

Abstract
The electron-donating properties of the axial His ligand to heme iron in cytochromes c (cyts c) are found to be correlated with midpoint reduction potential (Em) in variants of Hydrogenobacter thermophilus cytochrome c552 (Ht cyt c552) in which mutations have been made in and near the Cys-X-X-Cys-His (CXXCH) heme-binding motif. To probe the strength of the His-Fe(III) interaction, we have measured 13C nuclear magnetic resonance (NMR) chemical shifts for 13CN− bound to heme iron trans to the axial His in Ht Fe(III) cyt c552 variants. We observe a linear relationship between these 13C chemical shifts and Em, indicating that the His-Fe(III) bond strength correlates with Em. To probe a conserved hydrogen bonding interaction between the axial His Hδ1 and the backbone carbonyl of a Pro residue, we measured the pKa of the axial His Hδ1 proton (pKa(2)), which we propose to relate to the His-Fe(III) interaction, reduction potential and local electrostatic effects. The observed linear relationship between the axial His 13Cβ chemical shift and Em is proposed to reflect histidinate (anionic) character of the ligand. A linear relationship also is seen between the average heme methyl 1H chemical shift and Em which may reflect variation in axial His electron-donating properties or in the ruffling distortion of the heme plane. In summary, chemical shifts of the axial His and exogenous CN− bound trans to His are shown to be sensitive probes of the His-Fe(III) interaction in variants of Ht cyt c552 and display trends that correlate with Em.
Introduction
Iron-protoporphyrin IX (heme) is a widely distributed cofactor, and proteins containing heme are critical to an array of functions, including cellular respiration, photosynthesis, cellular signaling, gene regulation and oxygen sensing.1–2 A number of factors are recognized to modulate heme function including the type and number of heme axial ligands,3–5 hydrogen-bonding interactions with heme substituents and ligands,4,6–9 dynamics in the heme pocket,10–11 and heme conformation.12–14 His is the most common axial ligand found in heme proteins,15–16 and His-ligated heme proteins perform a wide variety of functions, including O2 transport and storage (globins),10,17 substrate oxidation and hydrogen peroxide reduction (peroxidases),18 and electron transfer (cytochromes).19–20 An unresolved question is how such variation in function and reactivity can be achieved in proteins containing the same redox cofactor and His-ligation at the active site. For example, proteins with bis-His axial ligation often function in electron transfer and exhibit a large range (~0.8 V) of midpoint reduction potentials (Em) despite conservation of heme ligation.1,15,16,21–22
Electron-donating properties of iron-coordinating His residues play an important role in determining the reactivity of heme-containing proteins.3,5,23 The amount of histidinate (His−), or anionic, character of the proximal His ligand contributes to determining relative stability of different oxidation states, and therefore to determining potential and reactivity.6–7,24–25 It has been proposed that the strength of the hydrogen-bonding interaction with the proximal His ring NH is related to the His− character, and mediates the strength of the Fe-N bond.1,4,6–7,18,26 In peroxidases, for instance, the His ring NH has a strong hydrogen bond to an asparate or glutamate side chain. In globins and many cytochromes, a weaker hydrogen bond is formed to a backbone carbonyl oxygen. A strong hydrogen bond to the His ring NH has been proposed to contribute to the strength of the electron donation to the heme iron, stabilizing the higher oxidation state necessary for catalysis in peroxidases.1,6,18,26–27
An additional variable in modulation of heme protein reactivity is the type of heme. Heme b and heme c are the most common types of heme found in proteins, and are differentiated from one another only by the covalent attachment of heme c to the polypeptide.22 Covalent attachment of heme c is generally via a conserved Cys-X-X-Cys-His (CXXCH) motif, in which two cysteines form thioether linkages to the heme, X-X represents any two amino acids other than Cys, and His serves as the proximal axial ligand to the heme iron (Figure 1). Significant interest has been expressed in the functional importance of the covalent attachment, which comes at a high biochemical cost (since heme c is synthesized from heme b).28–33 It is particularly notable that heme c displays a greater range of reduction potentials in nature relative to heme b when comparing heme proteins with the same ligand set.21–22,33 We hypothesize that the CXXCH heme-binding motif plays a role in making subtle adjustments in second-sphere interactions with the axial His in c-type heme proteins that may have substantial functional implications.11, 22, 34
Figure 1.
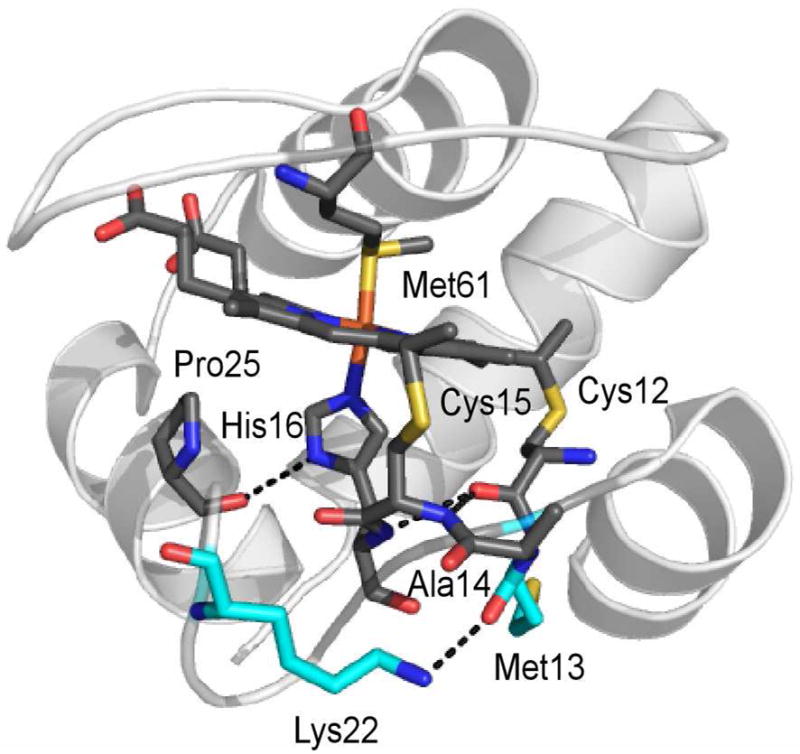
Heme, axial ligands, Lys22, Pro25, and CXXCH motif in Ht cyt c552. Hydrogen bonding interactions (heavy-atom distances of less than 3 Å) are shown with dashed lines. Shown in cyan are the two residues selected for mutation, Met13 and Lys22. The hydrogen bonding interaction between the axial His ligand and the backbone carbonyl of Pro is highly conserved in Class I cyts c.
NMR is a powerful technique for investigating the interactions between heme, the coordinating ligands, and the surrounding protein environment. In Fe(III), S = 1/2 heme proteins, the unpaired electron can cause chemical shifts of active-site nuclei to be shifted outside of the diamagnetic region. The resulting observed chemical shift, δobs can be expressed as:
| (1) |
where δdia is the shift in a diamagnetic reference molecule and δhf is the contribution to the observed shift by the interaction between the unpaired electron and nucleus.17,35–37 The δhf can be further broken down into a through-bond Fermi contact (FC) component (δcon), and a through-space pseudocontact (PC), or dipolar, component (δpc). The FC component of the observed chemical shift results from through-bond interactions transferring spin density to the nucleus being observed, and is highly sensitive to bonding interactions within the active site. Proton NMR has been used for the investigation of His-Fe(III) interactions, and His ring proton chemical shifts are proposed to be indicators of His− character of the axial His ligand.1,6–7,11,38–39 In work on 13CN−-Fe(III) ligated heme proteins, Fujii and coworkers have proposed that the chemical shift of 13C bound trans to His reports on the strength of the electron donation from His to Fe(III).40–42
In this work, we use NMR to investigate the His-Fe interaction in cytochromes c (cyts c), heme c-containing proteins. We measure and analyze hyperfine shifts in a series of variants of Hydrogenobacter thermophilus cyt c552 (Ht cyt c552) in which mutations have been made on the “proximal” side of the heme near and within the CXXCH heme-binding motif (residues 12–16). Analysis of the structure of Ht cyt c552 near the CXXCH motif and comparison to the structure of the homologous Pseudomonas aeruginosa (Pa) cyt c551 guided the selection of variants.11,43–44 The amide protons of Cys15 and His16 are positioned to form hydrogen bonds to the carbonyl oxygen of Cys12 (Figure 1). One of the ‘X’ residues, Met13, was targeted for mutation and replaced with Val, the corresponding residue in Pa cyt c551. The Lys residue at position 22 is positioned near Pro25, which hydrogen bonds with His16, and the CXXCH segment. We anticipated that replacement of the charged Lys residue with a hydrophobic Met (the corresponding residue in Pa cyt c551) would stabilize the CXXCH structure. Mutations of these residues in Ht cyt c552 were found in a previous study to significantly decrease the hydrogen exchange rates for Cys15 and His16 within the CXXCH motif, consistent with stabilizing the hydrogen bonding interactions involving these residues.11 Changes in Em by up to 81 mV have been observed for the series of variants (Table 1).11 We have hypothesized that hydrogen-bonding interactions in the heme pocket, both within the CXXCH motif and between the axial His (His16) Hδ1 and the Pro25 backbone carbonyl, modulate the His− character in these proteins which we assess via His side chain hyperfine shifts (Figure 1).11 To probe hydrogen bonding between His16 Hδ1 and Pro25 CO, we have determined pKa values of the axial His Hδ1 proton (pKa(2)). To further investigate how these mutations affect the His-Fe(III) interaction we have measured 13C chemical shifts of cyanide (13CN−) bound trans to the axial His.45 This study illustrates that chemical shifts of axial ligand nuclei are powerful probes of subtle changes in heme protein active sites.
Table 1.
Chemical shifts in part per million (ppm) and νCO (cm−1) frequency for Ht cyt c552 variants
| Ht cyt c552 variant | Ema | Proximal His16 | Distal Met61 | Heme methyl | Distal 13CNb | Distal COb | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fe(III) Hδ1a | Fe(II) Hδ1 | Fe(III) Nδ1a | Fe(II) Nδ1 | Fe(III) Cβ | δ 1 pKa(2) | Fe(III) εC1H3 | Fe(III) Avg 1H3 | Fe(III) 13C | Fe(II) νCO | ||
| Wild-type | 236 ± 2 | 13.2 | 8.9 | 182.3 | 168.4 | 26.2 | 10.11 ± 0.05 | −17.4 | 21.8 | −3948 | 1974.1 |
| M13V | 177 ± 1 | 11.8 | 8.9 | 170.5 | 167.6 | 25.2 | 10.26 ± 0.04 | −19.5 | 21.3 | −3895 | 1972.1 |
| K22M | 199 ± 1 | 12.6 | 8.9 | 181.3 | 168.7 | 25.6 | 10.51 ± 0.04 | −17.4 | 21.7 | −3902 | 1973.0 |
| M13V/K22M | 155 ± 2 | 11.2 | 8.9 | 167.1 | 167.7 | 24.3 | 10.62 ± 0.06 | −19.4 | 21.0 | −3863 | 1970.2 |
Reported in Michel, et al.11
These measurements made on the Met61 to Ala derivatives to allow exogenous ligand to bind
Experimental Section
Protein Expression and Purification
The plasmid pSCH55261A46 (Ampr) was used as a template for site-directed mutagenesis of Ht cyt c552. Mutants (Ht-M13V/M61A, Ht-K22M/M61A, and Ht-M13V/K22M/M61A) were prepared using the QuikChange II kit (Stratagene). All Ht cyt c552 variants were expressed and purified as previously described.11,46–47 Uniformly 15N-labeled samples were prepared by expression on minimal medium containing [15N, 99%]NH4Cl (Cambridge Isotope Laboratories) as the sole nitrogen source as previously described.11,48
NMR Spectroscopy to Detect Axial His Nuclei and Heme Methyl Protons
Detection of the His16 Cβ-Hβ cross peaks, heme methyl protons and Met61 Cε1H3 protons used oxidized Ht cyt c552, Ht-M13V, Ht-K22M, and Ht-M13V/K22M samples (1–3 mM protein, 50 mM NaPi, 10% D2O, 5-fold excess K3[Fe(CN)6]). Designations for axial ligand and heme methyl nuclei are shown in Figure 2. Spectra were collected on a Varian INOVA 500-MHz spectrometer (operating at 499.839 MHz for 1H) at 299 K. 1-D 1H spectra of samples at pH 7.0 were collected as previously described and assigned using standard methods, assisted by literature assignments.11,47 2-D 1H-13C (natural abundance) heteronuclear multiple quantum coherence (HMQC) spectra of samples at pH 7.0 were acquired with 2048 points over a spectral width of 40 kHz in the 1H dimension and 1024 increments over a spectral width of 50 kHz in the 13C dimension, with 64 scans and a recycle time of 0.55 s. 13C chemical shifts were indirectly referenced to 1H2O. 1H-15N heteronuclear single quantum coherence (HSQC) spectra of uniformly 15N-labeled Ht cyt c552, Ht-M13V, Ht-K22M, and Ht-M13V/K22M (1–3 mM protein, 50 mM NaPi, 10% D2O, 5-fold excess K3[Fe(CN)6]) were acquired with 1024 points over a spectral width of 12 kHz in the 1H dimension and 128 points over a spectral width of 8 kHz in the 15N dimension, with 8 scans and a recycle time of 1.5 s. The Hδ1-Nδ1 cross peak of the axial His was enhanced by using a relatively high JNH value (135 Hz), which selectively increases the intensity of cross peaks for nuclei that have short relaxation times.11 A series of HSQC spectra were collected for each variant over a pH range of 5.3 to 12.2 for pKa(2) determination. For Ht cyt c552 wild-type and Ht-M13V/K22M, the titration was started at acidic pH (~5.3). Because there was little change observed in the chemical shift between pH 5 and pH ~9, for the single mutants the titration was started at pH 6.55 (Ht-K22M) and pH 6.97 (Ht-M13V). To adjust the pH of these samples, μL amounts of 1 M NaOH were added. At pH values > 10.9 for wild-type and > 11.3 for Ht-M13V, the number of transients was increased from 8 to 32 to enhance detection of the axial His Hδ1-Nδ1 cross peak. 1-D 1H and 2-D 1H-15N HSQC spectra of all samples were examined to verify structural integrity of the samples as pH was increased. For the ferrous proteins (pH 7.0), samples were reduced with 5-fold excess sodium dithionite (Sigma) and HSQC spectra were obtained for each variant, with parameters as reported above but with a JNH of 94 Hz. 2-D spectra were processed with NMRPipe49 and assigned using SPARKY.50
Figure 2.
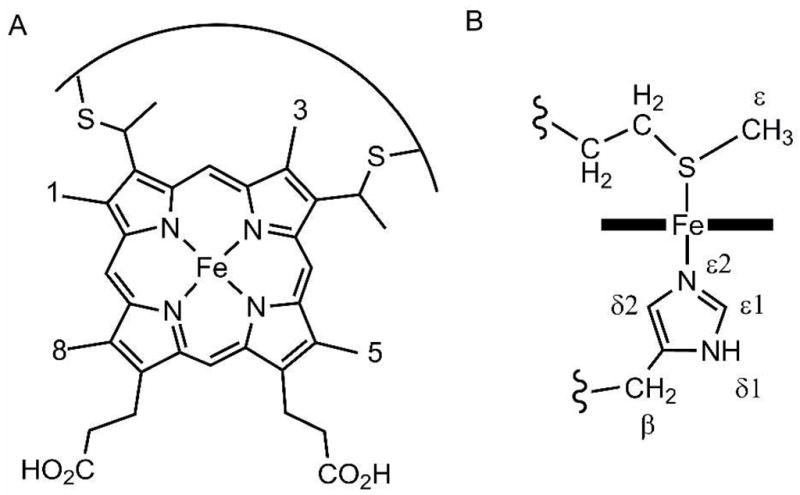
A) Heme c, with heme methyls 1, 3, 5, and 8 labeled. B) His and Met nomenclature conventions used herein.
NMR Spectroscopy of Fe(III)-CN− Derivatives
Oxidized Ht-M61A-13CN, Ht-M13V/M61A-13CN, Ht-K22M/M61A-13CN, and Ht-M13V/K22M/M61A-13CN samples (2–3 mM protein, 50 mM NaPi, 10% or 99.9% D2O) were prepared by adding 10-fold excess Na13CN (Cambridge Isotope Laboratories) and adjusting the pH* (uncorrected) to 9.0 with μL amounts of 1 M NaOH. Cyanide binding was monitored by UV-vis spectroscopy and 1-D 1H NMR. 1-D 13C spectra were collected on a Varian INOVA 600-MHz spectrometer (operating at 599.999 MHz for 1H), using a broadband probe. 13C NMR spectra were obtained with a recycle time of 75 ms; to obtain an acceptable signal-to-noise ratio for the 13C signals 106 transients were required. 1-D spectra were processed using SpinWorks.
IR Spectroscopy of Fe(II)-CO Derivatives
IR spectra of reduced Ht-M61A-CO, Ht-M13V/M61A-CO, Ht-K22M/M61A-CO, and Ht-M13V/K22M/M61A-CO (1.5–2.0 mM protein, 50 mM NaPi) were collected on a Shimadzu FTIR spectrometer. Samples were deoxygenated and then reduced using a 2-fold excess of sodium dithionite. Carbon monoxide gas was flowed over the sample for 10 to 15 min. CO binding was monitored by UV-vis. A baseline spectrum was obtained with the same concentration of protein (not CO-ligated), after which the CO-ligated sample was transferred to a CaF2 IR cell using a gas-tight syringe. A total of 600 scans at 2 cm−1 resolution with a window of 1750–2050 cm−1 were taken for each variant.
Pseudocontact Shift Calculations
The PC shifts for Ht cyt c552 nuclei were approximated using the relationship δpc = (1/12πr3)*[(Δχax*3cos(θ) − 1) + (3/2)*(Δχrh*sin2(θ)cos(2φ)], where Δχax and Δχrh are axial and rhombic magnetic susceptibility anisotropies51 and r, θ, and φ are the polar coordinates for the nucleus’ position in the magnetic frame. Structural coordinates from the published structure of Ht cyt c552 were used.52 These calculations were assisted by software written by Linghao Zhong.51
Results and Analysis
Analysis of Ht Cyt c552 Heme and Axial His Fermi Contact Shifts
The FC shift is a probe of spin density at the nucleus resulting from through-bond interaction with the paramagnetic center.17,36 Given the thorough resonance assignments of Ht cyt c552 and previous characterization of its magnetic anisotropy,51 it is possible to use Equation 1 to estimate the FC contribution to the observed chemical shifts in ferric Ht cyt c552 which is of value because the FC shift provides sensitive information on metal-ligand bonding.17,35,36 Using the chemical shifts in the Fe(II) oxidation state as a diamagnetic reference47 and previously described magnetic axes to approximate the PC shift contribution,51 we have estimated the FC contribution to the chemical shifts observed for selected heme and axial His nuclei in Fe(III) Ht cyt c552 (Table 2). The iron center has positive spin density,14 and the delocalization of that spin density can be observed from the FC contribution to the observed chemical shift, where the sign of the spin density alternates at each σ bond beyond the porphyrin core or on the ligating amino acid as a consequence of spin polarization.36,53 As expected for nuclei two bonds from the porphyrin core, the heme methyl 1H chemical shifts have positive FC contribution (the average for the 4 methyls is 21.8 ppm). Consistent with being three bonds from the iron, the Met61 Cε1H3 protons exhibit a large negative FC shift (−38.5 ppm). The downfield shift observed for His16 Hδ2 is consistent with a dominating positive PC contribution (36.9 ppm) and a smaller negative FC contribution (−20.4 ppm). His16 Hε1 δobs is shifted upfield because while the overall FC contribution (−23.6 ppm) is similar to that of Hδ2, there is a smaller (though still positive) PC contribution (6.6 ppm). The spin density, and therefore FC shift, at Hδ2 and Hε1 are both expected to be negative, and of approximately the same magnitude, because of spin polarization through three σ bonds from the positive spin density at the Fe center. The PC shift depends on the location of the nucleus with respect to the axes of the magnetic anisotropy tensor and the magnetic anisotropy. The large difference in PC shift between Hδ2 and Hε1 arises from the rhombic anisotropy and the position of each nucleus relative to the magnetic axes which are determined largely by orientations of both the axial His and Met.54 The FC contribution to the His chemical shifts is attenuated more bonds away from the Fe center, which makes estimation of the FC and PC contributions to observed chemical shifts less reliable. Based on the patterns observed for the other His nuclei, however, the sign of the FC contribution to the His Nδ1 chemical shift should be negative and to the His Hδ1 shift should be positive. It is important to note that the PC shifts determined from magnetic axes for nuclei very close to the iron have large uncertainties because of their high sensitivity to position within the magnetic axes. Thus the PC and FC shifts stated above should be treated as indicative of magnitude and sign of shifts rather than as precise values.
Table 2.
Estimates of FC and PC contributions to selected chemical shifts in Ht cyt c552
| nucleus | Fe(III) | Fe(II) | δhf | δpc | δcon |
|---|---|---|---|---|---|
| His16 1Hδ2 | 17.2 | 0.8b | 16.4 | 36.9 | −20.4 |
| His16 1Hε1 | −16.4a | 0.6b | −17.0 | 6.6 | −23.6 |
| Met61 Cε1H3 | −17.4 | −2.9b | −14.5 | 24.0 | −38.5 |
| Heme 8-C1H3 | 24.0b | 3.5b | 20.5 | −2.3 | 22.9 |
| Heme 5-C1H3 | 23.2b | 3.3b | 19.9 | −2.2 | 22.1 |
| Heme 3-C1H3 | 21.8b | 3.9b | 18.0 | −3.4 | 21.4 |
| Heme 1-C1H3 | 18.5b | 3.7b | 14.9 | −2.2 | 17.1 |
| Average Heme C1H3 | 21.9 | 3.6 | 18.3 | −2.5 | 20.8 |
Analysis of Chemical Shifts in the Variants
The chemical shifts of axial His ring protons have been proposed to reflect histidinate character of axial His ligands in paramagnetic heme proteins.7,11,25,55–56 The His16 Cβ chemical shift for Ht cyt c552 wild-type is downfield relative to the chemical shift of the double mutant (Table 1), and the Cβ chemical shifts correlate linearly with the reduction potential observed in these variants (0.023 ± 0.004 ppm/mV; Fig. 4C). These results suggest that relative shifts of the axial His Cβ reflect the relative histidinate character in heme containing proteins when comparing proteins of the same ligand set. The impact of the mutations on the Fe(II) oxidation state was investigated by using 1H-15N HSQC to measure the His16 Hδ1-Nδ1 cross peak for the ferrous species. Formation of hydrogen bonds has been observed in some systems to cause proton chemical shift differences of 15–20 ppm,57–58 and as a function of hydrogen bond strength in diamagnetic proteins, amide proton chemical shift changes of up to 4 ppm are expected.58–59 His ring nitrogen chemical shift differences of ~10 ppm have been observed when the His ring NH proton forms a hydrogen bonding interaction in model complexes.60 Here the His16 Hδ1-Nδ1 chemical shifts in the ferrous oxidation state exhibit almost no change across the series of variants (Table 1), indicating that the mutations have little impact on the axial His δNH in the ferrous proteins.
Figure 4.

A) 1-D 13C spectra for 1) Ht-M13V/K22M/M61A-13CN, 2) Ht-M13V/M61A-13CN, 3) Ht-K22M/M61A-13CN, and 4) Ht-M61A-13CN. B) Plot of 13C cyanide chemical shift of the 13CN derivative of Met-to-Ala variants vs. reduction potential for Ht cyt c552 variants. C) Plot of His16 13Cβ chemical shifts vs. reduction potential for Ht cyt c552 variants. D) Plot of average heme methyl 1H chemical shifts vs. reduction potential for Ht cyt c552 variants.
Heme methyl proton chemical shifts are dominated by the FC shift, and provide information about the delocalization of the unpaired electron in the porphyrin ring.61 The orientation and donor properties of the axial ligands as well as heme distortions from planarity can impact the heme methyl 1H chemical shifts.14,53–54,56,62–63 The average heme methyl proton chemical shift reflects the spin density at the β-pyrrole positions (Table 1).14,53 The average heme methyl proton chemical shift has a linear correlation with Em in the variants (Figure 4D). The axial Met61 Cε1H3 protons exhibit upfield chemical shifts that are dominated by a FC shift contribution. The mutants exhibit increased upfield shifts for the axial Met61 Cε1H3 (Table 1). The changes observed in the average heme methyl proton chemical shift and axial Met61 Cε1H3 shift are consistent with decreased spin density on porphyrin ring substituents and increased spin density on the axial Met in the variants with lower Em.
pH Dependence of Proximal His Hδ1 and Nδ1 Chemical Shifts in Ferric Species
To probe the relationship between the conserved hydrogen bond between His16 Hδ1 and Pro25 CO and the reduction potential, measurements of the His16 Hδ1-Nδ1 cross peak for the Fe(III) species over a range of pH values were performed with 1H-15N HSQC. As the pH is increased for all Ht cyt c552 variants, the His16 Hδ1-Nδ1 cross peak shifts downfield in both the 1H and 15N dimensions (Figure 3). The cross peak for wild-type begins to exhibit modified chemical shifts at a lower pH value (pH ~9.1) relative to the cross peak in Ht-M13V/K22M (pH ~9.9); for the single mutants the transition starts at intermediate pH values (Supporting Information). As the pH is increased, the loss of intensity of the cross peak also begins at lower pH values in wild-type relative to the double mutant (Figure 3). The His16 Hδ1-Nδ1 cross peak decreases in intensity until it is no longer observable.
Figure 3.
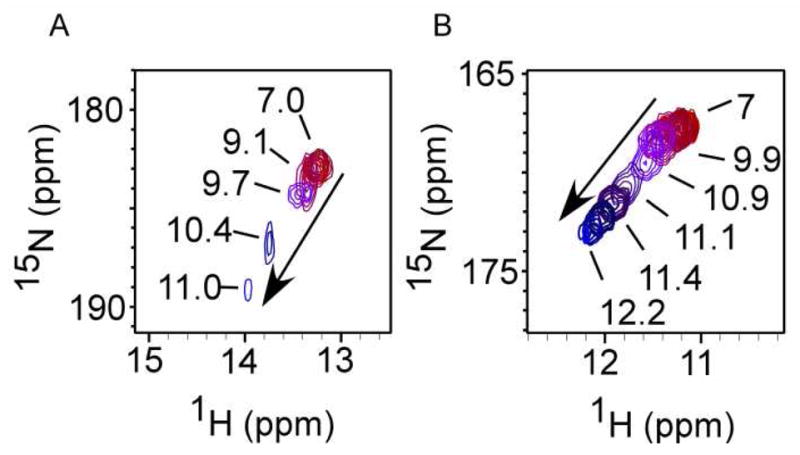
Overlay of series of 1H-15N HSQC spectra of Hδ1-Nδ1 cross peak for A) Ht cyt c552 wild type and B) Ht-M13V/K22M at increasing pH values (arrow). In wild-type the chemical shift begins changing at a lower pH value than that of Ht-M13V/K22M.
Histidine side chains have two pKa values, one corresponding to the first deprotonation event (HisH+ to neutral His, pKa(1)) and the second corresponding to the full deprotonation (neutral His to His−, pKa(2)). To determine pKa(2) from the pH dependence of the His16 Hδ1 and Nδ1 chemical shifts, the observed chemical shifts were plotted versus pH and fit using KaleidaGraph (Synergy Software) according to the following relationship:
| (2) |
where δobs is the observed chemical shift at each pH value, δB is the chemical shift at the highest pH observed, and Δδ represents the titration shift of the signal from low to high pH.64 We find that the pKa(2) for the axial His in cyts c is low (10.11 to 10.62) relative to the pKa(2) for deprotonation of neutral imidazole (14.5) and in a similar range to the pKa(2) determined for imidazole coordinated to ferrimyoglobin (10.45).65 For the series of variants, the pKa(2) for Ht cyt c552 wild-type is lower than that of Ht-M13V/K22M (Table 1). The pKa(2) values determined for the single mutants are intermediate in value. In contrast with the correlating trends observed in chemical shifts and reduction potential, the correlation between pKa(2) and Em is poor (Supporting Figure S1). A possible reason is that variants with the neutral Met in place of the positively charged Lys at position 22 (Ht-K22M and Ht-M13V/K22M) have increased pKa(2) values because of electrostatic effects. Considering the possible effect of charge on axial His pKa(2), overall these results are consistent with a stronger His16 Hδ1-to-Pro25 CO hydrogen bonding interaction in the mutants relative to wild-type, and thus stronger hydrogen bonding in the lower potential variants. These results indicate that the hydrogen bonding interaction between His16 Hδ1 and the backbone carbonyl of Pro25 is influenced by the hydrogen bonding network in the CXXCH motif which is modified in these mutants as demonstrated in earlier work.11
Fe(II)-CO Stretch
IR measurements were taken of the νCO for Fe(II)-CO ligated protein for the series of variants incorporating a Met61Ala mutation to open the 6th coordination site. The νCO decreases slightly for the CO derivative of reduced Ht-M13V/K22M/M61A relative to Ht-M61A (Table 1). These results indicate a slight decrease in the CO vibrational frequency as Em decreases.
Fe(III)-13CN− Chemical Shifts
The 13C shift of ferric heme-coordinated 13CN− bound trans to a His has been proposed to reflect the strength of the His-Fe(III) bond.40–41 Here, 13CN− is bound to ferric heme for the series of variants incorporating the Met61Ala mutation, and 1-D 13C NMR was utilized to observe the chemical shifts. The 13C resonances of Fe(III)-13CN− are found to be far upfield (~−4000 ppm) and extremely broad (~5000 Hz). The 13C signal for Ht-M61A is shifted upfield by 85 ppm relative to Ht-M61A/M13V/K22M (Table 1; Figure 4A). The chemical shift correlates linearly with Em in the series of variants (−1.00 ± 0.13 ppm/mV) (Figure 4B). These results indicate decreased σ bonding between Fe(III) and CN− in variants with lower reduction potential, and reflect the trans influence of a stronger Fe(III)-N(His) σ bonding interaction. These results are consistent with His16 having more His− character in the lower potential Ht-M13V/K22M relative to wild-type, and increased electron donation from the axial His to Fe(III).
Discussion
Heme Reactivity Modulation by Histidinate Character and Hydrogen Bonding
The amount of His− character of the proximal His in heme proteins has received considerable attention for its proposed role in modulating heme reactivity. The strength of hydrogen bonding interactions between the proximal His axial ligand and a hydrogen bond acceptor is one of the factors determining His− character, and has been proposed to play a functional role in many heme containing proteins.8,9,66 In peroxidases, for instance, a strong hydrogen bond between the proximal His and (often) an Asp increases the electron density at the heme iron, and promotes O-O bond heterolysis via the ‘push effect’.6–7,25,27,41,67 Less well investigated is the functional significance and variability of the hydrogen bonding interaction found in proteins with a backbone carbonyl acting as the hydrogen bond acceptor. The hydrogen bonding interaction in globins and cyts c could participate in fine-tuning the Em of the heme, but as it involves a backbone atom as the hydrogen bond acceptor, it is difficult to manipulate experimentally. Previous results suggest that cyt c variants with a more rigid CXXCH motif have a stronger His-Fe(III) interaction.11 Here, we explore this relationship in more depth to lend insight into how subtle differences in heme pocket architecture may influence heme redox potential.
Chemical shifts of axial His ligand nuclei have been used to probe the imidazolate character and hydrogen bonding interactions in heme model complexes and in peroxidase and globin heme proteins.4,6–7,24,55,68 Banci and coworkers demonstrated that the chemical shifts of proximal His protons Hε1 and Hδ1 in CN−-ligated derivatives correlate to reduction potentials observed of horseradish peroxidase (HRP), cytochrome c peroxidase (CcP) and lignin peroxidase (LiP). Specifically, as reduction potential decreases across the series of peroxidases (LiP > CcP > HRP; -130 mV > -194 mV > -278 mV), both Hε1 (−9.0 ppm > −20.6 > −29.9 ppm) and Hδ1 (14.0 ppm > 10.2 ppm > 9.9 ppm) shift upfield.7 The chemical shifts observed have been proposed to correlate to the hydrogen bonding interaction of the proximal His. In cyts c, we have been unable to observe these chemical shifts in the CN−-ligated derivatives, which precludes direct comparison with work on peroxidases and globins. In Fe(III) His-Met cyts c, however, we observe that the axial His Hε1 and Hδ1 chemical shifts tend to move upfield in variants with lower reduction potential (Table 1),11 consistent with an increase in spin density on these nuclei and a stronger His-Fe(III) bond. The Nδ1 chemical shifts for the Fe(III) oxidation state are observed to move upfield in variants with lower Em (Table 1),11 also consistent with an increase in spin density on the Nδ1 atom. In the Fe(II) oxidation state the Hδ1 and Nδ1 chemical shifts do not change appreciably (Table 1), indicating that these mutations have a greater effect on the Fe(III) oxidation state than on the Fe(II) oxidation state. The observation of Nδ1 chemical shifts that exhibit differences in Fe(III)cyt c variants with different reduction potentials shows the utility of using 15N chemical shifts in His-ligated heme proteins to investigate His− character. The His16 Cβ chemical shifts move upfield as reduction potential decreases, indicating that the axial His Cβ chemical shift in heme proteins also may be useful as a probe nucleus for changes in His-Fe(III) interactions. Because it is shifted away from congested regions of the spectrum, observation of this nucleus usually requires no isotopic labeling, which may make it more experimentally tractable for observation in a variety of heme proteins. In summary, changes observed in the chemical shifts of axial His nuclei reflect increased His-Fe(III) interaction in variants with lower Em.
Fe(II)-CO Stretch
The well-documented correlation between νFeC and νCO in CO-ligated heme proteins reflects backbonding between Fe(II) dπ orbitals and unoccupied CO π* orbitals.69–70 As Fe-C π bond strength increases, CO bond order decreases, and these changes can be observed in the linear inverse correlation between the vibrational frequencies. CO bound to iron in heme proteins is subject to both distal and proximal interactions.69,71 In 20 pig and sperm whale myoglobin variants, Phillips et al. observed νCO and νFeC measurements from 1916 to 1984 cm−1 and 526 to 478 cm−1, and explain the variation observed by changes in polarity and electrostatics in the distal heme pocket for these variants.72 While there have been a number of studies investigating the effects of changes in the proximal heme pocket to νCO and νFeC, it can be difficult to parse out the varying effects of σ bonding, π bonding and π back-bonding.71,73–73 It has been observed that donor strength of the ligand trans to CO in the proximal pocket impacts νCO.69,74 When His is the proximal ligand, hydrogen bonding interactions have been examined for both heme proteins and model compounds.71,73,75 Franzen has investigated the effects of modifying the hydrogen bonding interaction of the proximal His in models of the peroxidase active site. Stronger hydrogen bonding to the proximal His can be correlated with increased Fe(II)-Nε2(His) σ bond strength and decreased Fe(II)-C bond strength for CO bound trans to the His, and also correlated with increased CO π back bonding interactions with Fe(II).73–74 Franzen therefore proposes that the νCO stretch for Fe(II) heme proteins is sensitive to the histidinate character of the trans axial His, and should decrease as His− increases.73 Consistent with this proposal, comparison of the Fe(II)-CO vibrational frequencies of HRP relative to CcP reveals that the lower potential peroxidase (HRP) exhibits higher νFeC and lower νCO.7,70,76–77 In recent DFT results by Daskalakis and Varotsis, however, it was determined that the νCO changes for model complexes can be quite small for slight variations in His− character.71 The differences observed between fully deprotonated His (νCO 1952 cm−1) and neutral His (νCO 1993 cm−1) was 41 cm−1, but the difference in νCO between neutral His with no hydrogen bond and His with 2 hydrogen bonding interactions at Hδ1 (νCO 1990 cm−1) was only 3 cm−1.71 Here we observe small but systematic changes in νCO for the Met61-to-Ala CO-ligated Fe(II) variants. The νCO changes are quite small relative to the experimental resolution and compared to the relative magnitude of changes observed in peroxidases. They may therefore reflect only slight alteration in CO bond order in the Fe(II) oxidation state of these variants, which is consistent with the proposal based on NMR chemical shifts that these mutations primarily affect the ferric oxidation state. Conclusions about metal-ligand interactions using data on only one oxidation state should be interpreted with caution, because changes observed between protein species or variants may reflect effects primarily in one oxidation state or the other, or even different effects on the different oxidation states.
Heme Methyl and Met61 Chemical Shifts
In the series of variants under investigation here, we observe upfield chemical shift changes for the average of the heme methyl shifts (Table 1). La Mar and coworkers observed similar changes in model heme complexes, in which the deprotonation of an axial imidazole resulted in upfield chemical shifts for the heme methyl protons. They propose that the upfield shift for the average heme methyl chemical shift is consistent with a decrease in porphyrin-to-Feπ spin delocalization, which is expected if there is increased σ donation from His− to Fe(III).24 The observations here can be interpreted as a decrease in the spin density at the heme methyl protons, reflecting a decrease in the Feπ donation to the porphyrin e(π) orbital, consistent with an increase in His-Fe(III) interaction. Another interpretation is that the decrease in the average heme methyl 1H chemical shift reflects an increase in the ruffling of the porphyrin plane, which we would expect to correlate with a decrease in reduction potential.14,63 The upfield trend in chemical shift for Met61 Cε1H3 as Em decreases for the series of variants may also be expected with increased heme ruffling, although it may also reflect changes in the axial His bonding. The interpretation of changes in hyperfine shifts must be made with caution, as multiple active-site changes may affect shifts.
Axial His pKa Values
The average pKa(1) value for HisH+ in proteins is 6.6 ± 1.0, with 90% of the values found in the pH range 5.0 to 8.1.78–79 Measurements of pKa(1) values for individual His residues are often undertaken in order to understand the role of His in enzymatic mechanisms.78–81 In general pKa(1) is found to be very low in His-ligated to metals; a protonated HisH+ is not expected to coordinate to the metal. The pKa(2) for imidazole (Im) is approximately 14.5 in solution.65 There are few reports of pKa(2) in proteins because the presence of a fully deprotonated His is unusual except for metal-coordinated His residues. The pKa(2) imidazole bound to the heme in myoglobin is 10.34, i.e., approximately 4 units lower than the value in solution.65,82 The pKa(2) of heme-ligated His has been infrequently measured because it can be difficult to observe the His ligand across a range of pH values in Fe(III) heme proteins.64,83–85 Yu and Smith note that in Fe(III) Rhodospirillum rubrum cyt c2 the proximal His Hδ1 is still observable up to pH 8.5, but could not be measured at higher pH values because of spectral overlap.85 Wolff and coworkers used diamagnetic gallium-protoporphyrin IX-substituted Serratia marcescens HasA to measure the pKa(2) of a His with a strong hydrogen bonding interaction with the proximal Tyr heme axial ligand (pKa of ~9.7).64 The axial His ligand in HasA is found to have a pKa(2) of ~9.0.81 Here, we take advantage of 1H-15N HSQC to directly monitor the His16 Hδ1-Nδ1 cross peak as a function of pH in the Fe(III) oxidation state. It should be noted that simultaneous changes are also observed in the chemical shifts of the Met61 Cε1H3 and the heme methyl protons in these variants as a function of pH. Since the His16 Hδ1-Nδ1 cross peak decreases in intensity at high pH until it is no longer observable, but the Met61 Cε1H3 and the heme methyl protons continue to be detected, we attribute the changes observed to deprotonation of His16 Hδ1. The increasing pKa(2) values for the Ht cyt c552 variants suggest that the hydrogen bond between His16 Hδ1 and Pro25 backbone carbonyl plays a significant functional role in cyts c. Notably, pKa(2) broadly tends to increase as Em decreases for this series of variants (Table 1; Supporting Information) and this supports the proposal that the hydrogen bonding interaction at the proximal His is important in modulation of Em.
13CN− Chemical Shifts
Diatomic ligands have been extensively used to probe features of the heme environment in heme containing proteins. Fujii and Yoshida reported a small, 36 ppm range for the 13CN− chemical shifts among three species of mitochondrial cyts c (horse heart, bovine heart and yeast).41 These 13C chemical shifts exhibit little species variation within the experimental error, in spite of differences in Em values for horse heart (260 mV) and yeast cyt c (290 mV).21,86 Nonaka et al. used both 13C and 15N chemical shifts to probe the proximal His ligand electron donation and the hydrogen bonding interactions on the distal side of the protein to elucidate details of the push-pull mechanism of peroxidase heme proteins.42 We expect that the structure of the distal pocket of the heme is the same for all of the variants under investigation here since mutations were made on the proximal side. Therefore, the changes that are observed in the Fe(III)-13CN− chemical shift should reflect changes induced by modification of the proximal heme pocket.
The Fe(III)-CN− interaction is dominated by σ donation from the cyanide lone pair to the metal dz2 orbital.87–88 It is well-established that π back-bonding in Fe(III)-CN− is very weak and contributes minimally to the bonding interaction.88 Chemical shifts observed in heme 13CN−-ligated proteins are found to be very far upfield, which is consistent with a negative spin density, a consequence of spin polarization through the σ bond. The 13C chemical shifts measured for this series of variants are in the same range as those found in other Fe(III)-13CN− heme proteins examined by Fujii and coworkers.40–42 The 13C chemical shift is dominated by a FC contribution and can be used to report on the electron donating effect of the trans axial ligand (through a trans ligand influence). As His becomes a stronger σ donor, the CN− σ bond with Fe(III) is weakened. This results in a smaller FC contribution for 13CN− because the spin density on the 13C nucleus decreases. The Fe(III)-13CN− interaction is strongest in the case when the His-Fe(III) interaction is weakest. A larger δhf (further upfield) occurs for Ht-M61A-13CN−, which is consistent with the least His− character and the highest reduction potential in the series of variants. Thus, the 13CN− carbon chemical shift is sensitive to differences in the His-Fe(III) interaction among mutants with relatively small differences in redox potential, demonstrating the high sensitivity of this nucleus as a probe of the heme active site.
Conclusion
Subtle changes in the heme environment can have large effects on heme reactivity, but can be difficult to observe experimentally. NMR spectra of paramagnetic species, however, are exquisitely sensitive to small but functionally relevant variations in metalloprotein active sites. The existence of extensive chemical shift assignments for a number of heme proteins allows us to interpret hyperfine shifts in paramagnetic cytochromes in detail. Chemical shifts of axial His ring nuclei and of 13CN− bound trans to the axial His reveal variations in the His-Fe(III) interaction brought about by introduction of mutations in the heme pocket. Greater His− character will stabilize the Fe(III) oxidation state relative to the Fe(II) oxidation state, and NMR allows for detection of changes in spin density at particular heme axial ligand nuclei. The observed changes in average heme methyl proton chemical shifts also are consistent with increased heme out-of-plane ruffling playing a concomitant role in the decreases in redox potential in the variants. The effects of variations in and near the CXXCH motif on Em observed in this study support a functional basis for use of heme c. In addition, these results provide insight into how the protein environment tunes heme reactivity in heme proteins more generally.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM63170 to K.L.B.) S.E.J.B. acknowledges an Agnes M. and George Messersmith Graduate Fellowship. We thank Dr. Scott Kennedy (University of Rochester) for assistance with collection of 13C NMR data.
Footnotes
Supporting Information Available. Heme methyl 1H chemical shifts, 1Hδ1 and 15Nδ1 chemical shifts as a function of pH values for each variant, representative pKa(2) fits, and a plot of pKa(2) vs. Em. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Paoli M, Marles-Wright J, Smith A. DNA Cell Biol. 2002;21:271–280. doi: 10.1089/104454902753759690. [DOI] [PubMed] [Google Scholar]
- 2.Chiarugi P, Fiaschi T. Cell Signal. 2007;19:672–682. doi: 10.1016/j.cellsig.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Rubio I, Braun M, Gromov I, Thöny-Meyer L, Schweige A. Biophys J. 2007:92. doi: 10.1529/biophysj.106.098277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banci L, Rosato A, Turano P. J Biol Inorg Chem. 1996;1:364–367. [Google Scholar]
- 5.Takahashi A, Kurahashi T, Fujii H. Inorg Chem. 2009;48:2614–2625. doi: 10.1021/ic802123m. [DOI] [PubMed] [Google Scholar]
- 6.La Mar GN, de Ropp JS, Chacko VP, Satterlee JD, Erman JE. Biochim Biophys Acta. 1982;708:317–325. doi: 10.1016/0167-4838(82)90443-5. [DOI] [PubMed] [Google Scholar]
- 7.Banci L, Bertini I, Turano P, Tien M, Kirk TK. Proc Natl Acad Sci USA. 1991;88:6956–6960. doi: 10.1073/pnas.88.16.6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rousseau DG, Rousseau DL. J Struct Biol. 1992;109:13–17. doi: 10.1016/1047-8477(92)90062-f. [DOI] [PubMed] [Google Scholar]
- 9.Quinn R, Mercer-Smith J, Burstyn JN, Valentine JS. J Am Chem Soc. 1984;106:4136–4144. [Google Scholar]
- 10.Laberge M, Yonetani T. IUBMB Life. 2007;59:528–534. doi: 10.1080/15216540701222914. [DOI] [PubMed] [Google Scholar]
- 11.Michel LV, Ye T, Bowman SEJ, Levin BD, Hahn MA, Russell BS, Elliott SJ, Bren KL. Biochemistry. 2007;46:11753–11760. doi: 10.1021/bi701177j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shelnutt JA, Song XZ, Ma JG, Jia SL, Jentzen W, Medforth CJ. Chem Soc Rev. 1998;27:31–41. [Google Scholar]
- 13.Jentzen W, Ma JG, Shelnutt JA. Biophys J. 1998;74:753–763. doi: 10.1016/S0006-3495(98)74000-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liptak MD, Wen X, Bren KL. J Am Chem Soc. 2010;126 doi: 10.1021/ja102098p. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fufezan C, Zhang J, Gunner MR. Proteins. 2008;73:690–704. doi: 10.1002/prot.22097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merlino A, Vergara A, Sica F, Mazzarella L. Mar Gen. 2009;2:51–56. doi: 10.1016/j.margen.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 17.La Mar GN, Satterlee JD, De Ropp JS. Nuclear Magnetic Resonance of Hemoproteins. In: Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Vol. 5. 2000. pp. 185–298. [Google Scholar]
- 18.Poulos TL. J Biol Inorg Chem. 1996;1:356–363. [Google Scholar]
- 19.Moore GR, Pettigrew GW. Cytochromes c: Evolutionary, Structural, and Physicochemical Aspects. Springer-Verlag; Berlin: 1990. [Google Scholar]
- 20.Pettigrew GW, Moore GR. Cytochromes c: Biological Aspects. Springer-Verlag; Berlin: 1987. [Google Scholar]
- 21.Reedy CJ, Elvekrog MM, Gibney BR. Nucleic Acids Res. 2008;36:D307–313. doi: 10.1093/nar/gkm814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bowman SEJ, Bren KL. Nat Prod Rep. 2008;25:1118–1130. doi: 10.1039/b717196j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park KD, Guo K, Adebodun F, Chiu ML, Sligar SG, Oldfield E. Biochemistry. 1991;30:2333–2347. doi: 10.1021/bi00223a007. [DOI] [PubMed] [Google Scholar]
- 24.Chacko VP, La Mar GN. J Am Chem Soc. 1982;104:7002–7007. [Google Scholar]
- 25.Banci L, Bertini I, Kuan IC, Tien M, Turano P, Vila AJ. Biochemistry. 1993;32:13483–13489. doi: 10.1021/bi00212a013. [DOI] [PubMed] [Google Scholar]
- 26.La Mar GN, De Ropp JS, Smith KM, Langry KC. J Biol Chem. 1980;255:6646–6652. [PubMed] [Google Scholar]
- 27.Dawson JH. Science. 1988;240:433–439. doi: 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]
- 28.Stevens JM, Daltrop O, Allen JWA, Ferguson SJ. Acc Chem Res. 2004;37:999–1007. doi: 10.1021/ar030266l. [DOI] [PubMed] [Google Scholar]
- 29.Allen JWA, Ginger ML, Ferguson SJ. Biochem Soc Trans. 2005;33:145–146. doi: 10.1042/BST0330145. [DOI] [PubMed] [Google Scholar]
- 30.Barker PD, Ferguson SJ. Structure. 1999;7:R281–R290. doi: 10.1016/s0969-2126(00)88334-3. [DOI] [PubMed] [Google Scholar]
- 31.Page MD, Sambongi Y, Ferguson SJ. Trends Biochem Sci. 1998;23:103–108. doi: 10.1016/s0968-0004(98)01173-6. [DOI] [PubMed] [Google Scholar]
- 32.Kranz R, Lill R, Goldman B, Bonnard G, Merchant S. Mol Microbiol. 1998;29:383–396. doi: 10.1046/j.1365-2958.1998.00869.x. [DOI] [PubMed] [Google Scholar]
- 33.Zheng Z, Gunner MR. Proteins. 2009;75:719–734. doi: 10.1002/prot.22282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cowley AB, Kennedy ML, Silchenko S, Lukat-Rodgers GS, Rodgers KR, Benson DR. Inorg Chem. 2006;45:9985–10001. doi: 10.1021/ic052205k. [DOI] [PubMed] [Google Scholar]
- 35.Walker FA. Coord Chem Rev. 1999;185–186:471–534. [Google Scholar]
- 36.Ubbink M, Worrall JAR, Canters GW, Groenen EJJ, Huber M. Ann Rev Biophy Biom. 2002;31:393–422. doi: 10.1146/annurev.biophys.31.091701.171000. [DOI] [PubMed] [Google Scholar]
- 37.Bren KL. Nuclear Magnetic Resonance (NMR) Spectroscopy of Metallobiomolecules. In: Scott RA, Lukehart CM, editors. Applications of Physical Methods to Inorganic and Bioinorganic Chemistry. Wiley; 2007. pp. 357–384. [Google Scholar]
- 38.Choudhury K, Sundaramoorthy M, Hickman A, Yonetani T, Woehl E, Dunn MF, Poulos TL. J Biol Chem. 1994;269:20239–20249. [PubMed] [Google Scholar]
- 39.de Ropp JS, Sham S, Asokan A, Newmyer S, de Montellano PRO, La Mar GN. J Am Chem Soc. 2002;124:11029–11037. doi: 10.1021/ja020176w. [DOI] [PubMed] [Google Scholar]
- 40.Fujii H. J Am Chem Soc. 2002;124:5936–5937. doi: 10.1021/ja025737y. [DOI] [PubMed] [Google Scholar]
- 41.Fujii H, Yoshida T. Inorg Chem. 2006;45:6816–6827. doi: 10.1021/ic0607383. [DOI] [PubMed] [Google Scholar]
- 42.Nonaka D, Wariishi H, Fujii H. Biochemistry. 2009;48:898–905. doi: 10.1021/bi802030a. [DOI] [PubMed] [Google Scholar]
- 43.Travaglini-Allocatelli C, Gianni S, Dubey VK, Borgia A, Di Matteo A, Bonivento D, Cutruzzolà F, Bren KL, Brunori M. J Biol Chem. 2005;280:25729–25734. doi: 10.1074/jbc.M502628200. [DOI] [PubMed] [Google Scholar]
- 44.Matsuura Y, Takano T, Dickerson RE. J Mol Biol. 1982;156:389–409. doi: 10.1016/0022-2836(82)90335-7. [DOI] [PubMed] [Google Scholar]
- 45.Massari AM, McClain BL, Finkelstein IJ, Lee AP, Reynolds HL, Bren KL, Fayer MD. J Phys Chem B. 2006;110:18803–18810. doi: 10.1021/jp054959q. [DOI] [PubMed] [Google Scholar]
- 46.Massari AM, Finkelstein IJ, McClain BL, Goj A, Wen X, Bren KL, Loring RF, Fayer MD. J Am Chem Soc. 2005;127:14279–14289. doi: 10.1021/ja053627w. [DOI] [PubMed] [Google Scholar]
- 47.Karan EF, Russell BS, Bren KL. J Biol Inorg Chem. 2002;7:260–272. doi: 10.1007/s007750100292. [DOI] [PubMed] [Google Scholar]
- 49.Morar AS, Kakouras D, Young GB, Boyd J, Pielak GJ. J Biol Inorg Chem. 1999;4:220–222. doi: 10.1007/s007750050307. [DOI] [PubMed] [Google Scholar]
- 49.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 50.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- 51.Zhong LH, Wen X, Rabinowitz TM, Russell BS, Karan EF, Bren KL. Proc Natl Acad Sci USA. 2004;101:8637–8642. doi: 10.1073/pnas.0402033101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hasegawa J, Yoshida T, Yamazaki T, Sambongi Y, Yu YH, Igarashi Y, Kodama T, Yamazaki K, Kyogoku Y, Kobayashi Y. Biochemistry. 1998;37:9641–9649. doi: 10.1021/bi9803067. [DOI] [PubMed] [Google Scholar]
- 53.Walker FA. Inorg Chem. 2003;42:4526–4544. doi: 10.1021/ic026245p. [DOI] [PubMed] [Google Scholar]
- 54.Shokhirev NV, Walker FA. J Am Chem Soc. 1998;120:981–990. [Google Scholar]
- 55.Bougault CM, Dou Y, Ikeda-Saito M, Langry KC, Smith KM, La Mar GN. J Am Chem Soc. 1998;120:2113–2123. [Google Scholar]
- 56.La Mar GN, Dalichow F, Zhao X, Dou Y, Ikeda-Saito M, Chiu ML, Sligar SG. J Biol Chem. 1994;269:29629. [PubMed] [Google Scholar]
- 57.Hibbert F, Emsley J. Adv Phys Org Chem. 1990;26:255–279. [Google Scholar]
- 58.Dingley AJ, Cordier F, Grzesiek S. J Magn Reson. 2001;13:103–127. [Google Scholar]
- 59.Wagner G, Pardi A, Wüthrich K. J Am Chem Soc. 1983;105:5948–5949. [Google Scholar]
- 60.Bachovichin WW. Magn Reson Chem. 2001;39:S199–S213. [Google Scholar]
- 61.Shokhirev NV, Walker FA. J Biol Inorg Chem. 1998;3:581–594. [Google Scholar]
- 62.Bertini I, Luchinat C, Parigi G, Walker FA. J Biol Inorg Chem. 1999;4:515–519. doi: 10.1007/s007750050337. [DOI] [PubMed] [Google Scholar]
- 63.Shokhirev TK, Shokhirev NV, Berry RE, Zhang HJ, Walker FA. J Biol Inorg Chem. 2008;13:941–959. doi: 10.1007/s00775-008-0381-8. [DOI] [PubMed] [Google Scholar]
- 64.Wolff N, Deniau C, Letoffe S, Simenel C, Kumar V, Stojiljkovic I, Wandersman C, Delepierre M, Lecroisey A. Protein Sci. 2002;11:757–765. doi: 10.1110/ps.3630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.George P, Hanania GIH, Irvine DH, Abu-Issa I. J Chem Soc. 1964:5689–5694. [Google Scholar]
- 66.O’Brien P, Sweigart DA. Inorg Chem. 1985;24:1405–1409. [Google Scholar]
- 67.Goodin DB, McRee DE. Biochemistry. 1993;32:3313–3324. [PubMed] [Google Scholar]
- 68.La Mar GN, Davis NL, Johnson RD, Smith WS, Hauksson JB, Budd DL, Dalichow F, Langry KC, Morris IK, Smith KM. J Am Chem Soc. 1993;115:3869–3876. [Google Scholar]
- 69.Spiro TG, Wasbotten IH. J Inorg Biochem. 2005;99:34–44. doi: 10.1016/j.jinorgbio.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 70.Vogel KM, Kozlowski PM, Zgierski MZ, Spiro TG. Inorg Chim Acta. 2000;297:11–17. [Google Scholar]
- 71.Daskalakis V, Varotis C. Int J Mol Sci. 2009;10:4137–4156. doi: 10.3390/ijms10094137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Phillips GN, Teodoro ML, Li T, Smith B, Olson JS. J Phys Chem B. 1999;103:8817–8829. [Google Scholar]
- 73.Franzen S. J Am Chem Soc. 2001;123:12578–12589. doi: 10.1021/ja0108988. [DOI] [PubMed] [Google Scholar]
- 74.Xu C, Ibrahim M, Spiro TG. Biochemistry. 2008;47:2379–2387. doi: 10.1021/bi702254y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Decatur SM, DePillis GD, Boxer SG. Biochemistry. 1996;35:3925–3932. doi: 10.1021/bi952625t. [DOI] [PubMed] [Google Scholar]
- 76.Evangelista-Kirkup R, Smulevich G, Spiro TG. Biochemistry. 1986;25:4420–4425. doi: 10.1021/bi00363a037. [DOI] [PubMed] [Google Scholar]
- 77.Smulevich G, Evangelista-Kirkup R, English A, Spiro TG. Biochemistry. 1986;25:4426–4430. doi: 10.1021/bi00363a038. [DOI] [PubMed] [Google Scholar]
- 78.Grimsley GR, Scholtz JM, Pace CN. Protein Sci. 2009;18:247–251. doi: 10.1002/pro.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Markley JL. Acc Chem Res. 1975;8:70–80. [Google Scholar]
- 80.Huang ZJ, Lin ZJ, Song C. J Phys Chem A. 2007;111:4340–4352. doi: 10.1021/jp067280a. [DOI] [PubMed] [Google Scholar]
- 81.Farrell D, Miranda ES, Webb H, Georgi N, Crowley PB, McIntosh LP, Nielsen JE. Proteins. 2010;78:843–857. doi: 10.1002/prot.22611. [DOI] [PubMed] [Google Scholar]
- 82.Gadsby PMA, Thomson AJ. FEBS Lett. 1982;150m:59–63. [Google Scholar]
- 83.Bashford D, Case DA, Dalvit C, Tennant L, Wright PE. Biochemistry. 1993;32:8045–8056. doi: 10.1021/bi00082a027. [DOI] [PubMed] [Google Scholar]
- 84.Bhattacharya S, Sukits SF, MacLaughlin KL, Lecomte JT. Biophys J. 1997;73:3230–3240. doi: 10.1016/S0006-3495(97)78348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu LP, Smith GM. Biochemistry. 1990;29:2920–2925. doi: 10.1021/bi00464a005. [DOI] [PubMed] [Google Scholar]
- 86.Taniguchi VT, Sailasutascott N, Anson FC, Gray HB. Pure Appl Chem. 1980;52:2275–2281. [Google Scholar]
- 87.Cheng RJ, Lee CH, Chao CW. Chem Comm. 2009:2526–2528. doi: 10.1039/b901116a. [DOI] [PubMed] [Google Scholar]
- 88.Kushkuley B, Stavrov SS. Biochim Biophys Acta. 1997;1341:238–250. doi: 10.1016/s0167-4838(97)00082-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.