

Summary
Anthracycline antibiotics have saved the lives of many cancer victims in the 50 plus years since their discovery. A major limitation of their use is the dose-limiting cardiotoxicity. Efforts focusing on understanding the biochemical basis for anthracycline cardiac effects have provided several strategies currently in clinical use: limit dose exposure; encapsulate anthracyclines in liposomes to reduce myocardial uptake; administer concurrently with the iron chelator dexrazoxane to reduce free iron-catalyzed reactive oxygen species formation; modification of anthracycline structure in an effort to reduce myocardial toxicity. In spite of these efforts, anthracycline-induced heart failure continues to occur with consequences for both morbidity and mortality. Our inability to predict and prevent anthracycline cardiotoxicity is in part due to the fact that the molecular and cellular mechanisms remain controversial and incompletely understood. Studies examining the effects of anthracyclines in cardiac myocytes in vitro and small animals in vivo have demonstrated several forms of cardiac injury, and it remains unclear how these translate to the clinical setting. Given the clinical evidence that myocyte death occurs after anthracycline exposure in the form of elevations in serum troponin, myocyte cell death appears to be a probable mechanism for anthracycline-induced cardiac injury. Other mechanisms of myocyte injury include the development of cellular ‘sarcopenia’ characterized by disruption of normal sarcomere structure. Anthracyclines suppress expression of several cardiac transcription factors, and this may play a role in the development of myocyte death as well as sarcopenia. Degradation of the giant myofilament protein titin may represent an important proximal step that leads to accelerated myofilament degradation. An interesting interaction has been noted clinically between anthracyclines and newer cancer therapies that target the erbB2 receptor tyrosine kinase. There is now evidence that erbB2 signaling in response to the ligand neuregulin regulates anthracycline uptake into cells via the multidrug-resistance protein. Therefore upregulation of cardiac neuregulin signaling may be one strategy to limit myocardial anthracycline injury. Moreover, assessing an individual’s risk for anthracycline injury may be improved by having some measure of endogenous activity of this and other myocardial protective signals.
Overview of Anthracycline cardiotoxicity
Anthracycline antibiotics are highly effective and widely used cytotoxic agents with applications in the treatment of multiple cancers. The mechanisms of cytotoxicity of anthracyclines in cancer cells are diverse including (reviewed in [1]): 1) inhibition of both DNA replication and RNA transcription; 2) free radical generation, leading to DNA damage or lipid peroxidation; 3) DNA alkylation; 4) DNA cross-linking; 5) interference with DNA unwinding of DNA strand separation and helicase activity; 6) direct membrane damage due to lipid oxidation; 7) inhibition of topoisomerase II. In response to some or all of these effects, tumor cell growth is inhibited and cells are more likely to die by one or more mechanisms.
A major limitation of anthracycline use is a cumulative dose-dependent cardiac toxicity. Cardiotoxicity has long been considered to occur by mechanisms other than those mediating their antitumor effectiveness, a concept that raises hope for development of strategies for protecting the heart while not diminishing tumor response [2]. Early strategies to prevent cardiac toxicity included reductions in single doses of anthracyclines as well prolonging the infusion of drug to limit peak serum concentrations [3]. However, in spite of these efforts the cardiotoxicity remains [4].
Multiple mechanisms of anthracycline-induced cardiac cellular injury have been proposed based upon studies in animals and cell culture systems, and it remains unclear which of these are at work in the clinical context of anthracycline use (see Figure). Most mechanisms proposed involve oxidative stress induced by the anthracyclines, though it is not clear why this would result in preferential toxicity to the myocardium. Anthracyclines induce membrane damage via lipid peroxidation in all tissues, including the heart [5]. While formation of reactive oxygen species is induced by the quinone moiety of anthracyclines, oxidative stress can also occur via induction of nitric oxide synthase, leading to nitric oxide and peroxynitrite formation [6]. This mechanism has been linked to nitration and inactivation of key enzymes in the heart including myofibrillar creatine kinase [7]. Anthracyclines also cause impairment of membrane binding, assembly, and enzymatic activity of mitochondrial creatine kinase, though the consequences of this function are yet unclear [8]. In the heart, like other tissue, anthracyclines intercalate into nucleic acids, causing suppression of DNA, RNA and protein synthesis [9]. Some transcriptional regulatory proteins that appear important for regulation of cardiac specific genes are particularly susceptible to anthracyclines [10-12]. Impaired synthesis of myofilament proteins, which in the presence of anthracycline accelerated myofilament degradation [13], leads to a net negative balance of sarcomeric proteins, a condition we have termed ‘cardiac sarcopenia’. Myocyte cell death by both apoptosis and necrosis has also been implicated, and the net loss of cells may further contribute to ‘cardiac wasting’. Finally, anthracyclines induce changes in adrenergic function and adenylate cyclase[14, 15], as well as abnormalities in Ca2+ handling[16], all critical for the dynamic regulation of cardiac function. The extent to which each of these contributes to the dose-dependent clinical heart failure in anthracycline treated patient remains controversial.
Figure.
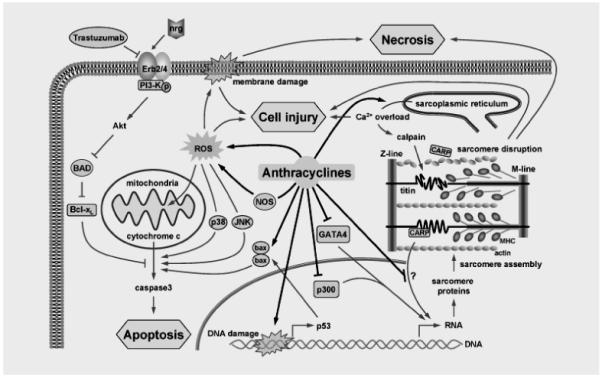
Anthracyclines promote myocyte injury and death via multiple mechanisms. This is balanced by intracellular signaling (e.g. GATA4, Akt) that is responsive to paracrine factors such as neuregulin (nrg). Trastuzumab (Herceptin) increases the susceptibility to anthracyclines by targeting the erb2 receptor. Anthracyclines also disrupt cardiac sarcomere structure by accelerating the degradation of key sarcomeric proteins (e.g. titin) and inhibit new sarcomere protein synthesis via alteration of transcriptional regulation. See text for details.
Anthracyclines induce Myocyte Cell Death
An attractive mechanism for anthracycline-induced cardiotoxicity is the hypothesis that with each exposure to anthracyclines there is myocyte death. Given the limited regenerative capacity of the heart, cumulative toxicity eventually surpasses a threshold of damage that triggers a more generic process of ventricular remodeling common to multiple forms of cardiac injury. Endomyocardial biopsies obtained from human patients within hours after anthracycline exposure shows electron micrographic evidence of mitochondrial swelling and chromatin contraction, hallmarks of apoptosis [17]. Detection of myocyte death by measurement of released cardiac sarcomeric troponins corroborate that myocyte loss occurs after anthracycline exposure [18].
Experimental studies also demonstrate that anthracyclines can induce myocyte death, and these experimental systems have been utilized to examine potential mechanisms for these effects. Anthracycline treatment of cultured cardiac myocytes induces cell death in a concentration-dependent manner [19, 20]. The mechanism of cell death varies with anthracycline concentration with apoptosis occurring at lower and necrosis at higher concentrations [20]. Furthermore, work in animals and humans have demonstrated that apoptotic cell death occurs after in vivo exposure to anthracyclines [21-23].
As discussed above, anthracyclines generate cellular oxidative stress, and this may be a proximal step leading to myocyte cell death. Hydroxyl radical formation in the intact heart increases in the presence of anthracyclines and can be inhibited by treatment with superoxide dismutase, catalase and the iron chelator dexrazoxane [24]. Both dexrazoxane and a superoxide dismutase mimetic can suppress anthracycline induced cell death [20]. Overexpression of manganese-SOD in mice demonstrates early protection from mitochondrial damage and decreased markers of myocyte death in the serum [25]. Interestingly the necrosis of myocytes that occurs at high concentrations of anthracyclines is not suppressed by antioxidants.
These experimental results correlate with clinical data. Dexrazoxane is the only drug approved by the FDA to prevent anthracycline-induced heart injury [26]. Dexrazoxane suppresses anthracycline-induced troponin elevation, a marker of myocyte cell death, as well as late cardiotoxicity [27]. That dexrazoxane can also inhibit anthracycline-induced myocyte death in vitro supports the concept that oxidative stress induced myocyte apoptosis is a proximal event in the development of anthracycline cardiotoxicity.
Cellular stress, and specifically oxidative stress, activates a host of signaling pathways that regulate cell fate, and these pathways likely modulate the response of the heart to anthracycline exposure. MAPKs deliver extracellular signals from activated receptors to effector proteins and regulate cell processes such as survival and proliferation [28]. In cardiac myocytes, ERK is activated by growth factors/hypertrophic agents as well as oxidant stress (for review see [29]). The SAPKs JNK and p38 are activated by cellular stresses and correlate with cardiac myocyte apoptosis [30]. ROS production can also activate these pathways [31], in particular JNK and p38 [32], supporting the association of anthracyclines and SAPK mediated apoptosis.
The PI-3K/Akt pathway has been shown to regulate the survival of cardiac myocytes from insults such as transient ischemia [33]. Akt is activated in response to many different growth ligands such as insulin-like growth factor-1 (IGF-1)[34-37], and neuregulin [38]. Activation of PI3k/Akt signaling suppresses anthracycline induced apoptosis and inhibition of PI3K/Akt (treatment of LY294002 or transfection of a dominant negative Akt mutant) increases anthracycline-induced apoptosis [38].
Anthracycline induced apoptosis in the heart appears to occur via a mitochondrial pathway involving Bax, cytochrome c and caspase-3 [39, 40]. Cytochrome c is normally located in the mitochondria. Production of ROS induces cytochrome c release, suggesting anthracycline-induced ROS generation may trigger apoptosis through this mechanism. Recent studies have demonstrated release of cytochrome c after anthracycline treatment in myocytes [39]. Myopathic hearts from anthracycline-treatment have presented with increased Bax/Bcl-2 ratio [41], similar to what has been seen after ischemia-reperfusion. Activated caspase-3 is also seen on anthracycline-induced cardiomyopathy [42] similar to reperfused rat hearts [43].
Another trigger for apoptosis is likely DNA damage. Anthracyclines are planar and intercalate between base pairs in DNA. This non-covalent interaction inhibits DNA and RNA synthesis and leads to DNA oxidant damage [44]. DNA damage at levels below that necessary to trigger cell death may lead to cumulative cardiotoxicity via accumulation of DNA mutations. The consequences of cumulative nuclear DNA damage in cardiac myocytes warrant further investigation.
One additional mechanism by which these DNA-damaging effects of anthracyclines could lead to cardiotoxicity is via suppressing expression and/or activity of transcription factors known to modulate sarcomere synthesis as well as cell survival. GATA4 is a member of a zinc finger transcription factor family that regulates myocyte differentiation, sarcomere synthesis, and survival. GATA4 regulates several cardiac specific genes[45, 46] as well as antiapoptotic genes[47]. Treatment with anthracyclines down-regulates GATA4 expression in cardiac myocytes and overexpression of GATA4 attenuates anthracycline induced apoptosis [48]. Increasing GATA4 expression by alpha-adrenergic agonists suppresses anthracycline cardiotoxicity[49]. This has led to the hypothesis that pharmacologic activation of alpha-adrenergic receptors may reduce anthracycline cardiac injury. Conceptually this idea is obviously unappealing as the adrenergic system in general is known to promote adverse outcome after cardiac injury. Moreover, carvedilol has α1 blocking activity and yet is able to reduce anthracycline injury [50]. However, specific activation of α1-adrenergic receptors is a potent hypertrophic stimulus, and this may be cardioprotective under some forms of cardiac stress [49]. Hence, prevention of Adriamycin (doxorubicin) toxicity with an α1-adrenergic agonist may be worth exploring.
Anthracycline induce sarcomere disruption
Many of the effects of anthracyclines on cardiac cells that have been implicated in the induction of cardiac myocyte cell death may also play a role in the induction of other cellular phenotypes such as disruption of cardiac sarcomeric structure. Ultrastructural changes to myocyte sarcomeres seen in endomyocardial biopsies of anthracycline treated patients as well as rodents include loss of myofibrils, disarray of myofibrils of the sarcomere, dilation of the sarcoplasmic reticulum, swelling of the mitochondria and cytoplasmic vacuolization [51-53]. An early step in the breakdown of sarcomeres after anthracycline exposure is the degradation of titin. Titin is a giant myofilament protein and an integral part of the sarcomere in striated muscle. Titin spans a half-sarcomere, from M-line to Z-disk, and functions to modulate the passive and restoring forces of the myocytes through its elastic elements [54]. Titin serves as a scaffold for assembly of myofilamentous proteins into sarcomeres [55]. Titin integrity is critical for the dynamic regulation of contractile function via length-dependent changes in the sensitivity of actin-myosin cross-bridges to calcium [56]. The loss of intact titin has been demonstrated in ischemic and failing human hearts, implicating titin disruption in the pathogenesis of progressive ventricular dysfunction in these conditions. Anthracycline exposure results in accelerated titin degradation via calpain proteolytic activity. At least in vitro, titin degradation leads to sarcomere disorganization [13].
Accelerated degradation is only part of the equation that will lead to the cardiac “sarcopenia” typical of anthracycline associated heart failure. Suppression of sarcomere protein synthesis by anthracyclines also contributes. The mechanism involved likely includes anthracycline binding to and disrupting nucleic acid synthesis, suppression of critical signaling pathways, as well as suppression of transcription factor activity as discussed above.
In addition to GATA4, a transcriptional regulator that has been implicated but poorly characterized, is cardiac ankyrin repeat protein (CARP, aka Ankrd1). CARP was also named cardiac adriamycin-responsive protein due to the sensitivity of its mRNA to anthracycline treatment [11]. Although CARP function is incompletely understood, its localization to the I-band is essential for normal sarcomere organization [57]. CARP is regulated in part by GATA4 expression [11]. Suppression of CARP expression using siRNA is sufficient to induce myofibrillar disarray similar to that observed with anthracycline treatment (unpublished data). CARP protein is rapidly degraded in myocytes after anthracycline treatment, similar to the transcriptional coactivator p300 [58]. Thus down-regulation and/or degradation of CARP, GATA4 and p300 may all contribute to the sarcopenia that develops after anthracycline exposure.
Modification of anthracycline toxicity by erbB2 signaling
Newer cancer therapeutics have been developed that target specific oncogenes involved in regulation of cancer growth. One of these, trastuzumab (Herceptin) appears to increase the susceptibility of the heart to anthracyclines, and this experience provides some new insight into the mechanisms of anthracycline cardiotoxicity. Trastuzumab targets the erbB2 (HER2/neu) oncogene, a receptor tyrosine kinase in the erbB family. There are four members, erbB1 (a.k.a. EGFR) to erbB4 [59]. ErbB2 is a ligand-less 185Kd transmembrane protein with a tyrosine kinase in the cytoplasmic domain that is normally activated by heterodimerization with erbB1, erbB3, or erbB4 [60]. ErbB2 is overexpressed in 25-30% of human breast cancer and this is associated with enhanced tumor aggressiveness and a high risk of relapse and death [61]. Several monoclonal antibodies directed against the erbB2 ectodomain that specifically inhibit the growth of tumor cell lines overexpressing erbB2 have been developed. One of them, 4D5 was fully cloned [62] and the Fc portion replaced with human Fc to create trastuzumab [63]. Trastuzumab’s antitumor activity against erbB2-positive human breast tumor cells in laboratory models led to clinical trials that ultimately demonstrated a clinical benefit in women with erbB2-overexpressing breast cancers [64]. On the basis of trastuzumab clinical efficacy, this antibody was approved in 1998 for treatment of patients with erbB2 overexpressing metastatic breast cancer.
A fraction of patients treated with trastuzumab develop clinical heart failure with a decrease in cardiac contractile function [64]. This was most notable in patients receiving concurrent anthracyclines, where cardiac dysfunction or symptomatic heart failure was reported at ~4-fold higher rates in patients who received trastuzumab than in those who received anthracycline and cyclophosphamide alone. While initially there was some concern that the effect of trastuzumab on the heart should limit its use [65], more recent clinical trials have demonstrated lower rates of treatment-associated heart failure. Moreover, experience with treatment of those patients who develop heart failure with standard medical therapy suggests that the contractile dysfunction is often reversible[66]. Nevertheless, this experience has triggered further research into the molecular mechanisms of anthracycline-induced cardiotoxicity that may lead to strategies for cardioprotection.
Studies in animals and cell culture have provided some insight into the mechanisms for trastuzumab’s effects on the heart. Gene targeting studies in mice have demonstrated that erbB2, the target of trastuzumab, is essential for cardiac development [67], and conditional deletion of erbB2 in mice leads to the development of a dilated cardiomyopathy [68, 69]. Thus erbB2 is essential for maintenance of normal cardiac structure and function. In vitro studies with cardiac myocytes provide further insights regarding the cellular and molecular mechanisms for trastuzmab’s effects. ErbB2 is expressed in adult cardiac myocytes, along with erbB4, and transmits growth and survival signals in response to the ligand neuregulin-1 [70]. In response to anthracyclines, there is myofilament degradation [13], as well as cell death by both apoptosis and necrosis [20], and both of these effects are suppressed by neuregulin treatment [71]. ErbB2 and erbB4 localize in the transverse tubules of ventricular myocytes [72]. Collectively these data suggest that one role of erbB2/4 signaling is to dynamically regulate sarcomere structure, perhaps in response to stress. In this paradigm, suppression of erbB2 signaling by trastuzumab may accelerate the net breakdown of sarcomeric proteins induced by anthracyclines, increasing the likelihood of cardiac dysfunction and the clinical syndrome of heart failure.
Strategies to limit Anthracycline Cardiotoxicity
At present, there is no generally accepted method to provide selective protection of the heart from damage induced by anthracyclines. Modification of anthracycline structure has resulted in compounds with reduced cardiotoxicity, allowing for higher dose administration. However, toxicity remains a problem. The iron chelator dexrazoxane reduces the incidence of contractile dysfunction in cancer victims treated with anthracyclines [73]. However, the possibility that dexrazoxane reduces tumor response rates has prevented dexrazoxane administration from becoming a universal practice [74]. Current guidelines from the American Society of Clinical Oncology, Chemotherapy, and Radiotherapy Expert Panel support the use of dexrazoxane in patients who have received high dose of Doxorubicin (Dox, over 300mg/m2) for metastatic breast cancer and who may benefit from continued Dox treatment or who have received over 300mg/m2 Dox for treatment of malignancies other than breast cancer or who responded to previous anthracycline-based chemotherapy and may benefit from continued therapy with Epirubicin for advanced breast cancer.
The observation that suppressing erbB2 signaling with trastuzumab increases the likelihood of anthracycline cardiotoxicity leads to the hypothesis that augmentation of erbB2 signaling prior to or at the time of anthracycline exposure would be cardioprotective. Certainly in vitro studies with recombinant neuregulin support this hypothesis [38]. Recent work with the heterozygous neuregulin-1 knockout mouse also supports this hypothesis [75]. Pharmacological strategies such as systemic delivery of neuregulin does reduce cardiotoxicity of anthracyclines in mice [75], though the pleiotrophic growth effects of this ligand, included in some cancers, makes this suboptimal. Other pharmacologic agents to consider along these lines include other growth factors that have demonstrated cytoprotection from anthracyclines such as insulin-like growth factor-1, or cardiotrophin-1[76]. Like neuregulin, however, there are concerns that these agents may promote cancer growth. Obviously the ideal strategy would be one that locally activates neuregulin and/or other cardioprotective ligands or the downstream effector mechanisms in the heart. Local overexpression of downstream effector kinases such as Akt [77] or transcription factors such as GATA4 [78] protect mice heart from anthracyclines. However these tools are still far away from clinical utility.
One strategy yet to be explored is to use endogenous ligands and kinases to create a window of cardioprotection. Focusing on neuregulin, for example, we know that there is robust expression of this ligand in the heart, though it appears to have minimal activity at baseline. The levels of expression of neuregulin indicate that it serves some physiological function beyond cardiac development, though this is yet to be fully understood. If we knew the physiological mechanisms for activation of neuregulin in the heart, one could envision a scenario where subjecting a patient to those conditions prior to anthracycline exposure would create a window of cardioprotection. There is now evidence that erbB2 signaling in response to the ligand neuregulin regulates anthracycline efflux out of the cells via the multidrug-resistance protein [79]. Therefore upregulation of cardiac neuregulin signaling may be one strategy to limit myocardial anthracycline injury.
Based upon our experimental work in rat and human skeletal muscle [80, 81], one could hypothesize that physiologic cardiac stress in the form of exercise will increase neuregulin/erbB signaling. In skeletal muscle, neuregulin-1 and the erbB receptors are known to be expressed where they serve a function at the neuromuscular junction to regulate myoblast differentiation as well as formation and maintenance of the post-synaptic endplate [82]. We demonstrated that these proteins are localized in the adult skeletal muscle at the neuromuscular junction, and that exercise is a potent activator of neuregulin release, with subsequent activation of erbB phosphorylation [80]. The proximal mechanisms for neuregulin release during exercise remain incompletely characterized. Interestingly, an exercise training regimen in people increases the expression of erbB3 receptors in skeletal muscle [81]. Arguably increased erbB3 expression will increase sensitivity of muscle to neuregulin action. The demonstration that neuregulin increases skeletal muscle glucose uptake suggests an interesting hypothesis, that exercise-regulated glucose uptake in skeletal muscle is mediated in part by neuregulin.
Thus an attractive hypothesis is that a period of exercise sufficient to activate neuregulin (and/or other cardioprotective signaling) can protect a cancer patient’s heart during anthracycline exposure [83]. Certainly periodic exercise has been known for a long time to reduce risk of ischemic cardiovascular events (e.g. [84, 85]). In healthy individuals as well as those with chronic disease, exercise has been shown to improve quality of life and reduce all-cause mortality [86, 87]. Epidemiologic studies suggesting that periodic exercise is associated with lower risk of the development of post-menopausal breast cancer [88]. Several recent randomized trials examined whether similar benefits are found in breast cancer patients undergoing conventional adjuvant chemotherapy. The results have provided preliminary evidence that exercise training is a feasible intervention that may attenuate a broad range of deleterious symptoms (e.g., functional decline, fatigue, nausea) associated with cytotoxic therapy, leading to clinically relevant improvements in quality of life [89-91].
The hypothesis that exercise will protect the heart from anthracycline-induced damage has been examined in animals. More than 20 years ago Kanter et al. demonstrated that swim training in rats decreased the histopathologic damage induced by anthracylines [92]. The authors suggested that the exercise effect they observed in rats occurred via changes in antioxidant activity. They reported increased levels of catalase, SOD and glutathione peroxidase in blood, liver and heart in swim-trained rats [92]. There are many other potential mechanisms by which forced strenuous exercise might protect the heart including the well-known effects of strenuous exercise on expression of heat shock proteins (e.g. [93]), which are cardioprotective against anthracycline cardiotoxicity [94]. Finally the effects of exercise on adrenergic tone might also promote cardioprotection based upon the finding that pharmacologic administration of the alpha-adrenergic agonist phenylephrine is able to prevent anthracycline cardiotoxicity in mice [49]. Whether exercise is protecting the heart of patients being treated with therapies that have known cardiovascular effects including anthracyclines, radiation therapy, and trastuzumab has not been specifically examined.
ACKNOWLEDGEMENTS
We thank Vicki Kivett for administrative assistance with this manuscript. This work was supported by grants HL068144 (DBS) and K01AG024056 (CCL) from the National Institutes of Health and an Established Investigator Award from the American Heart Association to DBS, American Heart Association Beginning Grant-In-Aid to XP, Amyloidosis Foundation Research Grant to XP. CCL is a Stahlman scholar at Vanderbilt University.
Abbreviations
- SOD
Superoxide dismutase
- ERK
extracellular singal-regulated kinase
- SAPK
stress-activated protein kinase
- JNK
c-Jun N-terminal kinase
- ROS
Reactive oxygen species
- PI3K
Phosphoinositide-3-kinase
- Akt
protein kinase family, a.k.a. protein kinase B
- GATA4
gene name, member of GATA family of zinc-finger transcription factors
- IGF
Insulin-like growth factor
- CARP
cardiac ankyrin repeat protein and cardiac adriamycin-responsive protein
- Dox
Doxorubicin
- Nrg
Neuregulin
- siRNA
short interfering RNA
- erbB2
(a.k.a. HER2 – homology of EGF receptor, neu) erythroblastic leukemia viral oncogene homolog 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57(7):727–41. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 2.Minotti G, et al. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56(2):185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 3.Legha SS, et al. Reduction of doxorubicin cardiotoxicity by prolonged continuous intravenous infusion. Ann Intern Med. 1982;96(2):133–9. doi: 10.7326/0003-4819-96-2-133. [DOI] [PubMed] [Google Scholar]
- 4.Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97(11):2869–79. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- 5.Balanehru S, Nagarajan B. Intervention of adriamycin induced free radical damage. Biochem Int. 1992;28(4):735–44. [PubMed] [Google Scholar]
- 6.Fogli S, Nieri P, Breschi MC. The role of nitric oxide in anthracycline toxicity and prospects for pharmacologic prevention of cardiac damage. Faseb J. 2004;18(6):664–75. doi: 10.1096/fj.03-0724rev. [DOI] [PubMed] [Google Scholar]
- 7.Mihm MJ, Bauer JA. Peroxynitrite-induced inhibition and nitration of cardiac myofibrillar creatine kinase. Biochimie. 2002;84(10):1013–9. doi: 10.1016/s0300-9084(02)00005-6. [DOI] [PubMed] [Google Scholar]
- 8.Tokarska-Schlattner M, Wallimann T, Schlattner U. Multiple interference of anthracyclines with mitochondrial creatine kinases: preferential damage of the cardiac isoenzyme and its implications for drug cardiotoxicity. Mol Pharmacol. 2002;61(3):516–23. doi: 10.1124/mol.61.3.516. [DOI] [PubMed] [Google Scholar]
- 9.Olson RD, Mushlin PS. Doxorubicin cardiotoxicity: analysis of prevailing hypotheses. FASEB J. 1990;4(13):3076–86. [PubMed] [Google Scholar]
- 10.Ito H, et al. Doxorubicin selectively inhibits muscle gene expression in cardiac muscle cells in vivo and in vitro. Proc Natl Acad Sci U S A. 1990;87(11):4275–9. doi: 10.1073/pnas.87.11.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeyaseelan R, et al. A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J Biol Chem. 1997;272(36):22800–8. doi: 10.1074/jbc.272.36.22800. [DOI] [PubMed] [Google Scholar]
- 12.Jeyaseelan R, et al. Molecular mechanisms of doxorubicin-induced cardiomyopathy. Selective suppression of Reiske iron-sulfur protein, ADP/ATP translocase, and phosphofructokinase genes is associated with ATP depletion in rat cardiomyocytes. J Biol Chem. 1997;272(9):5828–32. doi: 10.1074/jbc.272.9.5828. [DOI] [PubMed] [Google Scholar]
- 13.Lim CC, et al. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J Biol Chem. 2004;279(9):8290–9. doi: 10.1074/jbc.M308033200. [DOI] [PubMed] [Google Scholar]
- 14.Fu M, et al. Properties of G-protein modulated receptor-adenylyl cyclase system in myocardium of spontaneously hypertensive rats treated with adriamycin. Int J Cardiol. 1994;44(1):9–18. doi: 10.1016/0167-5273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 15.Calderone A, de Champlain J, Rouleau JL. Adriamycin-induced changes to the myocardial beta-adrenergic system in the rabbit. J Mol Cell Cardiol. 1991;23(3):333–42. doi: 10.1016/0022-2828(91)90069-x. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi S, et al. Effects of in vitro and in vivo exposure to doxorubicin (adriamycin) on caffeine-induced Ca2+ release from sarcoplasmic reticulum and contractile protein function in ‘chemically-skinned’ rabbit ventricular trabeculae. Jpn J Pharmacol. 1998;76(4):405–13. doi: 10.1254/jjp.76.405. [DOI] [PubMed] [Google Scholar]
- 17.Unverferth DV, et al. The effect of first-dose doxorubicin on the cyclic nucleotide levels of the human myocardium. Toxicol Appl Pharmacol. 1981;60(1):151–4. doi: 10.1016/0041-008x(81)90145-9. [DOI] [PubMed] [Google Scholar]
- 18.Lipshultz SE, et al. Predictive value of cardiac troponin T in pediatric patients at risk for myocardial injury. Circulation. 1997;96(8):2641–8. doi: 10.1161/01.cir.96.8.2641. [DOI] [PubMed] [Google Scholar]
- 19.Arola OJ, et al. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60(7):1789–92. [PubMed] [Google Scholar]
- 20.Sawyer DB, et al. Daunorubicin-induced apoptosis in rat cardiac myocytes is inhibited by dexrazoxane. Circ Res. 1999;84(3):257–65. doi: 10.1161/01.res.84.3.257. [DOI] [PubMed] [Google Scholar]
- 21.Buzdar AU, et al. Early and delayed clinical cardiotoxicity of doxorubicin. Cancer. 1985;55(12):2761–5. doi: 10.1002/1097-0142(19850615)55:12<2761::aid-cncr2820551206>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 22.Gille L, Nohl H. Analyses of the molecular mechanism of adriamycin-induced cardiotoxicity. Free Radic Biol Med. 1997;23(5):775–82. doi: 10.1016/s0891-5849(97)00025-7. [DOI] [PubMed] [Google Scholar]
- 23.Pouna P, et al. Development of the model of rat isolated perfused heart for the evaluation of anthracycline cardiotoxicity and its circumvention. Br J Pharmacol. 1996;117(7):1593–9. doi: 10.1111/j.1476-5381.1996.tb15326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajagopalan S, et al. Adriamycin-induced free radical formation in the perfused rat heart: implications for cardiotoxicity. Cancer Res. 1988;48(17):4766–9. [PubMed] [Google Scholar]
- 25.Yen HC, et al. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest. 1996;98(5):1253–60. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiseman LR, Spencer CM. Dexrazoxane. A review of its use as a cardioprotective agent in patients receiving anthracycline-based chemotherapy. Drugs. 1998;56(3):385–403. doi: 10.2165/00003495-199856030-00009. [DOI] [PubMed] [Google Scholar]
- 27.Lipshultz SE, et al. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N Engl J Med. 2004;351(2):145–53. doi: 10.1056/NEJMoa035153. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W, et al. Role of mitogen-activated protein kinase in cardiac hypertrophy and heart failure. Exp Clin Cardiol. 2003;8(4):173–83. [PMC free article] [PubMed] [Google Scholar]
- 29.Lorenz K, et al. Cardiac hypertrophy: targeting Raf/MEK/ERK1/2-signaling. Int J Biochem Cell Biol. 2009;41(12):2351–5. doi: 10.1016/j.biocel.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Petrich BG, Wang Y. Stress-activated MAP kinases in cardiac remodeling and heart failure; new insights from transgenic studies. Trends Cardiovasc Med. 2004;14(2):50–5. doi: 10.1016/j.tcm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Laderoute KR, Webster KA. Hypoxia/reoxygenation stimulates Jun kinase activity through redox signaling in cardiac myocytes. Circ Res. 1997;80(3):336–44. doi: 10.1161/01.res.80.3.336. [DOI] [PubMed] [Google Scholar]
- 32.Xia Z, et al. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326–31. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 33.Matsui T, et al. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104(3):330–5. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- 34.Matsui T, Davidoff AJ. Assessment of PI-3 kinase and Akt in ischemic heart diseases in diabetes. Methods Mol Med. 2007;139:329–38. doi: 10.1007/978-1-59745-571-8_22. [DOI] [PubMed] [Google Scholar]
- 35.Chao W, et al. Strategic advantages of insulin-like growth factor-I expression for cardioprotection. J Gene Med. 2003;5(4):277–86. doi: 10.1002/jgm.347. [DOI] [PubMed] [Google Scholar]
- 36.Hong F, et al. Insulin-like growth factor-1 protects H9c2 cardiac myoblasts from oxidative stress-induced apoptosis via phosphatidylinositol 3-kinase and extracellular signal-regulated kinase pathways. Life Sci. 2001;68(10):1095–105. doi: 10.1016/s0024-3205(00)01012-2. [DOI] [PubMed] [Google Scholar]
- 37.Lai HC, et al. Insulin-like growth factor-1 prevents loss of electrochemical gradient in cardiac muscle mitochondria via activation of PI 3 kinase/Akt pathway. Mol Cell Endocrinol. 2003;205(1-2):99–106. doi: 10.1016/s0303-7207(03)00200-4. [DOI] [PubMed] [Google Scholar]
- 38.Fukazawa R, et al. Neuregulin-1 protects ventricular myocytes from anthracycline-induced apoptosis via erbB4-dependent activation of PI3-kinase/Akt. J Mol Cell Cardiol. 2003;35(12):1473–9. doi: 10.1016/j.yjmcc.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Childs AC, et al. Doxorubicin treatment in vivo causes cytochrome C release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl-2:Bax ratio. Cancer Res. 2002;62(16):4592–8. [PubMed] [Google Scholar]
- 40.An J, et al. ARC is a critical cardiomyocyte survival switch in doxorubicin cardiotoxicity. J Mol Med. 2009;87(4):401–10. doi: 10.1007/s00109-008-0434-z. [DOI] [PubMed] [Google Scholar]
- 41.Kleiner Y, et al. TVP1022 and propargylamine protect neonatal rat ventricular myocytes against doxorubicin-induced and serum starvation-induced cardiotoxicity. J Cardiovasc Pharmacol. 2008;52(3):268–77. doi: 10.1097/FJC.0b013e3181862441. [DOI] [PubMed] [Google Scholar]
- 42.Goswami SK, Das DK. Autophagy in the myocardium: Dying for survival? Exp Clin Cardiol. 2006;11(3):183–8. [PMC free article] [PubMed] [Google Scholar]
- 43.Jang YM, et al. Doxorubicin treatment in vivo activates caspase-12 mediated cardiac apoptosis in both male and female rats. FEBS Lett. 2004;577(3):483–90. doi: 10.1016/j.febslet.2004.10.053. [DOI] [PubMed] [Google Scholar]
- 44.Sorensen BS, et al. Mode of action of topoisomerase II-targeting agents at a specific DNA sequence. Uncoupling the DNA binding, cleavage and religation events. J Mol Biol. 1992;228(3):778–86. doi: 10.1016/0022-2836(92)90863-f. [DOI] [PubMed] [Google Scholar]
- 45.Charron F, Nemer M. GATA transcription factors and cardiac development. Semin Cell Dev Biol. 1999;10(1):85–91. doi: 10.1006/scdb.1998.0281. [DOI] [PubMed] [Google Scholar]
- 46.Charron F, et al. Cooperative interaction between GATA-4 and GATA-6 regulates myocardial gene expression. Mol Cell Biol. 1999;19(6):4355–65. doi: 10.1128/mcb.19.6.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grillot DA, et al. Genomic organization, promoter region analysis, and chromosome localization of the mouse bcl-x gene. J Immunol. 1997;158(10):4750–7. [PubMed] [Google Scholar]
- 48.Kim Y, et al. Anthracycline-induced suppression of GATA-4 transcription factor: implication in the regulation of cardiac myocyte apoptosis. Mol Pharmacol. 2003;63(2):368–77. doi: 10.1124/mol.63.2.368. [DOI] [PubMed] [Google Scholar]
- 49.Aries A, et al. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc Natl Acad Sci U S A. 2004;101(18):6975–80. doi: 10.1073/pnas.0401833101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cardinale D, et al. Anthracycline-induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. J Am Coll Cardiol. 2010;55(3):213–20. doi: 10.1016/j.jacc.2009.03.095. [DOI] [PubMed] [Google Scholar]
- 51.Rowan RA, Masek MA, Billingham ME. Ultrastructural morphometric analysis of endomyocardial biopsies. Idiopathic dilated cardiomyopathy, anthracycline cardiotoxicity, and normal myocardium. Am J Cardiovasc Pathol. 1988;2(2):137–44. [PubMed] [Google Scholar]
- 52.Mackay B, et al. Assessment of anthracycline cardiomyopathy by endomyocardial biopsy. Ultrastruct Pathol. 1994;18(1-2):203–11. doi: 10.3109/01913129409016291. [DOI] [PubMed] [Google Scholar]
- 53.Mortensen SA, Olsen HS, Baandrup U. Chronic anthracycline cardiotoxicity: haemodynamic and histopathological manifestations suggesting a restrictive endomyocardial disease. Br Heart J. 1986;55(3):274–82. doi: 10.1136/hrt.55.3.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Helmes M, Trombitas K, Granzier H. Titin develops restoring force in rat cardiac myocytes. Circ Res. 1996;79(3):619–26. doi: 10.1161/01.res.79.3.619. [DOI] [PubMed] [Google Scholar]
- 55.Wang SM, et al. Studies on cardiac myofibrillogenesis with antibodies to titin, actin, tropomyosin, and myosin. J Cell Biol. 1988;107(3):1075–83. doi: 10.1083/jcb.107.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Helmes M, et al. Titin determines the Frank-Starling relation in early diastole. J Gen Physiol. 2003;121(2):97–110. doi: 10.1085/jgp.20028652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bang ML, et al. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J Cell Biol. 2001;153(2):413–27. doi: 10.1083/jcb.153.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poizat C, et al. Proteasome-mediated degradation of the coactivator p300 impairs cardiac transcription. Mol Cell Biol. 2000;20(23):8643–54. doi: 10.1128/mcb.20.23.8643-8654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rajkumar T, Gullick WJ. The type I growth factor receptors in human breast cancer. Breast Cancer Res Treat. 1994;29(1):3–9. doi: 10.1007/BF00666177. [DOI] [PubMed] [Google Scholar]
- 60.Klapper LN, et al. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors. Adv Cancer Res. 2000;77:25–79. [PubMed] [Google Scholar]
- 61.Slamon DJ, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 62.Fendly BM, et al. The extracellular domain of HER2/neu is a potential immunogen for active specific immunotherapy of breast cancer. J Biol Response Mod. 1990;9(5):449–55. [PubMed] [Google Scholar]
- 63.Sliwkowski MX, et al. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin) Semin Oncol. 1999;26(4 Suppl 12):60–70. [PubMed] [Google Scholar]
- 64.Slamon DJ, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 65.Feldman AM, Lorell BH, Reis SE. Trastuzumab in the treatment of metastatic breast cancer: anticancer therapy versus cardiotoxicity. Circulation. 2000;102(3):272–4. doi: 10.1161/01.cir.102.3.272. [DOI] [PubMed] [Google Scholar]
- 66.Ewer MS, et al. Reversibility of trastuzumab-related cardiotoxicity: new insights based on clinical course and response to medical treatment. J Clin Oncol. 2005;23(31):7820–6. doi: 10.1200/JCO.2005.13.300. [DOI] [PubMed] [Google Scholar]
- 67.Negro A, Brar BK, Lee KF. Essential roles of Her2/erbB2 in cardiac development and function. Recent Prog Horm Res. 2004;59:1–12. doi: 10.1210/rp.59.1.1. [DOI] [PubMed] [Google Scholar]
- 68.Ozcelik C, et al. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc Natl Acad Sci U S A. 2002;99(13):8880–5. doi: 10.1073/pnas.122249299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crone SA, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med. 2002;8(5):459–65. doi: 10.1038/nm0502-459. [DOI] [PubMed] [Google Scholar]
- 70.Zhao YY, et al. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem. 1998;273(17):10261–9. doi: 10.1074/jbc.273.17.10261. [DOI] [PubMed] [Google Scholar]
- 71.Sawyer DB, et al. Modulation of anthracycline-induced myofibrillar disarray in rat ventricular myocytes by neuregulin-1beta and anti-erbB2: potential mechanism for trastuzumab-induced cardiotoxicity. Circulation. 2002;105(13):1551–4. doi: 10.1161/01.cir.0000013839.41224.1c. [DOI] [PubMed] [Google Scholar]
- 72.Chung KY, Walker JW. Interaction and inhibitory cross-talk between endothelin and ErbB receptors in the adult heart. Mol Pharmacol. 2007;71(6):1494–502. doi: 10.1124/mol.106.027599. [DOI] [PubMed] [Google Scholar]
- 73.Lipshultz SE. Dexrazoxane for protection against cardiotoxic effects of anthracyclines in children. J Clin Oncol. 1996;14(2):328–31. doi: 10.1200/JCO.1996.14.2.328. [DOI] [PubMed] [Google Scholar]
- 74.Swain SM, Vici P. The current and future role of dexrazoxane as a cardioprotectant in anthracycline treatment: expert panel review. J Cancer Res Clin Oncol. 2004;130(1):1–7. doi: 10.1007/s00432-003-0498-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu FF, et al. Heterozygous knockout of neuregulin-1 gene in mice exacerbates doxorubicin-induced heart failure. Am J Physiol Heart Circ Physiol. 2005;289(2):H660–6. doi: 10.1152/ajpheart.00268.2005. [DOI] [PubMed] [Google Scholar]
- 76.Kunisada K, et al. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97(1):315–9. doi: 10.1073/pnas.97.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suliman HB, et al. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest. 2007;117(12):3730–41. doi: 10.1172/JCI32967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suzuki YJ, Evans T. Regulation of cardiac myocyte apoptosis by the GATA-4 transcription factor. Life Sci. 2004;74(15):1829–38. doi: 10.1016/j.lfs.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 79.Peng XI, Basak, Zhong L, Chen B, Sawyer DB. Membrane Protein P-glycoprotein Expression and Activity in Cardiac Myocytes is regulated by ErbB2 Signaling. Circulation. 2009 Abstract for American Heart Association Basic Research Annual Meeting 2009. [Google Scholar]
- 80.Lebrasseur NK, et al. Regulation of neuregulin/ErbB signaling by contractile activity in skeletal muscle. Am J Physiol Cell Physiol. 2003;284(5):C1149–55. doi: 10.1152/ajpcell.00487.2002. [DOI] [PubMed] [Google Scholar]
- 81.LeBrasseur NK, et al. The expression of neuregulin and erbB receptors in human skeletal muscle: effects of progressive resistance training. Eur J Appl Physiol. 2005;94(4):371–5. doi: 10.1007/s00421-005-1333-4. [DOI] [PubMed] [Google Scholar]
- 82.Jo SA, et al. Neuregulins are concentrated at nerve-muscle synapses and activate ACh-receptor gene expression. Nature. 1995;373(6510):158–61. doi: 10.1038/373158a0. [DOI] [PubMed] [Google Scholar]
- 83.Peng X, et al. The Cardiotoxicology of Anthracycline Chemotherapeutics: TRANSLATING MOLECULAR MECHANISM INTO PREVENTATIVE MEDICINE. Mol Interv. 2005;5(3):163–71. doi: 10.1124/mi.5.3.6. [DOI] [PubMed] [Google Scholar]
- 84.Rechnitzer PA, et al. Ontario Exercise-Heart Collaborative Study Relation of exercise to the recurrence rate of myocardial infarction in men. Am J Cardiol. 1983;51(1):65–9. doi: 10.1016/s0002-9149(83)80012-5. [DOI] [PubMed] [Google Scholar]
- 85.Chandrashekhar Y, Anand IS. Exercise as a coronary protective factor. Am Heart J. 1991;122(6):1723–39. doi: 10.1016/0002-8703(91)90290-x. [DOI] [PubMed] [Google Scholar]
- 86.Physical Activity Guidelines Advisory Committee report, 2008. To the Secretary of Health and Human Services. Part A: executive summary. Nutr Rev. 2009;67(2):114–20. doi: 10.1111/j.1753-4887.2008.00136.x. [DOI] [PubMed] [Google Scholar]
- 87.Levine MN, Gafni A. Clinical decision making vs programme evaluation perspectives. Pharmacoeconomics. 1993;4(3):228–31. doi: 10.2165/00019053-199304030-00007. [DOI] [PubMed] [Google Scholar]
- 88.Wiseman M. The second World Cancer Research Fund/American Institute for Cancer Research expert report. Food, nutrition, physical activity, and the prevention of cancer: a global perspective. Proc Nutr Soc. 2008;67(3):253–6. doi: 10.1017/S002966510800712X. [DOI] [PubMed] [Google Scholar]
- 89.Mock V, et al. Exercise manages fatigue during breast cancer treatment: a randomized controlled trial. Psychooncology. 2005;14(6):464–77. doi: 10.1002/pon.863. [DOI] [PubMed] [Google Scholar]
- 90.Segal R, et al. Structured exercise improves physical functioning in women with stages I and II breast cancer: results of a randomized controlled trial. J Clin Oncol. 2001;19(3):657–65. doi: 10.1200/JCO.2001.19.3.657. [DOI] [PubMed] [Google Scholar]
- 91.Pinto BM, et al. Home-based physical activity intervention for breast cancer patients. J Clin Oncol. 2005;23(15):3577–87. doi: 10.1200/JCO.2005.03.080. [DOI] [PubMed] [Google Scholar]
- 92.Kanter MM, et al. Effect of exercise training on antioxidant enzymes and cardiotoxicity of doxorubicin. J Appl Physiol. 1985;59(4):1298–303. doi: 10.1152/jappl.1985.59.4.1298. [DOI] [PubMed] [Google Scholar]
- 93.Melling CW, Thorp DB, Noble EG. Regulation of myocardial heat shock protein 70 gene expression following exercise. J Mol Cell Cardiol. 2004;37(4):847–55. doi: 10.1016/j.yjmcc.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 94.Ito H, et al. Thermal preconditioning protects rat cardiac muscle cells from doxorubicin-induced apoptosis. Life Sci. 1999;64(9):755–61. doi: 10.1016/s0024-3205(98)00617-1. [DOI] [PubMed] [Google Scholar]