki value of 0.50±0.12 nM (mean±S.E.M., n=3), consistent with reversible non-covalent binding to the active site of the enzyme (Figure 4). This behaviour is again in contrast with that of bortezomib, which acts as a time-dependent slow–tight binding inhibitor that interacts covalently with the active site Thr1 residue, but which displays an equivalent Ki value for the β5 site of 0.56±0.072 nM (mean±S.E.M., n=3) (Figure 4 and [25]).
Figure 3. Structure of the capped tri-peptide screening hit, 5-methyl-N-[(S)-1-{(S)-1-[(S)-1-(4-methylbenzylamino)-1-oxo-4-phenylbutan-2-ylamino]-1-oxo-4-phenylbutan-2-ylamino}-1-oxo-4-phenylbutan-2-yl]pyrazine-2-carboxamide, compound 1.

Table 1. Active-site-selectivity of the peptide boronic acid, bortezomib, the capped tri-peptide screening hit, compound 1, and a series of fifteen capped di-peptides in vitro.
Purified human erythrocyte 20S and peripheral blood monocyte 20S (0.25 nM each) were used as a source of constitutive (c) and immunoproteasome (i) respectively to measure the peptidase activity of the β1 (caspase-like), β2 (trypsin-like) and β5 (chymotrypsin-like) sub-sites of each proteasome isoform. Assays were performed in the presence of 15 μM of the indicated fluorigenic peptide-AMC substrate, 12 nM PA28α and a titration of each inhibitor. As shown, and unlike bortezomib, compounds 1–16 were essentially inactive against the β1 and β2 sites of the constitutive proteasome and immunoproteasome. The IC50 values of the compounds against the β5c and β5i sites are mean values for at least three independent determinations.
| Human 20S–PA28 IC50 (nM) | ||||||
|---|---|---|---|---|---|---|
| Compound | β1c Z-LLE-AMC | β1i Ac-PAL-AMC | β2c Ac-KQL-AMC | β2i Ac-KQL-AMC | β5c Ac-WLA-AMC | β5i Ac-ANW-AMC |
| Bortezomib | 140 | 5.5 | 1500 | 940 | 8.2 | 3.3 |
| 1 | 28000 | >100000 | 57000 | >100000 | 16 | 8.9 |
| 2 | 83000 | >100000 | >100000 | >100000 | 1100 | 230 |
| 3 | 43000 | 65000 | 54000 | 47000 | 130 | 37 |
| 4 | 48000 | >100000 | >100000 | >100000 | 470 | 41 |
| 5 | 39000 | >100000 | >100000 | 70000 | 340 | 27 |
| 6 | >100000 | >100000 | >100000 | 44000 | 25 | 10 |
| 7 | 70000 | >100000 | >100000 | 91000 | 9100 | 1300 |
| 8 | 31000 | 83000 | 63000 | 47000 | 13 | 4.1 |
| 9 | 68000 | >100000 | >100000 | >100000 | 6.8 | 6.7 |
| 10 | 90000 | >100000 | >100000 | >100000 | 4.6 | 3.2 |
| 11 | >100000 | >100000 | >100000 | >100000 | 2.4 | 1.1 |
| 12 | 89000 | >100000 | 95000 | >100000 | 4.4 | 3.5 |
| 13 | 84000 | >100000 | 99000 | >100000 | 3.9 | 3.4 |
| 14 | 66000 | >100000 | >100000 | >100000 | 11 | 200 |
| 15 | 64000 | >100000 | >100000 | >100000 | 12 | 44 |
| 16 | >100000 | >100000 | >100000 | >100000 | 1.2 | 1.1 |
Figure 4. Kinetics of proteasome inhibition by compound 1 and bortezomib.

(A and B) The peptidase activity of human erythrocyte 20S (0.25 nM) was monitored over time using the fluorigenic substrate Suc-LLVY-AMC (10 μM) in the presence of 0.01% SDS and following the addition of various concentrations of either compound 1 or bortezomib, as indicated. (C) A non-linear least-fit of the fractional velocity of the reactions, vs/v0, as a function of inhibitor concentration, [I], was used to determine a Ki of 0.50±0.12 nM for compound 1 and 0.56±0.072 nM for bortezomib (means±S.E.M., n=3).
Cellular inhibition of proliferation, 4xUb-Luc degradation, NFκB-Luc activation by compound 1
On the basis of the high affinity and selectivity of compound 1 for the 20S β5 site in vitro, its activity was characterized further in cells as compared with that of bortezomib. Compound 1 inhibited the proliferation of MDA-MB-231 cells in a concentration-dependent manner with an LC50 (half-maximal lethal concentration) value of 150±23 nM (mean±S.E.M., n=3), as determined by an ATPlite assay at 72 h (see Supplementary Figure S1A at http://www.BiochemJ.org/bj/430/bj4300461add.htm). Indeed, viability assays performed across a panel of five cancer cell lines, comprising MDA-MB-231, Calu6, H460, HCT116 and HT29 cells, indicated that compound 1 has broad cytotoxic activity, with LC50 values ranging from 50 nM to 380 nM and being approx. 10–20-fold less potent than bortezomib (Table 2 and Supplementary Table S3 at http://www.BiochemJ.org/bj/430/bj4300461add.htm). In the screening assay, compound 1 caused up to an approx. 100-fold accumulation of the 4xUb-Luc reporter at 8 h [maximal effect of 52±16-fold, with an EC50 (half-maximally effective concentration) of 690±22 nM (mean±S.E.M., n=6)], showing an approx. 3-fold higher EC50 and one-seventh of the efficacy of bortezomib [maximal effect, 340±12-fold; EC50=240±21 nM (mean±S.E.M., n=102)] (Supplementary Figure S1B and Table 2). The activity of compound 1 was also assessed in the Proteasome-Glo™ assay that measures directly the peptidase activity of the β5 site of the 26S proteasome in cells using the luminogenic substrate Suc-LLVY-aminoluciferin. Treatment of Calu6 cells for 1 h with compound 1 caused a concentration-dependent inhibition of β5 activity in this assay with an IC50 (half-maximal inhibitory concentration) value of 53±16 nM (mean±S.E.M., n=3). Bortezomib was approx. 10-fold more potent, inhibiting hydrolysis of the chymotrypsin-like substrate with an IC50 of 3.9±0.49 nM (mean±S.E.M., n=8) (Supplementary Figure S1C and Table 2). Finally, we assessed the effect of compound 1 on activation of the transcription factor NFκB by TNF-α, which is dependent on proteasomal degradation of IκBα (inhibitory protein of NFκB α) following its rapid phosphorylation by the IκB kinase complex and ubiquitination by the SCFβTrCP (E3 ubiquitin-protein ligase complex Skp1–Cullin–F-box) [53]. Compound 1 inhibited TNFα-induced activation of NFκB-Luc in HEK-293 cells with an IC50 of 47±7.7 nM (mean±S.E.M., n=7), consistent with potent proteasome inhibition in these cells. Interestingly, it did not fully inhibit NFκB-Luc, even at saturating concentrations [maximal inhibition, 79±3.2% (mean ±S.E.M., n=7)], whereas bortezomib inhibited NFκB-Luc activity by 100% with an IC50 of 9.7±0.73 nM (mean±S.E.M., n=14) (Supplementary Figure S1D and Table 2). Collectively, these data indicate that selective inhibition of the β5 site of the proteasome by compound 1 in cells is sufficient to inhibit the degradation of the 4xUb-Luc reporter, TNF-α-dependent NFκB activity and the proliferation of cancer cells with potencies that are approximately one order of magnitude lower than those of bortezomib.
Table 2. Cell-based activity of bortezomib, compound 1 and fifteen capped di-peptide proteasome inhibitors.
The cell-based potencies of bortezomib and compounds 1–16 were assessed in a diverse panel of assays that monitor the short- and longer-term effects of proteasome inhibition in cells, as indicated. In particular, inhibition of the β5 activity of the proteasome was measured directly with the Proteasome-Glo™ assay using Suc-LLVY-aminoluciferin as substrate following incubation of Calu6 cells (10000 per well) with compound for 1 h. Inhibition of NFκB-luciferase activity in HEK-293 cells was determined by pre-incubation of the cells (10000 per well) with compound for 1 h followed by stimulation with 10 ng/ml TNF-α in the continued presence of compound for 3 h. The IC50 and percentage inhibition of NFκB-Luc activity are given. Accumulation of the 4xUb-Luc reporter in MDA-MB-231 cells was determined by incubation of the cells (10000 per well) with compound for 8 h and expressed as the fold accumulation of luciferase activity together with an EC50 value for this effect. The effects of the compounds on the viability of Calu6 cells (2000 per well) and H460 cells (1000 per well) were assessed by the ATPlite assay following incubation of the cell with compound for 72 h. The results are mean values for at least three independent experiments.
| Calu6 Proteasome-Glo™ β5 | HEK-293 NFκB-Luc | MDA231 4xUb-Luc | Cell viability | ||||
|---|---|---|---|---|---|---|---|
| Compound | IC50 (nM) | IC50 (nM) | Maximal inhibition (%) | EC50 (nM) | Maximal induction (fold) | Calu6 LC50 (nM) | H460 LC50 (nM) |
| Bortezomib | 3.9 | 9.7 | 100 | 240 | 340 | 5.2 | 13 |
| 1 | 53 | 47 | 79 | 690 | 52 | 130 | 380 |
| 2 | 48000 | >50000 | 28 | 1800 | 3 | >25000 | >25000 |
| 3 | 610 | 3100 | 84 | 2100 | 39 | 1700 | 3100 |
| 4 | 13000 | 24000 | 30 | 6900 | 12 | 13000 | 18000 |
| 5 | 12000 | >50000 | 16 | 4700 | 28 | 2000 | 2600 |
| 6 | 310 | 320 | 89 | 2100 | 76 | 1200 | 4300 |
| 7 | 5400 | 4400 | 86 | 10000 | 65 | 6600 | 17000 |
| 8 | 43 | 29 | 87 | 930 | 86 | 240 | 470 |
| 9 | 10 | 22 | 78 | 130 | 77 | 10 | 34 |
| 10 | 16 | 6.6 | 72 | 47 | 64 | 55 | 82 |
| 11 | 5.7 | 9.8 | 93 | 112 | 65 | 21 | 110 |
| 12 | 36 | 10 | 74 | 71 | 64 | 36 | 110 |
| 13 | 24 | 9.2 | 77 | 64 | 76 | 61 | 240 |
| 14 | 520 | 64 | 77 | 650 | 64 | 210 | 650 |
| 15 | 230 | 39 | 80 | 450 | 82 | 355 | 1000 |
| 16 | 9.5 | 12 | 92 | 31 | 75 | 9.6 | 44 |
X-ray crystallography of compound 1 in complex with the yeast 20S proteasome
To determine its binding mode, compound 1 was soaked into crystals of the yeast 20S core particle. X-ray diffraction data obtained at 3.1 Å resolution indicated that it only occupied the β5 sites of the proteasome (Figure 5A), consistent with selective inhibition of this active site in vitro. The C-terminal 4-methylbenzyl group (P1) was seen to occupy a well-defined S1 pocket, with the first homo-phenylalanine residue (P2) pointing toward solvent, the second (P3) occupying a well-defined S3 pocket and the third (P4) projecting into a much larger and ill-defined S4 pocket (Figure 5B). No electron density was observed corresponding to the N-terminal cap, presumably because this region of the molecule is disordered. The structure of compound 1 overlaid with that of bortezomib, as determined previously [26], suggests that, although the side chains of the two compounds differ in their orientations, most of the backbone hydrogen-bond interactions within the β5 site are the same (Figure 5C). Of note, in this structure as in others, residual electron density corresponding to Mes, the buffer in which the crystals were prepared, was observed near Thr1 in the β5 active site (Figure 5A). The main interaction between Mes and compound 1 involves hydrophobic contacts between the morpholino group of Mes and the homo-phenylalanine residue in the P2 position. For structures without ligand in the active site, no density for the morpholino group was observed (M.D. Sintchak, unpublished work), rather electron density at the position of the sulfur atom of Mes was seen. This sits at the same position as the nucleophilic water molecule referred to as NUK in a previous publication [54]. Importantly, although Mes was observed in the active site following crystallization, this buffer molecule neither affects the specific activity of 20S nor the affinity of compound 1 for the active site on the basis of Ki value determination (F.J. Bruzzese, unpublished work).
Figure 5. Electron-density map of compound 1 and occupancy of the specificity pockets of the 20S β5 subunit.

(A) A 3.1 Å sigmaA-weighted 2Fo-Fc electron-density map contoured at 1.0σ covering compound 1 (PDB entry 3MG4) and Mes in the β5/β6 active site of wild-type 20S is shown in stereo view, with residue positions P1–P4 indicated. Atoms are in ball-and-stick representation with carbon in yellow (compound 1) or salmon (Mes), nitrogen in blue and oxygen in red. The side chain atoms for Thr1 (β5) and Asp114 (β6) are shown in ball-and-stick representation, coloured as above, except for carbon in green. Protein atoms from 20S are shown in cartoon representation coloured by secondary structure with helices in cyan, β-strands in magenta and loops in salmon. The Figure was made using the PyMOL Molecular Graphics System (DeLano Scientific, Palo Alto, CA, U.S.A.). (B) The β5/β6 active site with compound 1 bound (coloured as in A), showing the molecular surface of 20S. Specificity pockets S1–S4 are indicated. (C) Superposition of compound 1 and bortezomib bound to the β5/β6 active site of 20S is shown, with positions P1–P4 indicated. Colour scheme is as described above, except bortezomib carbon atoms are coloured grey.
Optimization of capped di-peptide 20S inhibitors
The partial crystal structure of compound 1 (Figure 5) together with the report of a potent related tri-methoxy-phenylalanine-containing tri-peptide 20S inhibitor (Figure 1 and [38]) suggested that one homo-phenylalanine residue could be deleted, provided that a compensating hydrophobic N-terminal capping group could be incorporated in place of the pyrazine. A series of di-peptide-like libraries was therefore prepared by solution-phase parallel synthesis as outlined in the Experimental section (Scheme 2), denoting residues P1–P4 (Figure 6) according to the binding mode indicated by the structure of compound 1 (Figure 5). Given the close correlation in IC50 values of proteasome inhibitors from various chemical classes for human and yeast 20S (C. Tsu, unpublished work), compounds were optimized for in vitro β5 potency against human 20S, whereas crystal structures of representative examples were determined with the yeast 20S open-gate mutant in order to guide further library design.
Figure 6. Residue assignments of di-peptide inhibitors.

Optimization of compound 1 involved contracting the chain to a di-peptide of the general structure shown and systematically modifying the central amino acids and capping groups, as denoted by residues P1–P4 in a series of compound libraries prepared by solution-phase parallel synthesis (see the Experimental section).
The first library synthesized retained 4-methylbenzylamine as the C-terminal capping group at P1, contained varied amino acid residues at P2 and P3 and probed two alternative hydrophobic N-terminal caps at P4 with 348 compounds in total. All of these di-peptide analogues were less potent than the trimer compound 1, although several inhibited β5 activity with low and sub-micromolar potencies. For example, the truncated analogue of compound 1 with an N-terminal cap substitution, compound 2 (Figure 7), had a β5 IC50 value of 1.1 μM in vitro (Table 1). Substitution of the homo-phenylalanine residues of compound 2 by a tryptophan at P1 and by the di-methoxy-phenylalanine residue [38] at P2 gave compound 3 (Figure 7) with a β5 IC50 value of 130 nM (Table 1). In the next library, smaller replacements for the P3 homo-phenylalanine residue were sought in combination with alternative N-terminal P4 capping groups that provided compensating hydrophobic interactions within the large S4 binding pocket. Numerous compounds, such as compound 4 and the 4-benzyloxybenzoyl derivative compound 5, with small polar P3 residues, in this case a threonine residue (Figure 7), retained reasonable potency in vitro (β5 IC50 values of approx. 500 nM). Significant further improvements were effected by varying the capping group, with the indanone in compound 6 being among the optimal residues at P4 with this compound showing a β5 IC50 of 25 nM (Figure 7 and Table 1) and a Ki value of 13±1.4 nM (mean±S.E.M., n=3).
Figure 7. Chemical structures of capped di-peptide 20S β5 proteasome inhibitors.

The crystal structure of compound 6 bound to the β5 site of yeast 20S was determined at 2.6 Å resolution (Figure 8) and compared with that of bortezomib determined at 2.7 Å, a resolution comparable with that determined previously [26]. In the structure of bortezomib, in addition to the covalent and hydrogen-bonding interactions involving the boronate, backbone hydrogen-bonding interactions with the β5 site can be seen (Figure 8A). Interestingly, the same hydrogen-bonding interactions between the backbone of compound 6 and the β5 site are observed, including the presence of the bridging water molecule between the P3 carbonyl group and Ala49 and Ala50 (Figure 8A). Figures 8(B) and 8(C) show the occupancy of the binding pockets by compound 6, whereas Supplementary Figure S2 (at http://www.BiochemJ.org/bj/430/bj4300461add.htm) shows an alternative view to include the electron density of the water molecule. The 4′-methylbenzyl P1 residue occupies S1, with the methyl group occupying a small sub-pocket at the bottom of the cavity. The P2 homo-phenylalanine residue occupies S2 and the P3 threonine residue projects, to a small degree, towards the deep S3 binding pocket. The P2 homo-phenylalanine residue does not fill the S2 binding pocket, which is quite shallow, but likely confers potency by contributing a hydrophobic interaction at a ‘ledge’ formed from Gly47-Gly48 (Figure 8C). Of note, the P4 indanone in compound 6 is closely related to inhibitors reported previously [35,36], for example, CVT-659 (Figure 1B). Whereas the indanone carbonyl in these molecules was originally designed as a ‘putative electrophilic head group’ [35,36], the crystal structure of compound 6 places this moiety in the S4 binding pocket remote from the β5 catalytic Thr1 residue, thereby precluding such an interaction. In addition, the water molecule appears to be present in all higher-resolution structures (<2.6 Å), including those without ligand bound, such as that reported originally [54]. This water molecule was also observed in structures with inhibitors having IC50 values greater than 1 μM (M.D. Sintchak, unpublished work). Therefore it is unlikely that this single water-mediated hydrogen bond contributes significantly to potency.
Figure 8. Hydrogen-bonding interactions and crystal structure of compound 6 bound to the chymotrypsin-like site of the 20S proteasome with reference to bortezomib.

(A) Schematic representation of the β5/β6 active site of 20S with compound 6 (left-hand panel) (PDB entry 3MG6) and bortezomib (right-hand panel) (PDB entry 3MG0) bound. Key hydrogen bonds between the inhibitors and the protein are shown as dashed lines coloured magenta, with distances indicated in Å. Thus in the structure of bortezomib, the NH of the P1 leucine residue forms a hydrogen bond with the carbonyl of Gly47, whereas the carbonyl and amino groups of the P2 residue form hydrogen-bond interactions with Thr21. The carbonyl of the pyrazinoyl cap is involved in a network of hydrogen-bond interactions with Ala49, Ala50 and Asp114 that includes a bridging water molecule. Similar hydrogen-bonding interactions between the backbone of compound 6 and the β5 site are shown, including the presence of the bridging water molecule between the P3 carbonyl and Ala49 and Ala50; the NH of P3 makes the hydrogen-bond interaction with Asp114 that is formed by the pyrazine N in bortezomib. (B) 2.6 Å sigmaA-weighted 2Fo-Fc electron-density map contoured at 1.0σ covering compound 6 (PDB entry 3MG6) and MES is shown in the β5/β6 active site of yeast 20S open-gated mutant in stereo view, with positions P1–P4 indicated. The colour scheme is as described in Figure 5. (C) β5/β6 active site with compound 6 bound (coloured as described above), showing the molecular surface of 20S. Specificity pockets S1–S4 are indicated. The P2 binding ‘ledge’ is shown by shading of the molecular surface.
In the third library, optimization of the C-terminal P1 residue was explored. The requirement of the hydrogen-bonding interaction with Gly47 can be seen from N-methylation of P1 to give compound 7 (Figure 7), which diminished activity greatly (β5 IC50=9.1 μM; Table 1). However, comparable potency with compound 6 was observed for the 2′-chlorobenzyl derivative compound 8 (β5 IC50=13 nM; Table 1 and Figure 7). A crystal of compound 8 bound to 20S indicated an additional hydrophobic interaction between the ortho-halogen and a second ‘sub-pocket’ within the S1 site that cannot be accessed by the less active meta-substituted analogue ([42] and see Supplementary Figure S3 at http://www.BiochemJ.org/bj/430/bj4300461add.htm).
In the final library, the influence of a hydrophobic P3 residue was assessed, with emphasis on asparagine derivatives. As with the potency-enhancing N-terminal P4 capping groups described above, several low nanomolar inhibitors were identified, such as compounds 9 (β5 IC50=6.8 nM, Table 2) and 10 (β5 IC50=4.6 nM, Table 2) (Figure 7). Potent compounds such as 11 (β5 IC50=2.4 nM, Table 1) and 12 (β5 IC50=4.4 nM, Table 1) in which the homo-phenylalanine residue was replaced by an alanine residue and compound 13 (β5 IC50=3.9 nM, Table 1), in which 3-pyridyl was incorporated, were also identified (Figure 7). Similarly, the neopentyl-asparagine residue was able to compensate for sub-optimal interactions in S1, with compounds 14 and 15 showing good inhibitory activity (β5 IC50=11 and 12 nM respectively; Table 1 and Figure 7). Like compound 1, these di-peptides at 100 μM had negligible to modest activity against ten unrelated proteases (Supplementary Table S2), indicating that the most potent compounds are selective for the chymotrypsin-like site of the proteasome by 4 or 5 orders of magnitude.
The most potent proteasome inhibitor identified was compound 16 (Figure 7) with a β5 IC50=1.2±0.35 nM for the human enzyme (Table 1) and a Ki below the 20S concentration of 250 pM in the assay, indicating that it behaved as a non-covalent active-site titrant (F.J. Bruzzese, unpublished work). The X-ray structure of this compound bound to yeast 20S resolved at 2.6 Å is shown in Figure 9. The potentially electrophilic α-ketoamide N-terminal cap does not form any covalent interactions; it is positioned in the S4 binding pocket which is located away from the catalytic Thr1 and it also does not appear to interact with any residues in S4 (Figure 9A). Importantly, the high potency of compound 16 and all compounds with a P3 neopentyl-asparagine group can be accounted for by the near-optimal fit of this residue in the S3 binding pocket (Figure 9B). As with compound 8 (Supplementary Figure S3), the P1 ortho-chlorine of compound 16 is also likely to contribute to the potency by accessing the S1 sub-pocket (Figure 9A). Of note, however, the aromatic P2 contains only one methylene and therefore the ledge interaction cannot take place with the pyridyl projecting toward solvent (compare with Figure 8C).
Figure 9. Crystal structure of compound 16 bound to the chymotrypsin-like site of the 20S proteasome.

(A) β5/β6 active site with compound 16 (PDB entry 3MG8) bound to yeast open-gated 20S, showing the molecular surface of 20S (coloured as described in Figure 5). Selectivity pockets S1–S4 are indicated. (B) Close-up view of the S3 specificity pocket to illustrate occupancy by the neopentyl-asparagine residue of compound 16 in the β5/β6 active site of the 20S core particle (coloured as described above).
Relationship between 20S β5 potency and cellular activity of capped di-peptides
The effects of the di-peptides on 26S activity in cells were measured (Table 2) and compared with their 20S β5 IC50 values determined in vitro (Table 1 and Supplementary Figure S4 at http://www.BiochemJ.org/bj/430/bj4300461add.htm). Inhibition of the β5 site of the 26S proteasome was measured directly in Calu6 cells using the Proteasome-Glo™ assay and corresponded well with the inhibitory potencies determined in vitro with purified 20S. There was also a striking correlation between the IC50 values of the compounds for inhibition of NFκB-Luc activity in cells and 20S β5 activity in vitro, with five of the analogues, specifically compounds 10–13 and 16, showing inhibitory NFκB potencies in the range 6.6–12 nM, equivalent to or greater than that of bortezomib [NFκB-Luc IC50=13±2.0 nM (mean±S.E.M., n=6)]. Interestingly, the activity of NFκB-Luc was not fully inhibited even at saturating compound concentrations (Table 2). The di-peptides also stabilized the 4xUb-Luc reporter in MDA-MB-231 cells with EC50 values that tracked closely with their in vitro 20S β5 potencies (Table 2), although the maximal stabilization effects of approx. 100-fold were lower than those observed for bortezomib (Table 2), other boronates (J.L. Blank, unpublished work) and covalent inhibitors, such as salinosporamide A [15]. Finally, the cytotoxic LC50 values of these compounds also correlated well between cell lines (Table 2) and with their 20S β5 potencies (Table 1). Interestingly, none of the compounds were as potent as bortezomib in this assay, although several gave LC50 values that were approx. 100 nM or less in at least one cell line, specifically compounds 9–13 and 16 (Table 2). Significantly, each of these contained the neopentyl-asparagine substituent in P3 (Figure 8), indicating that this residue contributes to both in vitro and cell-based potency. Furthermore, since these compounds were also the most potent inhibitors of NFκB-Luc activity, these data strongly suggest that their effects on cell viability and NFκB activity are mediated by proteasome inhibition.
Effect of 20S β5 inhibition on bulk protein turnover
The majority of intracellular proteins are degraded by the proteasome, as demonstrated using inhibitors that irreversibly inactivate each of the 20S active sites [4]. The effect of selectively inhibiting the β5 site alone on the degradation of short-lived protein was assessed using compounds 10 and 16, two of the most potent cell-active di-peptides. As shown previously [41], bortezomib inhibits approx. 50% of bulk protein turnover in HCT116 cells (see Supplementary Figure S5 at http://www.BiochemJ.org/bj/430/bj4300461add.htm), an effect that is comparable with that observed with ML858, a synthetic version of salinosporamide A that irreversibly inhibits each of the 20S active sites [15] (T.A. Soucy, unpublished work). Under identical conditions, the di-peptides reduced protein turnover by approx. 20% (Supplementary Figure S5), indicating that inhibition of the β5 site alone results in partial inhibition of protein degradation.
Constitutive and immunoproteasome selectivity of di-peptide proteasome inhibitors
In addition to monitoring the β5 inhibitory potency of the capped di-peptides using purified human erythrocyte constitutive 20S proteasomes, IC50 values were also obtained for the β5i site of the immunoproteasome from human peripheral blood monocytes using the selective substrates Ac-WLA-AMC and Ac-ANW-AMC respectively (Table 1). Bortezomib, compound 1 and the majority of the capped di-peptides were non-selective in these assays, inhibiting the constitutive and immunoproteasome with similar IC50 values, although they tended to inhibit β5i with slightly greater potencies (Table 1). In common with compound 1, the di-peptides were essentially inactive with respect to inhibition of the β1i or β2i sites of the immunoproteasome. However, a limited number of compounds showed significantly greater selectivity for the immunoproteasome, with compounds 4 and 5 being the best examples, showing ~10-fold β5i selectivity [compound 4, β5i IC50=41±3.3 nM and β5c IC50=470±96 nM; compound 5, β5i IC50=27±2.7 nM and β5c IC50=340±31 nM (means±S.E.M., n=3)]. Conversely, compounds 14 and 15 showed the reverse selectivity, the former of these two compounds being 18-fold more selective for the constitutive proteasome [β5c IC50=11±2.3 nM; β5i IC50=200±62 nM (means±S.E.M., n=3)] and the latter 3.6-fold more selective [β5c IC50=12±2.8 nM (mean±S.E.M., n=4); β5i IC50=44±9.2 nM (mean±S.E.M., n=3)] (Table 1).
To determine whether this selectivity was maintained in cells, three B-cell lymphoma subtypes were used that differ in their proteasome isoform expression. As shown in Figure 10(A), OCI-Ly10 cells do not express the immunoproteasome, as demonstrated by the negligible hydrolysis of the β1i- and β5i-specific substrates Ac-PAL-AMC and Ac-ANW-AMC respectively, whereas Karpas-1106P express substantial β1i and β5i activities, indicating considerable enrichment of the immunoproteasome. The hydrolysis of the β5c substrate Ac-WLA-AMC supported this expression profile, although it is not entirely selective for β5c over β5i and therefore over-represents the amount of constitutive proteasome in Karpas-1106P cells. The chymotrypsin-like substrate Suc-LLVY-AMC is hydrolysed approximately equally well by the β5i and β5c sites in vitro, although cells enriched in the immunoproteasome can show approx. 2–3-fold higher activity with this substrate. On the basis of these data, Karpas-1106P cells are highly enriched for the immunoproteasome, whereas OCI-Ly10 cells only express the constitutive proteasome. WSU-DLCL2 cells are intermediate in this activity profile, indicating that they express a mixed population of the proteasome isoforms.
Figure 10. Proteasome-subtype selectivity of compounds 5 and 14 in B-cell lymphomas expressing different levels of the immunoproteasome.

(A) The activities of the β1 and β5 subunits of the constitutive (c) and immunoproteasome (i) were assessed in diffuse large B-cell lymphoma extracts (10 μg of protein per reaction) using recombinant PA28α (12 nM) and subtype-selective fluorigenic peptide substrates (100 μM), as indicated. As shown, the rank order of immunoproteasome expression in these cell lines was Karpas-1106>WSU-DLCL2≫LY10. (B and C) The B-cell lymphoma lines (20000 cells per well) were treated with the indicated concentrations of the immunoproteasome-selective compound 5 (B) or constitutive-selective compound 14 (C) for 1 h under cell culture conditions. Inhibition of the β5 site of the constitutive and immunoproteasome was then assessed in situ using the Proteasome-Glo™ assay with the chymotrypsin-like luminogenic substrate Suc-LLVY-aminoluciferin, as described in the Experimental section. As shown, the IC50 values of compound 5 in Karpas-1106, WSU-DLCL2 and LY10 cells were 200, 380 and 620 nM respectively; the IC50 values of compound 14 in Karpas-1106, WSU-DLCL2 and LY10 cells were 870, 120 and 19 nM respectively. Results are means±S.E.M. for triplicate determinations and are representative of three independent experiments, the results of which are summarized in Table 3.
We assessed the potency of the β5i- and β5c-selective compounds 5 and 14 respectively in the three B-cell lymphomas using the non-subtype-selective β5 substrate Suc-LLVY-aminoluciferin in the Proteasome-Glo™ assay. As shown in Figure 10(B) and Table 3, the IC50 values of the β5i-selective compound 5 correlated with immunoproteasome expression (i.e. the rank order of potency being Karpas-1106P>WSU-DLCL2>OCI-Ly10), although the differences were relatively modest (i.e. approx. ≤4-fold). However, the β5c-selective inhibitor compound 14, which was the most isoform-selective compound in vitro, displayed substantial selectivity for the constitutive proteasome in cells (Figure 10C). The rank order of effect observed (i.e. OCI-Ly10>WSU-DLCL2>Karpas-1106P) indicated an approx. 22-fold increase in inhibitory potency of compound 14 in cells that exclusively express the constitutive proteasome as compared with those that predominantly express the immunoproteasome. Importantly, bortezomib, which does not discriminate substantially between β5c and β5i sites in vitro (Table 1), gave IC50 values that were within 2-fold of each other in the three B-cell lymphomas (Table 3). These data therefore provide the first description of non-covalent inhibitors of the proteasome that can discriminate between the immunoproteasome and constitutive proteasome in vitro and in cells.
Table 3. Proteasome inhibition in B-cell lymphoma subtypes expressing differing levels of immunoproteasome by β5i- and β5c-selective compounds 5 and 14 respectively compared with bortezomib.
Inhibition of the β5 sites of the constitutive and immunoproteasome was assessed in the indicated B-cell lymphoma lines (20000 cells per well) in situ using the Proteasome-Glo™ assay. Cells were incubated with compound for 1 h and assays were performed with the chymotrypsin-like substrate Suc-LLVY-aminoluciferin as described in the Experimental section. The results are the means±S.E.M. for triplicate determinations.
| Proteasome-Glo™ β5 IC50 (nM) | |||
|---|---|---|---|
| Compound | OCI-Ly10 | WSU-DLCL2 | Karpas-1106P |
| 5 | 1300±400 | 320±120 | 310±150 |
| 14 | 39±7.6 | 190±76 | 850±240 |
| Bortezomib | 2.9±0.25 | 2.7±0.26 | 3.3±0.40 |
DISCUSSION
The present study has identified a new series of potent non-covalent proteasome inhibitors on the basis of a capped trimeric peptide derived from the unnatural amino acid S-homo-phenylalanine, first identified by a large-scale high-throughput cell-based screen for small-molecule inhibitors of the UPS. These capped di-peptides, prepared by high-throughput liquid-phase peptide synthesis methods, are entirely selective for the β5 site over the β1 and β2 sites of the 20S core particle, and over a panel of less closely related proteases. The most potent compounds show IC50 values in the single-digit nanomolar range for the human 20S β5 site in vitro, with the best example, compound 16, displaying an IC50 of 1.2 nM and a Ki below the enzyme concentration in the assay (0.25 nM 20S), indicating that it behaves as an active-site titrant. In this respect, compound 16 is remarkable in having a greater affinity for the β5 site of the proteasome than that of the covalent inhibitor bortezomib, which displays a Ki of 0.56 nM for this site.
Compound optimization was guided by X-ray crystallography of representative examples bound to the yeast 20S core particle, which established their non-covalent binding mode and provided a structural basis for potency-enhancing modifications. The crystal structures of compounds 1, 6, 8 and 16 bound to the 20S β5 active site are shown in Figures 5, 8, S2, and 9 respectively, and are overlaid in Supplementary Figure S6 (at http://www.BiochemJ.org/bj/430/bj4300461add.htm). The overlay confirms a common binding mode in which the P1 benzyl group occupies a well-defined S1 binding pocket, whereas the P2 residue points out towards the solvent, unless it is the homo-phenylalanine residue in which case a hydrophobic ‘ledge’ interaction can be accessed. The S3 binding pocket is also well-defined, whereas the S4 pocket is very large and can tolerate a range of capping groups that are likely to contribute to potency by picking up further hydrophobic interactions. This binding mode broadly corresponds to that proposed on the basis of a homology model [38], except that the crystal structures obtained in the present study place the N-terminal cap in a large S4 binding pocket rather than in small ‘accessory hydrophobic pockets’, designated previously AS1 and AS2 [38]. Importantly, the most potent compounds identified each contained a neopentyl-asparagine substituent in P3, which appears to provide a near-optimal fit for the S3 binding pocket of the β5 site (Figure 9B). This residue can also be found in the tri-peptide 2-keto-1,3,4-oxadiazoles (Figure 1A) that are slowly reversible and presumably covalent in nature [11].
Characterization of these compounds in cells demonstrated that they can functionally inhibit the 26S proteasome with potencies that correlated well with those determined in vitro using the purified enzyme (Table 2 and Supplementary Figure S4). Furthermore, since the analogues are essentially inactive against the β1 (caspase-like) and β2 (trypsin-like) sites of the pro-teasome (Table 1), these data indicate that inhibition of the β5 (chymotrypsin-like) site of the proteasome alone is sufficient to potently inhibit the degradation of the 4xUb-Luc reporter, activation of NFκB in response to TNF-α and the proliferation of cancer cells. Interestingly, despite the fact that the cellular IC50 values of these compounds spanned three orders of magnitude, with the most potent being in the single- or double-digit nanomolar range, neither the stabilization of 4xUb-Luc nor the inhibition of the NFκB pathway was complete, even at saturating compound concentrations (Table 2 and Supplementary Figure S1). These observations suggest that non-covalent inhibition of 20S β5 activity alone is not sufficient to fully inhibit the proteasomal degradation of either 4xUb-Luc or IκBα, at least over the time-scale of these cell-based assays. This is in contrast with the covalent inhibitors bortezomib and salinosporamide A, which completely block the degradation of these proteins in cells and can minimally inhibit both the β1 and β5 sites of the proteasome. Furthermore, estimates of bulk protein turnover in cells suggest that the overall catalytic rate of proteasome is reduced, but not abolished, by non-covalent inhibition of the β5 site (Supplementary Figure S5), although it remains a possibility such inhibition completely blocks the degradation of a subset of proteins that are only substrates of the β5 site and not the β1 and/or β2 sites of the proteasome. Importantly, however, prolonged inhibition of the β5 site alone appears to sufficient to inhibit cancer cell proliferation.
Proteasome inhibitors that are currently in clinical use do not discriminate between the chymotrypsin-like activities of the constitutive or immunoproteasome, and the potential therapeutic benefit of selective inhibition of either of these activities has not been demonstrated. However, recent evidence from preclinical studies with PR-957, a β5i-selective peptide epoxyketone analogue of carfilzomib [28], suggests that the anti-inflammatory effects of proteasome inhibitors, such as bortezomib, may be mediated by their inhibition of the chymotrypsin-like site of the immunoproteasome [8]. Our studies have identified compounds that differentially inhibit the β5 sites of the constitutive and immunoproteasome in vitro and in B-cell lymphomas. Of note, compound 5 that preferentially inhibited the β5i site contained the small threonine residue in P3 and the larger 4-methylbenzylamine aromatic cap in P1. Conversely, the two β5c-selective compounds 14 and 15 each contained a smaller residue at P1 and the bulkier potency-enhancing neopentyl-asparagine in P3 (Figure 8). This structure–activity relationship, albeit limited, is entirely consistent with selectivity data obtained from an approx. 6000 tri-peptide library screen for substrates that are preferentially hydrolysed by each active site of the constitutive and immunoproteasome ([55] and C. Tsu, unpublished work). For example, the tri-peptide substrate Ac-ANW-AMC, where P1 is a tryptophan residue and P3 is an alanine residue, is preferentially cleaved by the β5i subunit of the immunoproteasome, whereas Ac-WLA-AMC, where the P1 and P3 residues are reversed, is preferentially hydrolysed by the constitutive β5c subunit. The crystal structure of the immunoproteasome has not yet been determined to provide an explanation for these selectivity differences. However, the primary sequences of the mature forms of the human β5c and β5i subunits are 60.7% identical (see Supplementary Figure S7 at http://www.BiochemJ.org/bj/430/bj4300461add.htm). We have therefore mapped the sequence differences between the β5c and β5i subunits on to the bovine 20S crystal structure [56] and modelled compound 1 into the active site (see Supplementary Figure S8 at http://www.BiochemJ.org/bj/430/bj4300461add.htm). The majority of the sequence differences between β5c and β5i lie outside of the active site, whereas the residues that make contacts with compound 1 appear to be mainly the same, with some relatively minor exceptions, such as the replacement of Ala46 and Ser53 with Ser46 and Gln53 within the S1 binding pocket, Gly48 with Cys48 in the S2 ‘ledge’, and the reversal of the Ala27-Ser28 sequence with Ser27-Ala28 in the S3 pocket. Since only subtle differences exist between the ligand-interacting residues that form the β5c and β5i active sites, it is not possible to rationalize selectivity differences with respect to substrate preference or inhibitor sensitivity based on such an analysis. Clearly, crystal structures of the constitutive and immunoproteasomes in complex with selective inhibitors will help identify the structural features of each sub-site that contribute to selectivity. Finally, although a covalent immunoproteasome-selective inhibitor has been described recently [8], the function of the immunoproteasome in cancer cells is unknown. The identification of a new class of proteasome inhibitors, such as compounds 14 and 5, that preferentially inhibit the 20S β5c and β5i sites respectively and can thus discriminate between the constitutive and immunoproteasome in cells may help address their functional roles in various cellular models of disease including cancer.
Online data
AUTHOR CONTRIBUTION
Christopher Tsu, Paul Hales and Frank Bruzzese and designed and performed the biochemical characterization of the compounds. Cynthia Barrett, Jane Liu and Khristofer Garcia performed the cell-based assays and Teresa Soucy performed the protein turnover assays. Darshan Sappal generated the MDA231-4xUb-Luc cell line and performed the high-throughput library screen. Edward Olhava performed the re-synthesis of compound 1, and Christopher Blackburn, Kenneth Gigstad and Matthew Jones designed and performed the chemical syntheses of the capped di-peptides. Nancy Bump purified yeast 20S core particles for crystallization and Michael Sintchak prepared the crystals for X-ray crystallography and analysed the data. Paul Fleming and Lawrence Dick provided project oversight, participated in experimental design and data interpretation, and reviewed the manuscript. Jonathan Blank performed the high-throughput screen and the initial characterization of compound 1 in cells, contributed to experimental design and data analysis, and wrote the manuscript with Christopher Blackburn.
ACKNOWLEDGEMENTS
We are grateful to Dr Marion Schmidt and Dr Dan Finley for providing the yeast 20S expression strains, and to Dr Juan Gutierrez and Zhi Li their assistance with preparation of the yeast 20S enzymes. We also thank Alejandra Raimondi for formatting the 4xUb-Luc cell-based assay for the high-throughput screening, Ben Knight for his assistance with data processing and management, and Maria Dawn-Linsley, Kim Lincoln, Charles Gauthier, Dan Onea, Pam Ward, Ted Peters, Christina Majer and Elena Musotto for their help with assay automation and compound management. We gratefully acknowledge the assistance of Marjorie Solomon, David Lok, Lenny Dang and Qing Lu with the identification of compound 1 by MS. We thank Mingkun Fu who performed the high-resolution MS on the synthetic compounds. We acknowledge Abe Achab and Alla Mishechkina for their synthetic contributions towards development of the series. We are also grateful to Dr Gary Luker (Department of Microbiology and Immunology, University of Michigan Medical School, Ann Arbor, MI, U.S.A.) and Dr David Piwnica-Worms (Division of Biology and Biomedical Sciences, University of Washington, St Louis, MO, U.S.A.) from whom the 4xUb-Luc expression plasmid was licenced. We are also grateful to Dr Friedrich Feuerhake and Dr Margaret Shipp (Lymphoma Program, Dana-Faber Cancer Institute, Boston, MA, U.S.A.) for providing cell pellets derived from a panel of B-cell lymphoma subtypes to profile for immunoproteasome expression. Finally, we thank Mark Williamson and Dr Mark Rolfe for valuable discussions.
FUNDING
This work was supported by Millennium Pharmaceuticals, Inc. (Cambridge, MA, U.S.A.). All authors were employed by Millennium Pharmaceuticals at the time of their contribution to this work.
References
- 1.Hershko A., Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 2.Baumeister W., Walz J., Zuhl F., Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 3.Pickart C. M., Cohen B. Proteasomes and their kin: proteases in the machine age. Nat. Rev. Mol. Cell Biol. 2004;5:177–187. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- 4.Kisselev A. F, Goldberg A. L. Proteasome inhibitors: from research tools to drug candidates. Chem. Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 5.Borissenko L., Groll M. 20S Proteasome and its inhibitors: crystallographic knowledge for drug development. Chem. Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 6.Goldberg A. L. Functions of the proteasome: from protein degradation and immune surveillance to cancer chemotherapy. Biochem. Soc. Trans. 2007;35:12–17. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 7.Kloetzel P.-M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2001;2:179–187. doi: 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- 8.Muchamuel T., Basler M., Aujay M. A., Suzuki E., Kalim K. W., Lauer C., Sylvain C., Ring E. R., Shields J., Jiang J., et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009;15:781–787. doi: 10.1038/nm.1978. [DOI] [PubMed] [Google Scholar]
- 9.Groll M., Huber R., Moroder L. The persisting challenge of selective and specific proteasome inhibition. J. Pept. Sci. 2009;15:58–66. doi: 10.1002/psc.1107. [DOI] [PubMed] [Google Scholar]
- 10.García-Echeverria C. Peptide and peptide-like modulators of 20S proteasome enzymatic activity in cancer cells. Int. J. Pept. Res. Ther. 2006;12:49–64. doi: 10.1007/s10989-005-9001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rydzewski R. M., Burrill M., Mendonca R., Palmer J. T., Rice M., Tahilramani R., Bass K. E., Leung L., Gjerstad E., Janc J. W., Pan L. Optimization of subsite binding to the β5 subunit of the human 20S proteasome using vinyl sulfones and 2-keto-1,3,4-oxadiazoles: syntheses and cellular properties of potent, selective proteasome inhibitors. J. Med. Chem. 2006;49:2953–2968. doi: 10.1021/jm058289o. [DOI] [PubMed] [Google Scholar]
- 12.Adams J., Behnke M., Chen S., Cruickshank A. A., Dick L. R., Grenier L., Klunder J. M., Ma Y-T., Plamondon L., Stein R. L. Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998;8:333–338. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 13.Adams J., Palombella V. J., Sausville E. A., Johnson J., Destree A., Lazarus D. D., Maas J., Pien C. S., Prakash S., Elliott P. J. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59:2615–2622. [PubMed] [Google Scholar]
- 14.Hideshima T., Richardson P., Chauhan D., Palombella V. J., Elliott P. J., Adams J., Anderson K. C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–3076. [PubMed] [Google Scholar]
- 15.Williamson M. J., Blank J. L., Bruzzese F. J., Cao Y., Daniels J. S., Dick L. R., Labutti J., Mazzola A. M., Patil A. D., Reimer C. L., et al. Comparison of biochemical and biological effects of ML858 (salinosporamide A) and bortezomib. Mol. Cancer Ther. 2006;5:3052–3061. doi: 10.1158/1535-7163.MCT-06-0185. [DOI] [PubMed] [Google Scholar]
- 16.Richardson P. G., Barlogie B., Berenson J., Singhal S., Jagannath S., Irwin D., Rajkumar S. V., Srkalovic G., Alsina M., Alexanian R., et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Eng. J. Med. 2003;348:2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 17.Richardson P. G., Sonneveld P., Schuster M. W., Irwin D., Stadtmauer E. A., Facon T., Harousseau J.-L., Ben-Yehuda D., Lonial S., Goldschmidt H., et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Eng. J. Med. 2005;352:2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 18.Richardson P. G., Sonneveld P., Schuster M. W., Irwin D., Stadtmauer E. A., Facon T., Harousseau J.-L., Ben-Yehuda D., Lonial S., Goldschmidt H., et al. Extended follow-up of a phase 3 trial in relapsed multiple myeloma: final time-to-event results of the APEX trial. Blood. 2007;110:3557–3560. doi: 10.1182/blood-2006-08-036947. [DOI] [PubMed] [Google Scholar]
- 19.San Miguel J. F., Schlag R., Khuageva N. K., Dimopoulos MA., Shpilberg O., Kropff M., Spicka I., Petrucci M. T., Palumbo A., Samoilova O. S., et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N. Eng. J. Med. 2008;359:906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 20.Fisher R. I., Bernstein S. H., Kahl B., Djulbegovic B., Robertson M. J., de Vos S., Epner E., Krishnan A., Leonard J. P., Lonial S., et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2006;24:4867–4874. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 21.Voorhees P. M., Orlowski R. Z. The proteasome and proteasome inhibitors in cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2006;46:189–213. doi: 10.1146/annurev.pharmtox.46.120604.141300. [DOI] [PubMed] [Google Scholar]
- 22.Richardson P. G., Mitsiades C., Hideshima T., Anderson K. C. Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu. Rev. Med. 2006;57:33–47. doi: 10.1146/annurev.med.57.042905.122625. [DOI] [PubMed] [Google Scholar]
- 23.Orlowski R. Z., Kuhn D. J. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin. Cancer Res. 2008;14:1649–1657. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 24.Piva R., Ruggeri B., Williams M., Costa G., Tamagno I., Ferrero D., Giai V., Coscia M., Peola S., Massaia M., et al. CEP-18770: a novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood. 2008;111:2765–2775. doi: 10.1182/blood-2007-07-100651. [DOI] [PubMed] [Google Scholar]
- 25.Kupperman E., Lee E. C., Cao Y., Bannerman B., Fitzgerald M., Berger A., Yu J., Yang Y., Bruzzese F., Liu J., et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70:1970–1980. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- 26.Groll M., Berkers C. R., Ploegh H. L., Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 27.Chauhan D., Catley L., Li G., Podar K., Hideshima T., Velankar M., Mitsiades C., Mitsiades N., Yasui H., Letai A., et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with a mechanism distinct from bortezomib. Cancer Cell. 2005;8:407–419. doi: 10.1016/j.ccr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 28.Kuhn D. J., Chen Q., Voorhees P. M., Strader J. S., Shenk K. D., Sun C. M., Demo S. D., Bennett M. K., van Leeuwen F. W. B., Chanan-Khan A. A., Orlowski R. Z. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubuquitin–proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007;110:3281–3290. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Groll M., Huber R., Potts B. C. M. Crystal structures of salinoporamide A (NPI-0047) in complex with the 20S porteasome reveal important consequences of β-lactone ring opening and a mechanism for irreversible binding. J. Am. Chem. Soc. 2006;128:5136–5141. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 30.Groll M., Kim K. B., Kairies N., Huber R., Crews C. M. Crystal structure of expoxomicin:20S proteasome reveals a molecular basis for selectivity of α′,β′-epoxyketone proteasome inhibitors. J. Am. Chem. Soc. 2000;122:1237–1238. [Google Scholar]
- 31.Koguchi Y., Kohno J., Nishio M., Takahashi K., Okuda T., Ohnuki T., Komatsubara S. TMC-95A, B, C, and D, novel proteasome inhibitors produced by Apiospora montagnei Sacc. TC 1093. Taxonomy, production, isolation, and biological activities. J. Antibiot. 2000;53:105–109. doi: 10.7164/antibiotics.53.105. [DOI] [PubMed] [Google Scholar]
- 32.Groll M., Koguchi Y., Huber R., Kohno J. Crystal structure of the 20 S proteasome:TMC-95A complex: a non-covalent proteasome inhibitor. J. Mol. Biol. 2002;311:543–548. doi: 10.1006/jmbi.2001.4869. [DOI] [PubMed] [Google Scholar]
- 33.Groll M., Gotz M., Kaiser M., Weyher E., Moroder L. TMC-95-based inhibitor design provides evidence for the catalytic versatility of the proteasome. Chem. Biol. 2006;13:607–614. doi: 10.1016/j.chembiol.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Basse N., Piguel S., Papapostolou D., Ferrier-Berthelot A., Richey N., Pagano M., Sarthou P., Sobczak-Thépot J., Reboud-Ravaux M., Vidal J. Linear TMC-95-based proteasome inhibitors. J. Med. Chem. 2007;50:2842–2850. doi: 10.1021/jm0701324. [DOI] [PubMed] [Google Scholar]
- 35.Lum R. T., Kerwar S. S., Meyer S. M., Nelson M. G., Schow S. R., Shiffman D., Wick M. M., Joy A. A new structural class of proteasome inhibitors that prevent NF-κB activation. Biochem. Pharmacol. 1998;55:1391–1397. doi: 10.1016/s0006-2952(97)00655-2. [DOI] [PubMed] [Google Scholar]
- 36.Lum R. T., Nelson M. G., Joly A., Horsma A. G., Lee G., Meyer S. M., Wick M. M., Schow S. R. Selective inhibition of the chymotrypsin-like activity of the 20S proteasome by 5-methoxy-1-indanone di-peptide benzamides. Bioorg. Med Chem. Lett. 1998;8:209–214. doi: 10.1016/s0960-894x(98)00015-8. [DOI] [PubMed] [Google Scholar]
- 37.García-Echeverría C., Imbach P., France D., Furst P., Lang M., Noorani M., Scholz D., Zimmermann J., Furet P. A new class of selective and non-covalent inhibitors of the chymotrypsin-like activity of the 20S proteasome. Bioorg. Med. Chem. Lett. 2001;11:1317–1319. doi: 10.1016/s0960-894x(01)00205-0. [DOI] [PubMed] [Google Scholar]
- 38.Furet P., Imbach P., Noorani M., Koeppler J., Laumen K., Lang M., Guagnano V., Fuerst P., Roesel J., García-Echeverría C. Entry into a new class of potent proteasome inhibitors having high antiproliferative activity by structure-based design. J. Med. Chem. 2004;47:4810–4813. doi: 10.1021/jm049660v. [DOI] [PubMed] [Google Scholar]
- 39.Luker G. D., Pica C. M., Song J., Luker K. E., Piwnica-Worms D. Imaging 26S proteasome activity and inhibition in living mice. Nat. Med. 2003;9:969–973. doi: 10.1038/nm894. [DOI] [PubMed] [Google Scholar]
- 40.Lightcap E. S., McCormack T. A., Pien C. S., Chau V., Adams J., Elliott P. J. Proteasome inhibition measurements: clinical applications. Clin. Chem. 2000;46:673–683. [PubMed] [Google Scholar]
- 41.Soucy T. A., Smith P. G., Milhollen M. A., Berger A. J., Gavin J. M., Adhikari S., Brownell J. E., Burke K. E., Cardin D. P., Critchley S., et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–737. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 42.Blackburn C., Achab A., Blank J., Bump N., Bruzzese F., Dick L., Fleming P., Garcia K., Gigstad K., Hales P., et al. Identification and optimization of a series of non-covalent proteasome inhibitors guided by X-ray crystallography; 36th Northeast Regional Meeting of the American Chemical Society; 2009. Hartford, CT, U.S.A., October 7–10 2009, abstract NERM-055. [Google Scholar]
- 43.Groll M., Bajorek M., Köhler A., Moroder L., Rubin D. M., Huber R., Glickman M. H., Finley D. A gated channel into the proteasome core particle. Nat. Stuct. Biol. 2000;7:1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 44.Leggett D. S., Hanna J., Borodovsky A., Crosas B., Schmidt M., Baker R. T., Walz T., Ploegh H., Finley D. Multiple associated proteins regulate proteasome structure and function. Mol. Cell. 2002;10:495–507. doi: 10.1016/s1097-2765(02)00638-x. [DOI] [PubMed] [Google Scholar]
- 45.Leggett D. S., Glickman M. H., Finley D. Purification of proteasomes, proteasome subcomplexes and proteasome-associated proteins from budding yeast. Methods Mol. Biol. 2005;301:57–70. doi: 10.1385/1-59259-895-1:057. [DOI] [PubMed] [Google Scholar]
- 46.Groll M., Huber R. Purification, crystallization, and X-ray analysis of yeast 20S proteasome. Methods Enzymol. 2005;398:329–336. doi: 10.1016/S0076-6879(05)98027-0. [DOI] [PubMed] [Google Scholar]
- 47.Leslie A. G. W. The integration of macromolecular data. Acta Crystallogr. D Biol. Crystallogr. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 48.Evans P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2005;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 49.Schüttelkopf A. W., van Aalten D. M. F. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Potterton E., Briggs P., Turkenburg M., Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 52.Murshudov G. N., Vagin A. A., Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 53.Kroll M., Margottin F., Kohl A., Renard P., Durand H., Concordet J.-P., Bachelerie F., Arenzana-Seisdedos F., Benarous R. Inducible degradation of IκBα by the proteasome requires interaction with the F-box protein h-βTrCP. J. Biol. Chem. 1999;274:7941–7945. doi: 10.1074/jbc.274.12.7941. [DOI] [PubMed] [Google Scholar]
- 54.Groll M., Ditzel L., Löwe J., Stock D., Bochtler M., Bartunik H. D., Huber R. Stucture of 20S proteasome from yeast at 2.4 Å resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 55.Lin G., Tsu C., Dick L., Zhou X. K., Nathan C. Distinct specificities of Mycobacterium tuberculosis and mammalian proteasomes for N-acetyl tripeptide substrates. J. Biol. Chem. 2008;283:34423–34431. doi: 10.1074/jbc.M805324200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Unno M., Mizushima T., Morimoto Y., Tomisugi Y., Tanaka K., Yasuoka N., Tsukihara T. The structure of the mammalian 20S proteasome at 2.75 Å resolution. Structure. 2002;10:609–618. doi: 10.1016/s0969-2126(02)00748-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.