

Abstract
Clinicians have long known that a substantial proportion of patients treated with high-dose glucocorticoids experience a variety of serious side effects, including metabolic syndrome, bone loss, and mood shifts, such as depressive symptomatology, manic or hypomanic symptoms, and even suicide. The reason for individual variability in expression or severity of these side effects is not clear. However, recent emerging literature is beginning to shed light on possible mechanisms of these effects. As an introduction to this volume, this chapter will review the basic biology of glucocorticoid release and molecular mechanisms of glucocorticoid receptor function, and will discuss how dysregulation of glucocorticoid action at all levels could contribute to such side effects. At the molecular level, glucocorticoid receptor polymorphisms may be associated either with receptor hypofunction or hyperfunction and could thus contribute to differential individual sensitivity to the effects of glucocorticoid treatment. Numerous factors regulate hypothalamic-pituitary-adrenal (HPA) axis responsiveness, which could also contribute to individual differences in glucocorticoid side effects. One of these is sex hormone status and the influence of estrogen and progesterone on HPA axis function and mood. Another is immune system activity, in which immune molecules, such as interleukins and cytokines, activate the HPA axis and alter brain function, including memory, cognition, and mood. The effects of cytokines in inducing sickness behaviors, which overlap with depressive symptomatology, could also contribute to individual differences in such symptomatology. Taken together, this knowledge will have important relevance for identifying at-risk patients to avoid or minimize such side effects when they are treated with glucocorticoids. A framework for assessment of patients is proposed that incorporates functional, physiological, and molecular biomarkers to identify subgroups of patients at risk for depressive symptomatology associated with glucocorticoid treatment, and for prevention of side effects, which in many cases can be life-threatening.
Keywords: cortisol, HPA axis, glucocorticoid resistance, depression, psychosis, inflammation, cytokines
Introduction
Since the discovery of the potent anti-inflammatory and immunosupressive effect of glucocorticoids in the 1940s by Philip Hench and the synthesis of cortisone by Edward Kendall and Tadeus Reichstein, glucocorticoids have been an important treatment for many autoimmune and inflammatory diseases. Glucocorticoids have been widely prescribed in routine clinical practice, and clinicians have long known their many side effects. These include metabolic syndrome, bone loss, psychiatric symptoms, and even suicide. Up to 20% of patients on high doses of glucocorticoids develop psychiatric disorders including depression, mania, and psychosis or a mixed affective state,1 and up to 75% report psychiatric symptoms, which can be reversed with cessation of glucocorticoid therapy.2 Although this association between glucocorticoids and mood has been known for decades, the potential increased risk for the development of mood disorders, psychosis, cognitive deficits, and other psychiatric symptoms due to use of this medication is often overlooked (see Wolkowitz et al.2a and Brown2b).3,4 Of note, psychiatric side effects due to use of glucocorticoids are often sudden in onset,5,6 and patients who are taking higher doses of glucocorticoids are at increased risk for developing these side effects.1 Some studies have shown that psychiatric side effects can be prevented by co-administration of mood stabilizers with glucocorticoids (see Wolkowitz et al.2a and Brown2b).7,8
Additionally, it has long been known that endogenous excess or dysregulation of glucocorticoids, as occurs in Cushing’s syndrome, is associated with mood changes, including depression and hypomanic symptomatology.9–12 Interestingly, Cushing’s syndrome can also be triggered by prolonged and topical application of high doses of corticosteroids that can lead to hypothalamic-pituitary-adrenal (HPA) axis dysregulation.13–15 Conversely, major depressive disorder (MDD) itself is associated with dysregulation of the HPA axis, although it is not clear whether the altered HPA axis function is primary or secondary to depression. HPA axis dysfunction reported in patients with depression is thought to be related, at least in part, to impaired feedback inhibition by endogenous glucocorticoids due to glucocorticoid resistance16 and/or to hypersecretion of hypothalamic corticotropin-releasing hormone (CRH).17,18 Thus, both exogenous use of high-dose steroids and endogenous excess of these hormones is associated with mood disorders or depressive symptomatology in some individuals; however, the mechanisms of this effect are unknown. Indeed, successful antidepressant treatment with reduction of depressive symptomatology is associated with resolution of impaired HPA axis function.19–21
It has been suggested that HPA axis normalization, either through the use of glucocorticoid receptor (GR) antagonists or agonists, could provide new therapeutic strategies for at least some cases of depression (see Wolkowitz et al.2a and Pariante21a).22,23 Some studies have shown an antidepressant effect with GR agonists, such as dexamethasone, prednisolone, and hydrocortisone.22,24,25 Conversely, randomized controlled trials have been performed with different GR antagonists and have shown an antidepressant effect in some subtypes of depression (see Wolkowitz et al.2a).23,26–28 Moreover, clinical studies have shown that CRH receptor type 1 (CRH-R1) antagonists have a psychotropic effect, unrelated to their neuroendocrine action, reducing anxiety and depressive symptoms (see Chrousos and Kino,28a Hauger et al.28b and Refojo and Holsboer28c).17,18
Emerging literature is beginning to shed light on possible mechanisms of the association between glucocorticoids and mood. This review will discuss how dysregulation of glucocorticoid action at all levels, from altered circulating hormone levels to genetic mutations of the GR, could contribute to potential individual differences in susceptibility to glucocorticoid-induced side effects.
At a molecular level, polymorphisms in the human GR gene may be associated either with receptor hypo- or hyperfunction, which could contribute to differential individual sensitivity to the effects of glucocorticoid treatment. Indeed, recent large population-based studies indicate that some polymorphisms and other gene variants of GR are associated with a variety of clinical syndromes, including cardiovascular disease, dementia, rheumatoid arthritis (RA), metabolic syndrome, depression, and bipolar disorder (see Manenschijn et al.28d and Spijker and Van Rossum28e).29–35 Some of these functionally relevant polymorphisms occur in a prevalence range of 15–20%, indicating a need for systematically addressing their clinical and therapeutic implications for glucocorticoid therapy and potential side effects.
Differential release of glucocorticoids from the adrenal glands, via activation of the HPA axis, has also been described in endogenous forms of depression and could potentially contribute to individual variability in side effects induced by glucocorticoid treatment.
Immune system–HPA axis interactions could also play a role in influencing HPA axis activity. Thus, immune molecules, such as interleukins and cytokines, activate the HPA axis, with the resultant release of glucocorticoids, which in turn modulates the immune response.36,37 In addition, cytokines can alter local glucocorticoid availability and GR function, therefore affecting an individual’s glucocorticoid sensitivity. Understanding such interactions will shed light on the association between disruptions of the HPA axis and conditions, such as RA and other autoimmune/inflammatory diseases,38 as well as depression. In addition, the excess cytokines released in such conditions may induce sickness behavior, a syndrome that overlaps with depressive symptomatology.39,40 These interactions suggest another potential mechanism for individual variation in susceptibility to glucocorticoid-induced mood symptomatology and may explain the high prevalence of comorbidity between inflammatory diseases and psychiatric disorders.
Further elucidation of these mechanisms will have important relevance for identifying patients at risk for developing glucocorticoid-induced behavioral alterations, and therefore will aid in avoiding or minimizing such side effects when these individuals are treated with glucocorticoids. Thus, it will have great relevance to any clinical specialty in which glucocorticoids are used, which in many cases can be life-threatening and can result in serious mood effects, including depression and suicidality.
Glucocorticoids: Release, Availability and Receptor Action
Glucocorticoids affect a variety of biological processes including growth, metabolism, development, behavior, immune function, and the stress response.41 Maintenance of appropriate levels of glucocorticoids is thus essential for homeostasis in many physiological systems.
Glucocorticoid Release
The glucocorticoids cortisol (humans and primates) and corticosterone (rodents) are steroid hormones synthesized from cholesterol in the zona fasciculata of the adrenal cortex. In humans, cortisol is the principal glucocorticoid, involved in the regulation of metabolism, the stress response, and immune function, while corticosterone is important mainly as an intermediate in the steroidogenic pathway to produce aldosterone.42 Production of circulating glucocorticoids is under control of the HPA axis, which is activated by psychogenic and physical stress as well as inflammatory stimuli (Fig. 1).38
Figure 1.

Diagram of the routes of communication between the brain and immune system, including the HPA axis, autonomic nervous system, and cytokine interactions with both of these systems. Central nervous system (CNS); sympathetic nervous system (SNS); peripheral nervous system (PNS); corticotropin-releasing hormone (CRH); arginine vasopressin (AVP); adrenocorticotropic hormone (ACTH). [Reprinted with modifications by permission from Ref. 137.]
Activation of the HPA axis begins with the release of CRH from the paraventricular nucleus (PVN) of the hypothalamus. CRH neurons of the PVN serve as a final common stress pathway by receiving converging inputs from multiple areas of the brain, allowing CRH to coordinate the behavioral, neuroendocrine, autonomic, and immune responses to stress. These afferent pathways include projections from ascending noradrenergic pathways from the brain stem relaying visceral sensory information; descending cortical and limbic pathways relaying cognitive and emotional information; thalamic connections relaying sensory information; intrahypothalamic connections receiving input from the brain stem and limbic structures; and circumventricular organs relaying blood-borne chemosensory signals.36 Therefore, hypothalamic CRH neurons are strategically situated to intercept and disperse signals from and to the body about the internal and external environment.
CRH-containing neurons in the PVN of the hypothalamus terminate in the median eminence. CRH is then released into the hypophysial-portal circulation and acts on CRH-R1 receptors on anterior pituitary corticotrophs stimulating secretion of adrenocorticotropic hormone (ACTH) into the general circulation and promoting synthesis of the ACTH precursor peptide proopiomelanocortin (POMC) in the corticotrophs. Under certain conditions, hypothalamic arginine vasopressin acts in synergy with CRH to stimulate ACTH release. Circulating ACTH then acts on the MC2-R (type 2 melanocortin receptor) in the adrenal cortex, stimulating secretion of glucocorticoids into the bloodstream.36,42
In order to maintain appropriate levels of circulating glucocorticoids, these hormones exert negative feedback on the hypothalamus and pituitary to inhibit the synthesis and secretion of CRH and POMC/ACTH, respectively.43 In addition, they also downregulate the expression of corticotrophic CRH-R1 mRNA, thus decreasing CRH-R1 number, which ultimately diminishes ACTH secretion. Finally, the hippocampus, which expresses a high density of GR, exerts negative feedback control on the PVN, thereby reducing HPA axis activity.44
Glucocorticoid Availability
Once glucocorticoids are released, several factors are responsible for the regulation of glucocorticoid availability in order to produce an appropriate glucocorticoid response. Such factors include corticosteroid binding globulin (CBG), the multidrug resistance transporter (MDR), and 11β-hydroxysteroid dehydrogenase (11β-HSD), all of which are locally regulated at the tissue and/or cell specific level (for review see Ref. 36). Moreover, these factors have been shown to be altered under conditions of immune activation (Fig. 2), as well as stress and antidepressant treatment.
Figure 2.
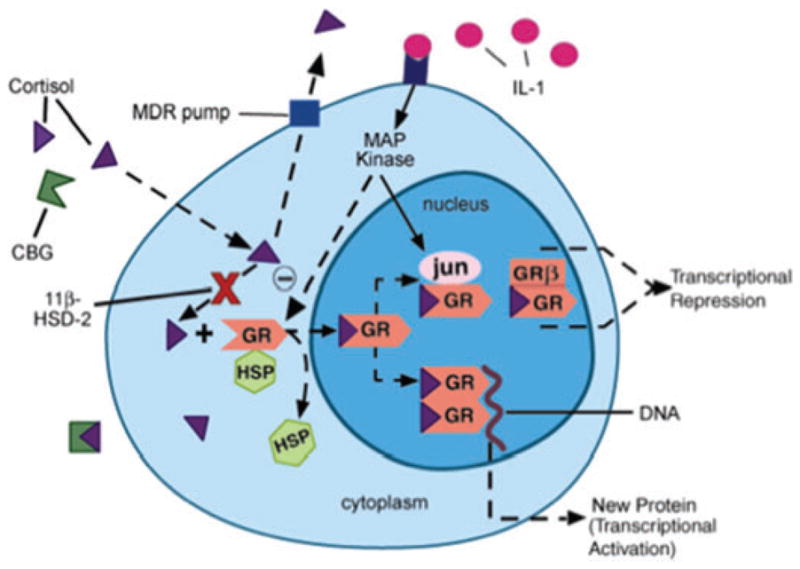
Local factors regulating glucocorticoid bioavailability and action. (1) Corticosteroid binding globulin (CBG), (2) multidrug resistance transporter (MDR pump), (3) 11β-hydroxysteroid dehydrogenase (11β-HSD), (4) glucocorticoid receptor (GR = GRα) nuclear translocation, (5) GR interaction with other transcription factors (AP-1 [jun/fos], NFκB) or MAP kinases, and (6) ratio of GRα:GRβ isoform expression. HSP, heat shock protein; MAP kinase, mitogen-activated protein kinase. (Reprinted by permission from Ref. 138.) (In color in Annals online.)
In order for glucocorticoids to exert their effects, they diffuse across the cell membrane and bind to cytosolic receptors. Only unbound glucocorticoids are capable of diffusing across the membrane; however, 90% of circulating glucocorticoids are bound to CBG.45 Therefore, the relative concentration of CBG is an important determinant of “free” and available glucocorticoid. Studies have shown that stress and hypercortisolemia are associated with lower CBG concentrations,46 whereas increased CBG levels have been reported in patients who responded to the antidepressant amitriptiline.47 Reduced levels of plasma CBG have also been found during various inflammatory conditions.36
Another important factor in the regulation of glucocorticoid availability is the MDR. The MDR P-glycoprotein (Pgp) is an ATP-dependent multidrug efflux pump expressed in several tissues, such as the brain, liver, kidney, intestine, and adrenal glands.48 At the blood–brain barrier, the MDR Pgp pump transports cortisol and the synthetic glucocorticoid dexamethasone, but not corticosterone, out of endothelial cells lining the brain.49 Therefore, modulation of MDR expression or function is also responsible for local glucocorticoid activity.
In vitro exposure to cytokines, such as interleukin (IL)-1, IL-6 and tumor necrosis factor (TNF-α), as well as an acute immune challenge in rodents, cause a decrease in MDR expression and/or function in multiple cell types. In contrast, patients with various autoimmune diseases, such as RA, colitis/Crohn’s disease, and lupus, tend to exhibit high lymphocytic MDR expression and/or activity, which positively correlates with disease activity in some cases (for review see Ref. 50). Greater MDR expression in immune cells may reduce glucocorticoid availability, thereby enhancing the synthesis/release of pro-inflammatory cytokines and exacerbating inflammatory responses. However, this increased MDR expression may be secondary to treatment with high-dose glucocorticoids.51,52 One possible reason for the discrepancy between the in vitro cytokine/animal studies and studies in humans with autoimmune disorders may be the differential effects of acute versus chronic inflammation on MDR expression. During acute inflammation, cytokines may downregulate MDR expression and increase local glucocorticoid concentrations, thereby limiting local inflammation and further cytokine release. In contrast, an upregulation of MDR expression may develop as a compensatory mechanism during chronic inflammatory conditions, and hence predispose humans toward autoimmune disease.
Altered MDR expression or function has also been shown to play a role in depression. Some studies have shown that different types of antidepressants increase glucocorticoid availability in cells by inhibiting MDR function and thereby possibly reducing glucocorticoid resistance (see Pariante21a).53,54 Furthermore, some studies have evaluated the relationship between MDR polymorphisms and therapeutic response to antidepressants; however, no association has yet been found.55,56
In addition, the enzyme 11β-HSD regulates glucocorticoid availability by acting as a shuttle in the conversion of glucocorticoids between its active and inactive forms. There are two isoforms of this enzyme. 11β-HSD-1 acts mainly as a reductase, converting inactive glucocorticoids (cortisone) to active glucocorticoids (cortisol), whereas 11β-HSD-2 acts as an oxidase/dehydrogenase to convert glucocorticoids from an active (cortisol) to inactive form (cortisone).57 While 11β-HSD-1 is widely expressed, with highest levels found in the liver, lung, adipose tissue and brain, 11β-HSD-2 is predominantly expressed in the aldosterone target tissues, such as the kidney, colon, and sweat glands (as well as the placenta), to prevent the binding of glucocorticoids to the mineralocorticoid receptor (MR). Pro-inflammatory cytokines, such as TNF-α and IL-1β, have been shown to upregulate 11β-HSD-1 and/or downregulate 11β-HSD-2 expression/activity in numerous cell types, favoring the formation of active glucocorticoids and counterbalancing inflammation.58 However, enhanced expression of 11β-HSD-2 or reduced expression of 11β-HSD-1 in immune cells would lead to reduced local levels of active glucocorticoid, thereby favoring a pro-inflammatory cytokine profile. This condition has been demonstrated in autoimmune patients and could be another possible mechanism underlying glucocorticoid resistance in such diseases (for more details see Ref. 50).
Regarding central nervous system function, glucocorticoids have been shown to influence mood, memory and neuronal integrity, where either too much or too little can exert deleterious effects.59 Accordingly, central 11β-HSD-1 expression is high in areas known to influence cognition and affect glucocorticoid negative feedback on the HPA axis, such as the prefrontal cortex, cerebellum, hippocampus, PVN of the hypothalamus, and pituitary gland. Moreover, inhibiting 11β-HSD-1 expression/function has been shown to improve cognitive functioning in aged mice and humans, who tend to exhibit elevated levels of circulating glucocorticoids.58 Although activity of 11β-HSD has been shown to be altered in patients with depression,60 inconsistent results have been reported.61
Glucocorticoid Receptor Action
Finally, glucocorticoids exert their activity by binding to cytosolic MRs and GRs that then translocate to the nucleus upon binding to their ligand.62,63 MRs have a high affinity for endogenous corticosteroids and have been associated with regulation of cortisol circadian fluctuations.21 Converserly, GRs have a lower affinity for endogenous corticosteroids, but a higher affinity for dexamethasone. Therefore, GRs play a role in regulating the peak morning response and stress response, as they are activated when endogenous glucocorticoid levels are higher.63
The GRs and MRs belong to a superfamily of nuclear hormone receptors, which can regulate gene expression by either direct interaction with specific promoter sequences called glucocorticoid response elements, or through protein–protein interactions with other transcription factors, such as NF-κB, AP-1, STAT, and NFAT (see Revollo and Cidlowski63a).64–67 The main anti-inflammatory effects of glucocorticoids are mediated through negative interactions with inflammatory-related transcription factors, such as NF-κB, AP-1, and STAT.68 Conversely, these transcription factors have been shown to inhibit GR function.69 In addition to being elevated in inflammatory disorders, NF-κB-DNA binding or activation of NF-κB responsive genes is elevated in depressed patients in response to a psychosocial stress and in subjects who are chronically stressed.70,71 This pathway may serve as another mechanism by which cytokines can confer a state of glucocorticoid resistance.
Sensitivity to glucocorticoid action at the level of the GR can be determined by factors, such as GR number, affinity, and function, including its ability to translocate to the nucleus and its interaction with other signal transduction pathways (as mentioned above).50,68 In addition, sensitivity to glucocorticoids depends on the expression of particular GR isoforms. There are multiple isoforms of GR arising from alternative splicing and translational events of a single gene (NR3C1): GRα, GRβ, and GR-P (for more details see Revollo and Cidlowski63a).62 GRα mediates glucocorticoid action, while GRβ and GR-P have been shown to mediate GRα activity and are unable to bind ligands. Generally, GRβ is expressed at very low levels compared to GRα; however, its expression is increased in tissues in inflammatory diseases, and it seems to be associated with decreased sensitivity to glucocorticoids.72
Increased GRβ expression may be related to impaired negative feedback mechanisms mediated by GRα activity and has been associated with development and/or aggravation of symptoms in various glucocorticoid-resistant diseases, such as asthma, colitis/Crohn’s, and RA.50 Moreover, changes in expression and stability of different GR isoforms can be triggered by inflammation73 and have also been associated with mutations or polymorphisms in the receptor.68,74,75
There have been approximately 550 single nucleotide polymorphism identified in the human GR gene. Studies have shown that some GR polymorphisms are related to increases (the N363S and BclI polymorphisms) or decreases (the ER22/23EK and 9β-A3669G polymorphisms) in glucocorticoid sensitivity and have been associated with altered susceptibility to metabolic disorders, cardiovascular disease, neuroendocrine responses to stress, and autoimmune and infectious disease, as well as altered cognition and affect (see Spijker and Van Rossum28e).33 Recently, genetic studies have shown that BclI and ER22/23EK polymorphisms are associated with increased susceptibility to develop major depression, and the ER22/23EK polymorphism is associated with a faster clinical response to antidepressant treatment (see Manenschijn et al.28d and Spijker and Van Rossum28e).30 In addition, the polymorphism in the 5′ region of the GR gene (NR3C1-1 in the promoter region) has been associated with increased vulnerability to depression (see Claes75a).35
Overall, a number of studies using different methodologies have reported that GR function is impaired in patients with depression. A lack of response of the GR to changes in ligand availability76 and reduced inhibition of immune function and cytokines production after exposure to dexamethasone, indicating reduced GR sensitivity, have been shown in patients with depression.77–80 Recently, impaired GR signaling has been proposed as a primary factor that could lead to alterations in HPA axis function, such as hypercortisolism, reduced negative feedback of cortisol, and increased production of CRH, all of which have been described in some patients with depression (see below).81,82 Interestingly, in patients with depression, GR sensitivity and HPA axis activity tend to return to normal after clinical recovery of depressive symptomatology (see Pariante21a).19,77,83,84
HPA Axis in Depression
Hyperactivity of the HPA axis has been described since the late 1950s in patients with depression.85 Since then, many studies, using different methodologies, have shown that a subgroup of patients with depression exhibit increased levels of cortisol in plasma, 24-h urine and cerebrospinal fluid.82,86–88 In addition, increased cortisol secretion in response to ACTH and enlargement of the adrenal and pituitary glands have also been reported in patients with depression.89 Some authors have suggested that hypercortisolemia reported in patients with depression may be related to over-secretion of CRH.90,91 This evidence is supported by several findings in patients with depression, including increased concentration of CRH in the CSF91; enlargement of the pituitary gland92; blunted ACTH responses to CRH challenge87; down-regulation of CRH-R1 receptors in frontal brain regions of postmortem suicide patients93; and increased CRH expression in hypothalamic neurons of postmortem patients.94
In addition, studies have revealed that the circadian rhythm of cortisol secretion is impaired in patients with depression, as evidenced by an elevated nadir and hence flattening of the circadian rhythm95 and irregular cortisol secretion.86 Elevated levels of cortisol in some patients with depression can also result from a reduced negative feedback response to cortisol that is reflected by a nonsuppression response in the dexamethasone suppression test (DST).96 Indeed, a large meta-analysis showed that the DST is a potent indicator of a poor prognosis and is also a predictor of suicide in depression.96 However, due to the fact that dexamethasone has distinct pharmacodynamic and pharmacokinetic profiles from cortisol, and due to the low sensitivity of the DST in detecting patients with depression, new tests, such as the DEX/CRH challenge test and the prednisolone test, have been developed to evaluate the negative feedback of cortisol.87,97
The DEX/CRH test combines the DST test (oral administration of single dose of dexamethasone at 11 PM) and the CRH stimulation test (intravenous bolus of CRH on the following day at 3 PM). Studies have shown that the DEX/CRH test has a higher specificity than the DST in detecting HPA axis dysfunction in patients with depression compared with the DST alone.98 Patients with depression tend to exhibit a lack of inhibition of ACTH responses to CRH following dexamethasone pretreatment, which represents an impaired feedback inhibition at the level of the pituitary.82,87 Although it is an invasive and expensive methodology, studies have shown that this test could predict relapses of depression and poor outcome in patients under treatment.99,100
Recently, a new and noninvasive test using prednisolone, a synthetic glucocorticoid, has been developed by Pariante and colleagues.97 Some of the advantages of using this test are due to the similarity of prednisolone’s pharmacodynamics and pharmacokinetics to cortisol. Thus, prednisolone and cortisol have a similar affinity to both MR and GR (twofold higher; dexamethasone only binds to GR)101 and to CBG.102 In addition, both have comparable half-lives103 and a similar ability to access the brain. Studies using this test have shown that patients with depression have a normal suppression response to the prednisolone test concomitant with a reduced suppression response to the dexamethasone test. These findings reflect preserved MR activity but reduced GR sensitivity,104 suggesting a possible dissociation between GR and MR function in a subgroup of patients with depression.
Although evaluation of HPA axis responsiveness has been performed in patients with depression, conflicting results are still described. Moreover, only a percentage of patients with depression exhibit HPA axis hyperactivity. Factors that may contribute to these apparently controversial results include the presence of comorbidities with anxiety disorders,105 history of early life stress,106 subtypes of depression and severity of symptoms,107 and immune activation with excess of innate proinflammatory cytokine production.21,40 Finally, it is very likely that different subtypes of depression may be associated with distinct patterns of HPA axis dysregulation.
Indeed, studies have shown that HPA axis hyperactivity is associated with certain subtypes of depression. Thus, hypercortisolemia (particularly a higher nadir) is more common in patients with severe depression, both of melancholic and psychotic types.108,109 Impairment of glucocorticoid negative feedback has been described to be more intense in depressed patients with higher rates of anxiety disorder comorbidities.105 In addition, a recent population-based study showed that older depressed patients exhibit two distinct patterns of HPA axis activity. Those with HPA axis hypo-activity tended to be women with joint disease and higher rates of tobacco use, whereas HPA axis hyperactivity was more prevalent in males with cardiovascular diseases and higher use of nonsteroidal anti-inflammatory drugs.110 Of note, hypo-activity of the HPA axis has also been demonstrated in other psychiatric disorders, such as PTSD (see Yehuda110a).111
Alterations in the HPA axis could also be influenced by sex hormones. Studies have shown that female rats secrete higher levels of ACTH and glucocorticoids in response to a stressor than males.112 This gender difference could be related to the effect of estrogen in regulating CRH secretion by action through the estrogen receptor.113 The role of sex hormones in modulating the HPA axis and mood disorders is discussed in detail in Schmidt and Rubinow.113a,114,115
Finally, alterations of the HPA axis in psychiatric disorders could also be influenced by activation of the immune system through pro-inflammatory cytokines. As already discussed and discussed further below, cytokines can stimulate glucocorticoid release and also alter glucocorticoid availability and GR function, which, under certain conditions, may favor a state of glucocorticoid resistance (see Pariente21a and Pace and Miller115a).36,50
Cytokines and Depression
HPA Axis–Immune Interactions
There is a bidirectional communication between the immune system and the HPA axis,36–38 in which cytokines stimulate the HPA axis and the resulting release of glucocorticoids provides negative feedback control of the immune response, keeping inflammation in check. It is well established that glucocorticoids exert an important modulatory role on the immune system, both suppressing and enhancing a variety of immune functions.116,117 Glucocorticoids influence the balance between pro-inflammatory and anti-inflammatory cytokine secretion, as it inhibits the production of innate pro-inflammatory and Th1 cytokines (mainly inflammatory) and enhances the production of Th2 cytokines (mainly anti-inflammatory).118 Therefore, the degree of cortisol responsiveness has an important influence on susceptibility and resistance to inflammatory, autoimmune and infectious diseases. For instance, an impaired glucocorticoid response that results from inappropriately low concentrations of cortisol or impaired GR function can predispose the subject to autoimmune and inflammatory diseases, such as systemic lupus erythematosus and RA.37 Conversely, chronic activation of the HPA axis with prolonged elevation of cortisol, as occurs in chronic stress, or GR hypersensitivity, can lead to an inhibition of pro-inflammatory/Th1 immune responses and, therefore, predispose the subject to greater susceptibility to infection. It is possible that bidirectional communication between altered HPA axis function and immune function might also contribute to symptomatology in psychiatric disorders. Other conditions in which altered HPA axis and immune function co-exist include metabolic syndrome, atherosclerosis, insulin resistance, type II diabetes, and osteoporosis, all of which exhibit a high incidence of co-morbidity with depression.119,120
Cytokines have been shown to stimulate the HPA axis by acting at all three levels: the brain, pituitary, and adrenal gland.36,121 Since cytokines are large, soluble peptides, significant consideration has been given to the mechanism by which cytokines cross the blood–brain barrier. Peripheral cytokines can signal the brain by several different pathways, including: (a) stimulating afferent (vagal nerve) fibers that project to the nucleus tractus solitarius in the brain stem, thus activating the release of norepinephrine in the PVN; (b) passively crossing the blood–brain barrier at “leaky” regions where it is not intact, such as the circumventricular organs and activating neurons that project to the hypothalamus; (c) acting on endothelial cells of the brain vasculature and/or glial cells in the circumventricular organs, inducing the synthesis/release of secondary messengers, such as prostaglandins, nitric oxide, and central cytokines and (d) crossing the blood–brain barrier via active transport. Evidence exists to support all of these pathways, and they are not mutually exclusive.36,39
Although it is not known whether increased activity of the HPA axis is a primary cause or is secondary to depressive symptomatology, if it is causal, such activation by pro-inflammatory cytokines could conceivably contribute to depression. Studies have also shown that pro-inflammatory cytokines can induce glucocorticoid resistance by reducing glucocorticoid function.69 Interestingly, reduction of GR function and affinity induced by cytokines has been found in patients with inflammatory diseases, especially those exhibiting resistance to glucocorticoid therapy.69 Moreover, reduced GR function could contribute to enhanced pro-inflammatory cytokine production, which has been associated with the development of depressive symptomatology (see below). Such phenomena might explain the high incidence of depression in patients with inflammatory diseases.
Cytokines, Behavior, and Depression
Studies have suggested that cytokines may play a role in the pathophysiology and/or symptomatology of depression. Cytokines have been shown to induce a constellation of symptoms referred to as “sickness behavior,” which has many overlapping features with depression (lethargy, somnolence, fatigue, anhedonia, decreased appetite and locomotion, and cognitive deficits).39 Indeed, the use of interferon-α (IFN-α) therapy for hepatitis C and cancer has been associated with the development of depressive symptomatology in up to 50% of patients.122–125 Conversely, cytokine antagonists or knockout mice have been found to block these behavioral changes in rodents and reduce depression and fatigue in patients with autoimmune or inflammatory disorders.40,125,126 Furthermore, in a group of patients with depression, increased levels of pro-inflammatory cytokines have been described in different bodily fluids, including plasma,127 cerebrospinal fluid,128 and sweat,129 although conflicting results have been described (see Pace and Miller115a).130–133
Although cytokines may play a role in the pathophysiology of depression, only a percentage of patients who receive IFN-α therapy will develop depression. In addition, only a percentage of patients with depression exhibit increased levels of pro-inflammatory cytokines. The presence of inflammation and elevated cytokines could be a risk factor contributing to the development of depressive symptomatology in patients treated with high doses of glucocorticoids. Furthermore, other factors, such as glucocorticoid resistance or hypersensitivity, at the level of the GR could also contribute to depressive symptomatology. Thus, in order to reduce or prevent development of depression induced by IFN-α and glucocorticoid administration, a better understanding of individual risk factors should be acquired.
Biomarkers in Depression
Conflicting results from these studies indicate a need for defining patterns of a variety of biomarkers in patients with depression, in order to identify different subpopulations with different pathophysiologies. Such characterization will help to identify patients at risk, as well as to design more targeted therapeutic strategies and potentially to predict prognoses.
Recently, our group has validated a new and noninvasive methodology to simultaneously measure a large array of neuroimmune biomarkers in the sweat, using sweat patches coupled with recycling immunoaffinity chromatography (RIC).129,134 In addition, we have shown that sweat levels of neuroimmune biomarkers are strongly correlated with plasma levels in a group of healthy controls134 and in a group of patients with MDD mostly in remission.129 RIC is a single-antibody system that uses a set of high-performance liquid chromatographic immunoaffinity columns in series, and each column is immunospecific for a targeted, fluorescently labeled analyte. This highly sensitive method allows measurement of a large number of analytes in a very small volume of fluid.135 Applying this methodology, we have shown that women with MDD, even in clinical remission, exhibited higher levels of pro-inflammatory cytokines, and sympathetic (NPY) and sensory (substance P and CGRP) neuropeptides, but decreased parasympathetic neuropeptides (VIP) in plasma and in sweat compared with healthy controls. These findings suggest that women with MDD, even in clinical remission, exhibit alterations in neuroimune biomarkers that could predispose them to osteoporosis, diabetes, and cardiovascular disease.136 Furthermore, neuroimmune biomarker levels strongly correlated with symptoms of depression and anxiety, suggesting that symptom severity rather than disease classification per se may be related to these dysregulations in biomarker expression. In Table 1, we propose a framework for evaluations of important biomarkers of depression that should be addressed in future studies.
TABLE 1.
Molecular, Physiological, and Neuroimmune Biomarker Evaluation
| Evaluation | Test | References | |
|---|---|---|---|
| Molecular | Glucocorticoid availability | CBG MDR pump 11β-HSD | Deuschle et al., 2003; Pariante et al., 2003, 2008; Poor et al., 2004 |
| GR isoforms and polymorphisms | BclI, ER22/23EK, N363S NR3C1-1 | van Rossum, et al., 2006; van West et al., 2006 | |
| GR function | Sensitivity to DEX suppression of cytokine production ex vivo (whole blood/lymphocytes); sensitivity to DEX suppression of lymphocyte proliferation glucocorticoid binding assay | DeRijk et al., 2001; Miller et al., 2005; Lowy et al., 1984; Wodarz et al., 1991; Wassef et al., 1990 | |
| Physiological and Neuroimmune status | Functional endocrine tests | DST test; DEX/CRH test; Prednisolone test | Pariante et al., 2002 |
| Neural (autonomic and sensory) status | Heart rate variability neuropeptide levels in blood, sweat, and CSF | Thayer and Sternberg, 2006; Cizza et al., 2008 | |
| Immune system status | Cytokine levels in blood, sweat and CSF | Marques-Deak et al., 2006; Cizza et al., 2008 |
Corticosteroid binding globulin (CBG); multidrug resistance transporter (MDR pump); 11β-hydroxysteroid de-hydrogenase (11β-HSD); dexamethasone suppression test (DST); dexamethasone/corticotropin-releasing hormone (DEX/CRH); cerebrospinal fluid (CSF).
Conclusion
In summary, individual variability in susceptibility to the serious mood and depressive side effects of glucocorticoid therapy could stem from individual variations at all levels of glucocorticoid regulation. These include CRH signaling, glucocorticoid hormone levels, factors regulating local glucocorticoid availability, and ultimately, the GR, either through GR polymorphisms conferring hypo- or hypersensitivity of the GR or with variations in other related molecules necessary for GR function. In addition, the presence of immune activation with excess pro-inflammatory cytokines could also contribute to HPA dysfunction and behavioral changes that are cardinal features of depression. A systematic screening of these potential host factors in patients administered high-dose glucocorticoids might minimize the detrimental side effects induced by treatment with these agents. Subsequent chapters in this volume will address each of these potential host risk factors in greater detail.
Future Directions
Further studies should be performed to clarify the relationship between depression and glucocorticoids and to identify individual susceptibility factors that increase the risk for development of mood disorders. Simultaneous evaluation of the endocrine (HPA axis), nervous and immune systems, and their interactions, through the use of genetic techniques, functional endocrine tests and measurements of neuroimmune biomarkers, could help to elucidate the underpinning role of these systems in mood disorders.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.The Boston Collaborative Drug Surveillance Program. Acute adverse reactions to prednisone in relation to dosage. Clin Pharmacol Ther. 1972;13:694–698. doi: 10.1002/cpt1972135part1694. [DOI] [PubMed] [Google Scholar]
- 2.Wolkowitz OM. Prospective controlled studies of the behavioral and biological effects of exogenous corticosteroids. Psychoneuroendocrinology. 1994;19:233–255. doi: 10.1016/0306-4530(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 2a.Wolkowitz O, et al. Mood, memory, and mechanisms. Ann N Y Acad Sci. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [Google Scholar]
- 2b.Brown ES. Effects of glucocorticoids on mood, memory, and the hippocampus: treatment and preventive therapy. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.04981.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PubMed] [Google Scholar]
- 3.Wolkowitz OM, et al. The “steroid dementia syndrome”: an unrecognized complication of glucocorticoid treatment. Ann N Y Acad Sci. 2004;1032:191–194. doi: 10.1196/annals.1314.018. [DOI] [PubMed] [Google Scholar]
- 4.Wolkowitz OM, Lupien SJ, Bigler ED. The “steroid dementia syndrome”: a possible model of human glucocorticoid neurotoxicity. Neurocase. 2007;13:189–200. doi: 10.1080/13554790701475468. [DOI] [PubMed] [Google Scholar]
- 5.Brown ES, Suppes T. Mood symptoms during corticosteroid therapy: a review. Harv Rev Psychiatry. 1998;5:239–246. doi: 10.3109/10673229809000307. [DOI] [PubMed] [Google Scholar]
- 6.Lewis DA, Smith RE. Steroid-induced psychiatric syndromes. A report of 14 cases and a review of the literature. J Affect Disord. 1983;5:319–332. doi: 10.1016/0165-0327(83)90022-8. [DOI] [PubMed] [Google Scholar]
- 7.Brown ES, et al. Effect of phenytoin on mood and declarative memory during prescription corticosteroid therapy. Biol Psychiatry. 2005;57:543–548. doi: 10.1016/j.biopsych.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 8.Brown ES, et al. Attenuation of the effects of corticosteroids on declarative memory with lamot-rigine. Neuropsychopharmacology. 2008;33:2376–2385. doi: 10.1038/sj.npp.1301627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fava GA. Affective disorders and endocrine disease. New insights from psychosomatic studies. Psychosomatics. 1994;35:341–353. doi: 10.1016/S0033-3182(94)71755-2. [DOI] [PubMed] [Google Scholar]
- 10.Frank R, Doerr HG. Mania in a girl with Cushing’s disease. J Am Acad Child Adolesc Psychiatry. 1989;28:610–611. doi: 10.1097/00004583-198907000-00023. [DOI] [PubMed] [Google Scholar]
- 11.Ghadirian AM, Marcovitz S, Pearson Murphy BE. A case of seasonal bipolar disorder exacerbated by Cushing’s disease. Compr Psychiatry. 2005;46:155–158. doi: 10.1016/j.comppsych.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 12.Haskett RF. Diagnostic categorization of psychiatric disturbance in Cushing’s syndrome. Am J Psychiatry. 1985;142:911–916. doi: 10.1176/ajp.142.8.911. [DOI] [PubMed] [Google Scholar]
- 13.Borzyskowski M, Grant DB, Wells RS. Cushing’s syndrome induced by topical steroids used for the treatment of non-bullous ichthyosiform erythroderma. Clin Exp Dermatol. 1976;1:337–342. doi: 10.1111/j.1365-2230.1976.tb01440.x. [DOI] [PubMed] [Google Scholar]
- 14.Guven A, Gulumser O, Ozgen T. Cushing’s syndrome and adrenocortical insufficiency caused by topical steroids: misuse or abuse? J Pediatr Endocrinol Metab. 2007;20:1173–1182. doi: 10.1515/jpem.2007.20.11.1173. [DOI] [PubMed] [Google Scholar]
- 15.Ruiz-Maldonado R, et al. Cushing’s syndrome after topical application of corticosteroids. Am J Dis Child. 1982;136:274–275. doi: 10.1001/archpedi.1982.03970390088024. [DOI] [PubMed] [Google Scholar]
- 16.Juruena MF, Cleare AJ, Pariante CM. The hypothalamic pituitary adrenal axis, glucocorticoid receptor function and relevance to depression. Rev Bras Psiquiatr. 2004;26:189–201. doi: 10.1590/s1516-44462004000300009. [DOI] [PubMed] [Google Scholar]
- 17.Chrousos GP, Kino T. Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress. 2007;10:213–219. doi: 10.1080/10253890701292119. [DOI] [PubMed] [Google Scholar]
- 18.Holsboer F, Ising M. Central CRH system in depression and anxiety—evidence from clinical studies with CRH1 receptor antagonists. Eur J Pharmacol. 2008;583:350–357. doi: 10.1016/j.ejphar.2007.12.032. [DOI] [PubMed] [Google Scholar]
- 19.Calfa G, et al. Characterization and functional significance of glucocorticoid receptors in patients with major depression: modulation by antidepressant treatment. Psychoneuroendocrinology. 2003;28:687–701. doi: 10.1016/s0306-4530(02)00051-3. [DOI] [PubMed] [Google Scholar]
- 20.Linkowski P, et al. 24-hour profiles of adrenocorticotropin, cortisol, and growth hormone in major depressive illness: effect of antidepressant treatment. J Clin Endocrinol Metab. 1987;65:141–152. doi: 10.1210/jcem-65-1-141. [DOI] [PubMed] [Google Scholar]
- 21.Pariante CM. Glucocorticoid receptor function in vitro in patients with major depression. Stress. 2004;7:209–219. doi: 10.1080/10253890500069650. [DOI] [PubMed] [Google Scholar]
- 21a.Pariante CM. Risk factors for development of depression and psychosis: glucocorticoid receptors and pituitary implications for treatment with antidepressant and glucocorticoids. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.04978.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeBattista C, et al. Acute antidepressant effects of intravenous hydrocortisone and CRH in depressed patients: a double-blind, placebo-controlled study. Am J Psychiatry. 2000;157:1334–1337. doi: 10.1176/appi.ajp.157.8.1334. [DOI] [PubMed] [Google Scholar]
- 23.Wolkowitz OM, Reus VI. Treatment of depression with antiglucocorticoid drugs. Psychosom Med. 1999;61:698–711. doi: 10.1097/00006842-199909000-00011. [DOI] [PubMed] [Google Scholar]
- 24.Bouwer C, et al. Prednisone augmentation in treatment-resistant depression with fatigue and hypocortisolaemia: a case series. Depress Anxiety. 2000;12:44–50. doi: 10.1002/1520-6394(2000)12:1<44::AID-DA6>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 25.Dinan TG, et al. Dexamethasone augmentation in treatment-resistant depression. Acta Psychiatr Scand. 1997;95:58–61. doi: 10.1111/j.1600-0447.1997.tb00374.x. [DOI] [PubMed] [Google Scholar]
- 26.Reus VI, Wolkowitz OM. Antiglucocorticoid drugs in the treatment of depression. Expert Opin Investig Drugs. 2001;10:1789–1796. doi: 10.1517/13543784.10.10.1789. [DOI] [PubMed] [Google Scholar]
- 27.Schatzberg AF, Lindley S. Glucocorticoid antagonists in neuropsychotic disorders. Eur J Pharmacol. 2008;583:358–364. doi: 10.1016/j.ejphar.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 28.Wolkowitz OM, et al. Dexamethasone for depression. Am J Psychiatry. 1996;153:1112. doi: 10.1176/ajp.153.8.aj15381112. author reply 1112–1113. [DOI] [PubMed] [Google Scholar]
- 28a.Chrousos GP, Kino T. Glucocorticoid signaling in the cell: expanding clinical implications to complex human behavioral and somatic disorders. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.04988.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b.Hauger RL, et al. Role of CRF receptor signaling in stress vulnerability, anxiety, and depression. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.05011.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28c.Refojo D, Holsboer F. CRH signaling: molecular specificity for drug targeting in the central nervous system. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.04983.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PubMed] [Google Scholar]
- 28d.Manenschijn L, et al. Clinical features associated with glucocorticoid receptor polymorphisms: an overview. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.05013.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PubMed] [Google Scholar]
- 28e.Spijker AT, Van Rossum EFC. Glucocorticoid receptor polymorphisms in major depression: focus on glucocorticoid sensitivity and neurocognitive functioning. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.04985.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PubMed] [Google Scholar]
- 29.Van Den Akker EL, et al. Glucocorticoid receptor gene and risk of cardiovascular disease. Arch Intern Med. 2008;168:33–39. doi: 10.1001/archinternmed.2007.41. [DOI] [PubMed] [Google Scholar]
- 30.van Rossum EF, et al. Polymorphisms of the glucocorticoid receptor gene and major depression. Biol Psychiatry. 2006;59:681–688. doi: 10.1016/j.biopsych.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 31.van Rossum EF, et al. Glucocorticoid receptor variant and risk of dementia and white matter lesions. Neurobiol Aging. 2008;29:716–723. doi: 10.1016/j.neurobiolaging.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 32.van Rossum EF, et al. Identification of the BclI polymorphism in the glucocorticoid receptor gene: association with sensitivity to glucocorticoids in vivo and body mass index. Clin Endocrinol (Oxf) 2003;59:585–592. doi: 10.1046/j.1365-2265.2003.01888.x. [DOI] [PubMed] [Google Scholar]
- 33.van Rossum EF, Lamberts SW. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog Horm Res. 2004;59:333–357. doi: 10.1210/rp.59.1.333. [DOI] [PubMed] [Google Scholar]
- 34.van Rossum EF, Russcher H, Lamberts SW. Genetic polymorphisms and multifactorial diseases: facts and fallacies revealed by the glucocorticoid receptor gene. Trends Endocrinol Metab. 2005;16:445–450. doi: 10.1016/j.tem.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 35.van West D, et al. Glucocorticoid receptor gene-based SNP analysis in patients with recurrent major depression. Neuropsychopharmacology. 2006;31:620–627. doi: 10.1038/sj.npp.1300898. [DOI] [PubMed] [Google Scholar]
- 36.Silverman MN, et al. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) axis during viral infection. Viral Immunol. 2005;18:41–78. doi: 10.1089/vim.2005.18.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6:318–328. doi: 10.1038/nri1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marques-Deak A, Cizza G, Sternberg E. Brain-immune interactions and disease susceptibility. Mol Psychiatry. 2005;10:239–250. doi: 10.1038/sj.mp.4001643. [DOI] [PubMed] [Google Scholar]
- 39.Dantzer R, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 42.Jacobson L. Hypothalamic-pituitary-adrenocortical axis regulation. Endocrinol Metab Clin North Am. 2005;34:271–292. vii. doi: 10.1016/j.ecl.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 1984;5:1–24. doi: 10.1210/edrv-5-1-1. [DOI] [PubMed] [Google Scholar]
- 44.Herman JP, et al. Evidence for hippocampal regulation of neuroendocrine neurons of the hypothalamopituitary-adrenocortical axis. J Neurosci. 1989;9:3072–3082. doi: 10.1523/JNEUROSCI.09-09-03072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosner W. Plasma steroid-binding proteins. Endocrinol Metab Clin North Am. 1991;20:697–720. [PubMed] [Google Scholar]
- 46.Tinnikov AA, et al. Serum corticosteroid-binding globulin levels in children undergoing heart surgery. Steroids. 1993;58:536–539. doi: 10.1016/0039-128x(93)90031-h. [DOI] [PubMed] [Google Scholar]
- 47.Deuschle M, et al. Increased concentration of corticosteroid-binding globulin due to antidepressant treatment with amitritpyline, but not paroxetine. J Psychiatr Res. 2003;37:85–87. doi: 10.1016/s0022-3956(02)00066-3. [DOI] [PubMed] [Google Scholar]
- 48.Chin KV, et al. Regulation of mdr RNA levels in response to cytotoxic drugs in rodent cells. Cell Growth Differ. 1990;1:361–365. [PubMed] [Google Scholar]
- 49.Karssen AM, et al. Multidrug resistance P-glycoprotein hampers the access of cortisol but not of corticosterone to mouse and human brain. Endocrinology. 2001;142:2686–2694. doi: 10.1210/endo.142.6.8213. [DOI] [PubMed] [Google Scholar]
- 50.Silverman MN, Sternberg EM. Neuroendocrine-immune interactions in rheumatoid arthritis: mechanisms of glucocorticoid resistance. Neuroimmunology. 2008;15:19–28. doi: 10.1159/000135620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirano T, et al. MDR1 mRNA expressions in peripheral blood mononuclear cells of patients with ulcerative colitis in relation to glucocorticoid administration. J Clin Pharmacol. 2004;44:481–486. doi: 10.1177/0091270004264162. [DOI] [PubMed] [Google Scholar]
- 52.Maillefert JF, et al. Expression of the multidrug resistance glycoprotein 170 in the peripheral blood lymphocytes of rheumatoid arthritis patients. The percentage of lymphocytes expressing glycoprotein 170 is increased in patients treated with prednisolone. Br J Rheumatol. 1996;35:430–435. doi: 10.1093/rheumatology/35.5.430. [DOI] [PubMed] [Google Scholar]
- 53.Pariante CM. The role of multi-drug resistance p-glycoprotein in glucocorticoid function: studies in animals and relevance in humans. Eur J Pharmacol. 2008;583:263–271. doi: 10.1016/j.ejphar.2007.11.067. [DOI] [PubMed] [Google Scholar]
- 54.Pariante CM, et al. Antidepressant fluoxetine enhances glucocorticoid receptor function in vitro by modulating membrane steroid transporters. Br J Pharmacol. 2003;139:1111–1118. doi: 10.1038/sj.bjp.0705357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laika B, Leucht S, Steimer W. ABCB1 (P-glycoprotein/MDR1) gene G2677T/a sequence variation (polymorphism): lack of association with side effects and therapeutic response in depressed inpatients treated with amitriptyline. Clin Chem. 2006;52:893–895. doi: 10.1373/clinchem.2006.066605. [DOI] [PubMed] [Google Scholar]
- 56.Mihaljevic Peles A, et al. MDR1 gene polymorphism: therapeutic response to paroxetine among patients with major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1439–1444. doi: 10.1016/j.pnpbp.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 57.Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1-a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. doi: 10.1210/endo.142.4.8114. [DOI] [PubMed] [Google Scholar]
- 58.Chapman KE, Seckl JR. 11beta-HSD1, inflammation, metabolic disease and age-related cognitive (dys)function. Neurochem Res. 2008;33:624–636. doi: 10.1007/s11064-007-9504-9. [DOI] [PubMed] [Google Scholar]
- 59.McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 60.Poor V, et al. Urinary steroid metabolites and 11beta-hydroxysteroid dehydrogenase activity in patients with unipolar recurrent major depression. J Affect Disord. 2004;81:55–59. doi: 10.1016/S0165-0327(03)00199-X. [DOI] [PubMed] [Google Scholar]
- 61.Weber B, et al. Increased diurnal plasma concentrations of cortisone in depressed patients. J Clin Endocrinol Metab. 2000;85:1133–1136. doi: 10.1210/jcem.85.3.6469. [DOI] [PubMed] [Google Scholar]
- 62.Duma D, Jewell CM, Cidlowski JA. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol. 2006;102:11–21. doi: 10.1016/j.jsbmb.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 63.Joels M, et al. The coming out of the brain mineralocorticoid receptor. Trends Neurosci. 2008;31:1–7. doi: 10.1016/j.tins.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 63a.Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.04986.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PubMed] [Google Scholar]
- 64.Kassel O, Herrlich P. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol. 2007;275:13–29. doi: 10.1016/j.mce.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Liberman AC, et al. Glucocorticoids in the regulation of transcription factors that control cytokine synthesis. Cytokine Growth Factor Rev. 2007;18:45–56. doi: 10.1016/j.cytogfr.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 66.Liberman AC, et al. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein-protein interaction. FASEB J. 2007;21:1177–1188. doi: 10.1096/fj.06-7452com. [DOI] [PubMed] [Google Scholar]
- 67.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 68.Tait AS, Butts CL, Sternberg EM. The role of glucocorticoids and progestins in inflammatory, autoimmune, and infectious disease. J Leukoc Biol. 2008;84:924–931. doi: 10.1189/jlb.0208104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pace TW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun. 2007;21:9–19. doi: 10.1016/j.bbi.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller GE, et al. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry. 2008;64:266–272. doi: 10.1016/j.biopsych.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pace TW, et al. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry. 2006;163:1630–1633. doi: 10.1176/ajp.2006.163.9.1630. [DOI] [PubMed] [Google Scholar]
- 72.Colli LM, et al. Interindividual glucocorticoid sensitivity in young healthy subjects: the role of glucocorticoid receptor alpha and beta isoforms ratio. Horm Metab Res. 2007;39:425–429. doi: 10.1055/s-2007-980191. [DOI] [PubMed] [Google Scholar]
- 73.Tliba O, Cidlowski JA, Amrani Y. CD38 expression is insensitive to steroid action in cells treated with tumor necrosis factor-alpha and interferon-gamma by a mechanism involving the upregulation of the glucocorticoid receptor beta isoform. Mol Pharmacol. 2006;69:588–596. doi: 10.1124/mol.105.019679. [DOI] [PubMed] [Google Scholar]
- 74.Derijk RH, et al. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001;28:2383–2388. [PubMed] [Google Scholar]
- 75.Lee YM, et al. A mutation of the glucocorticoid receptor gene in patients with systemic lupus erythematosus. Tohoku J Exp Med. 2004;203:69–76. doi: 10.1620/tjem.203.69. [DOI] [PubMed] [Google Scholar]
- 75a.Claes S. Glucocorticoid receptor polymorphisms in major depression. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.05012.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PubMed] [Google Scholar]
- 76.Wassef A, et al. Mononuclear leukocyte glucocorticoid receptor binding characteristics and down-regulation in major depression. Psychoneuroendocrinology. 1990;15:59–68. doi: 10.1016/0306-4530(90)90047-d. [DOI] [PubMed] [Google Scholar]
- 77.Bauer ME, et al. Altered glucocorticoid immunoregulation in treatment resistant depression. Psychoneuroendocrinology. 2003;28:49–65. doi: 10.1016/s0306-4530(02)00009-4. [DOI] [PubMed] [Google Scholar]
- 78.Lowy MT, et al. Glucocorticoid resistance in depression: the dexamethasone suppression test and lymphocyte sensitivity to dexamethasone. Am J Psychiatry. 1984;141:1365–1370. doi: 10.1176/ajp.141.11.1365. [DOI] [PubMed] [Google Scholar]
- 79.Miller GE, et al. Clinical depression and regulation of the inflammatory response during acute stress. Psychosom Med. 2005;67:679–687. doi: 10.1097/01.psy.0000174172.82428.ce. [DOI] [PubMed] [Google Scholar]
- 80.Wodarz N, et al. Normal lymphocyte responsiveness to lectins but impaired sensitivity to in vitro glucocorticoids in major depression. J Affect Disord. 1991;22:241–248. doi: 10.1016/0165-0327(91)90070-9. [DOI] [PubMed] [Google Scholar]
- 81.Antonijevic IA. Depressive disorders—is it time to endorse different pathophysiologies? Psychoneuroendocrinology. 2006;31:1–15. doi: 10.1016/j.psyneuen.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 82.Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 83.Aihara M, et al. HPA axis dysfunction in unmedicated major depressive disorder and its normalization by pharmacotherapy correlates with alteration of neural activity in prefrontal cortex and limbic/paralimbic regions. Psychiatry Res. 2007;155:245–256. doi: 10.1016/j.pscychresns.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 84.Kaestner F, et al. Different activation patterns of proinflammatory cytokines in melancholic and non-melancholic major depression are associated with HPA axis activity. J Affect Disord. 2005;87:305–311. doi: 10.1016/j.jad.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 85.Michael RP, Gibbons JL. Interrelationships between the Endocrine System Neuropsychiatry. Int Rev Neurobiol. 1963;5:243–302. doi: 10.1016/s0074-7742(08)60597-8. [DOI] [PubMed] [Google Scholar]
- 86.Carroll BJ, et al. Pathophysiology of hypercortisolism in depression. Acta Psychiatr Scand Suppl. 2007;433:90–103. doi: 10.1111/j.1600-0447.2007.00967.x. [DOI] [PubMed] [Google Scholar]
- 87.Nemeroff CB. The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol Psychiatry. 1996;1:336–342. [PubMed] [Google Scholar]
- 88.Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- 89.Owens MJ, Nemeroff CB. The role of corticotropin-releasing factor in the pathophysiology of affective and anxiety disorders: laboratory and clinical studies. Ciba Found Symp. 1993;172:296–308. doi: 10.1002/9780470514368.ch15. discussion 308–316. [DOI] [PubMed] [Google Scholar]
- 90.Holsboer F. Corticotropin-releasing hormone modulators and depression. Curr Opin Investig Drugs. 2003;4:46–50. [PubMed] [Google Scholar]
- 91.Roy A, et al. CSF corticotropin-releasing hormone in depressed patients and normal control subjects. Am J Psychiatry. 1987;144:641–645. doi: 10.1176/ajp.144.5.641. [DOI] [PubMed] [Google Scholar]
- 92.Pariante CM, et al. Pituitary volume in psychosis. Br J Psychiatry. 2004;185:5–10. doi: 10.1192/bjp.185.1.5. [DOI] [PubMed] [Google Scholar]
- 93.Merali Z, et al. Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J Neurosci. 2004;24:1478–1485. doi: 10.1523/JNEUROSCI.4734-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raadsheer FC, et al. Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology. 1994;60:436–444. doi: 10.1159/000126778. [DOI] [PubMed] [Google Scholar]
- 95.Deuschle M, et al. Diurnal activity and pulsatility of the hypothalamus-pituitary-adrenal system in male depressed patients and healthy controls. J Clin Endocrinol Metab. 1997;82:234–238. doi: 10.1210/jcem.82.1.3689. [DOI] [PubMed] [Google Scholar]
- 96.Ribeiro SC, et al. The DST as a predictor of outcome in depression: a meta-analysis. Am J Psychiatry. 1993;150:1618–1629. doi: 10.1176/ajp.150.11.1618. [DOI] [PubMed] [Google Scholar]
- 97.Pariante CM, et al. A novel prednisolone suppression test for the hypothalamic-pituitary-adrenal axis. Biol Psychiatry. 2002;51:922–930. doi: 10.1016/s0006-3223(01)01314-2. [DOI] [PubMed] [Google Scholar]
- 98.Watson S, et al. The dex/CRH test—is it better than the DST? Psychoneuroendocrinology. 2006;31:889–894. doi: 10.1016/j.psyneuen.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 99.Hatzinger M, et al. The combined DEX-CRH test in treatment course and long-term outcome of major depression. J Psychiatr Res. 2002;36:287–297. doi: 10.1016/s0022-3956(02)00021-3. [DOI] [PubMed] [Google Scholar]
- 100.Zobel AW, et al. Cortisol response in the combined dexamethasone/CRH test as predictor of relapse in patients with remitted depression. A prospective study. J Psychiatr Res. 2001;35:83–94. doi: 10.1016/s0022-3956(01)00013-9. [DOI] [PubMed] [Google Scholar]
- 101.Lan NC, et al. Binding of steroids to mineralocorticoid receptors: implications for in vivo occupancy by glucocorticoids. J Clin Endocrinol Metab. 1982;54:332–342. doi: 10.1210/jcem-54-2-332. [DOI] [PubMed] [Google Scholar]
- 102.Pugeat MM, Dunn JF, Nisula BC. Transport of steroid hormones: interaction of 70 drugs with testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab. 1981;53:69–75. doi: 10.1210/jcem-53-1-69. [DOI] [PubMed] [Google Scholar]
- 103.Lac G, et al. Dexamethasone in resting and exercising men. II. Effects on adrenocortical hormones. J Appl Physiol. 1999;87:183–188. doi: 10.1152/jappl.1999.87.1.183. [DOI] [PubMed] [Google Scholar]
- 104.Juruena MF, et al. Different responses to dexamethasone and prednisolone in the same depressed patients. Psychopharmacology (Berl) 2006;189:225–235. doi: 10.1007/s00213-006-0555-4. [DOI] [PubMed] [Google Scholar]
- 105.Young EA, Breslau N. Saliva cortisol in posttraumatic stress disorder: a community epidemiologic study. Biol Psychiatry. 2004;56:205–209. doi: 10.1016/j.biopsych.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 106.Heim C, et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA. 2000;284:592–597. doi: 10.1001/jama.284.5.592. [DOI] [PubMed] [Google Scholar]
- 107.Mello AF, et al. Depression and stress: is there an endophenotype? Rev Bras Psiquiatr. 2007;29(Suppl 1):S13–S18. doi: 10.1590/s1516-44462007000500004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Belanoff JK, et al. An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol Psychiatry. 2002;52:386–392. doi: 10.1016/s0006-3223(02)01432-4. [DOI] [PubMed] [Google Scholar]
- 109.Schatzberg AF, et al. Neuropsychological deficits in psychotic versus nonpsychotic major depression and no mental illness. Am J Psychiatry. 2000;157:1095–1100. doi: 10.1176/appi.ajp.157.7.1095. [DOI] [PubMed] [Google Scholar]
- 110.Bremmer MA, et al. Major depression in late life is associated with both hypo- and hypercortisolemia. Biol Psychiatry. 2007;62:479–486. doi: 10.1016/j.biopsych.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 110a.Yehuda R. Status of glucocorticoid alterations in Post-traumatic stress disorder. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.04979.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PubMed] [Google Scholar]
- 111.Yehuda R. Clinical relevance of biologic findings in PTSD. Psychiatr Q. 2002;73:123–133. doi: 10.1023/a:1015055711424. [DOI] [PubMed] [Google Scholar]
- 112.Burgess LH, Handa RJ. Chronic estrogen-induced alterations in adrenocorticotropin and corticosterone secretion, and glucocorticoid receptor-mediated functions in female rats. Endocrinology. 1992;131:1261–1269. doi: 10.1210/endo.131.3.1324155. [DOI] [PubMed] [Google Scholar]
- 113.Miller WJ, et al. Estrogen receptor (ER)beta isoforms rather than ERalpha regulate corticotropin-releasing hormone promoter activity through an alternate pathway. J Neurosci. 2004;24:10628–10635. doi: 10.1523/JNEUROSCI.5540-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113a.Schmidt PJ, Rubinow DR. Sex hormones and mood in the perimenopause. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.04982.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Daly RC, et al. Concordant restoration of ovarian function and mood in peri-menopausal depression. Am J Psychiatry. 2003;160:1842–1846. doi: 10.1176/appi.ajp.160.10.1842. [DOI] [PubMed] [Google Scholar]
- 115.Roca CA, et al. Differential menstrual cycle regulation of hypothalamic-pituitary-adrenal axis in women with premenstrual syndrome and controls. J Clin Endocrinol Metab. 2003;88:3057–3063. doi: 10.1210/jc.2002-021570. [DOI] [PubMed] [Google Scholar]
- 115a.Pace TWW, Miller AH. Cytokines and glucocorticoid receptor signaling: relevance to major depression. Ann N Y Acad Sci. 2009 doi: 10.1111/j.1749-6632.2009.04984.x. Glucocorticoids and Mood: Clinical Manifestations, Risk Factors, and Molecular Mechanisms. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Adcock IM, Ito K. Molecular mechanisms of corticosteroid actions. Monaldi Arch Chest Dis. 2000;55:256–266. [PubMed] [Google Scholar]
- 117.Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond) 1998;94:557–572. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 118.Elenkov IJ. Glucocorticoids and the Th1/Th2 balance. Ann N Y Acad Sci. 2004;1024:138–146. doi: 10.1196/annals.1321.010. [DOI] [PubMed] [Google Scholar]
- 119.Black PH. The inflammatory response is an integral part of the stress response: implications for atherosclerosis, insulin resistance, type II diabetes and metabolic syndrome X. Brain Behav Immun. 2003;17:350–364. doi: 10.1016/s0889-1591(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 120.Kiecolt-Glaser JK, Glaser R. Depression and immune function: central pathways to morbidity and mortality. J Psychosom Res. 2002;53:873–876. doi: 10.1016/s0022-3999(02)00309-4. [DOI] [PubMed] [Google Scholar]
- 121.Silverman MN, et al. Characterization of an interleukin-6- and adrenocorticotropin-dependent, immune-to-adrenal pathway during viral infection. Endocrinology. 2004;145:3580–3589. doi: 10.1210/en.2003-1421. [DOI] [PubMed] [Google Scholar]
- 122.Capuron L, et al. Treatment of cytokine-induced depression. Brain Behav Immun. 2002;16:575–580. doi: 10.1016/s0889-1591(02)00007-7. [DOI] [PubMed] [Google Scholar]
- 123.Maddock C, et al. Psychopathological symptoms during interferon-alpha and ribavirin treatment: effects on virologic response. Mol Psychiatry. 2005;10:332–333. doi: 10.1038/sj.mp.4001634. [DOI] [PubMed] [Google Scholar]
- 124.Raison CL, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66:41–48. doi: 10.4088/jcp.v66n0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160:1554–1565. doi: 10.1176/appi.ajp.160.9.1554. [DOI] [PubMed] [Google Scholar]
- 126.Silverman MN, et al. Endogenous glucocorticoids protect against TNF-alpha-induced increases in anxiety-like behavior in virally infected mice. Mol Psychiatry. 2007;12:408–417. doi: 10.1038/sj.mp.4001921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maes M, et al. Increased plasma concentrations of interleukin-6, soluble interleukin-6, soluble interleukin-2 and transferrin receptor in major depression. J Affect Disord. 1995;34:301–309. doi: 10.1016/0165-0327(95)00028-l. [DOI] [PubMed] [Google Scholar]
- 128.Levine J, et al. Cerebrospinal cytokine levels in patients with acute depression. Neuropsychobiology. 1999;40:171–176. doi: 10.1159/000026615. [DOI] [PubMed] [Google Scholar]
- 129.Cizza G, et al. Elevated neuroimmune biomarkers in sweat patches and plasma of pre-menopausal women with major depressive disorder in remission: the P.O.W.E.R. study. Biol Psychiatry. 2008;64:907–911. doi: 10.1016/j.biopsych.2008.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carpenter LL, et al. Cerebrospinal fluid interleukin (IL)-6 in unipolar major depression. J Affect Disord. 2004;79:285–289. doi: 10.1016/S0165-0327(02)00460-3. [DOI] [PubMed] [Google Scholar]
- 131.Anisman H, et al. Endocrine and cytokine correlates of major depression and dysthymia with typical or atypical features. Mol Psychiatry. 1999;4:182–188. doi: 10.1038/sj.mp.4000436. [DOI] [PubMed] [Google Scholar]
- 132.Brambilla F, Monteleone P, Maj M. Interleukin-1beta and tumor necrosis factor-alpha in children with major depressive disorder or dysthymia. J Affect Disord. 2004;78:273–277. doi: 10.1016/S0165-0327(02)00315-4. [DOI] [PubMed] [Google Scholar]
- 133.Marques-Deak AH, et al. Cytokine profiles in women with different subtypes of major depressive disorder. J Psychiatr Res. 2007;41:152–159. doi: 10.1016/j.jpsychires.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 134.Marques-Deak A, et al. Measurement of cytokines in sweat patches and plasma in healthy women: validation in a controlled study. J Immunol Methods. 2006;315:99–109. doi: 10.1016/j.jim.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 135.Phillips TM. Multi-analyte analysis of biological fluids with a recycling immunoaffinity column array. J Biochem Biophys Methods. 2001;49:253–262. doi: 10.1016/s0165-022x(01)00202-0. [DOI] [PubMed] [Google Scholar]
- 136.Eskandari F, et al. Low bone mass in pre-menopausal women with depression. Arch Intern Med. 2007;167:2329–2336. doi: 10.1001/archinte.167.21.2329. [DOI] [PubMed] [Google Scholar]
- 137.Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol. 2002;20:125–163. doi: 10.1146/annurev.immunol.20.082401.104914. [DOI] [PubMed] [Google Scholar]
- 138.Silverman MN, Pearce BD, Miller AH. Cytokines and HPA axis regulation. In: Kronfol Z, editor. Cytokines and Mental Health. Kluwer Academic Publishers; Norwell, MA: 2003. pp. 85–122. [Google Scholar]
- 139.Thayer JF, Sternberg E. Beyond heart rate variability: vagal regulation of allostatic systems. Ann N Y Acad Sci. 2006;1088:361–372. doi: 10.1196/annals.1366.014. [DOI] [PubMed] [Google Scholar]