

Abstract
Nuclear factor erythroid 2–related factor 2 (Nrf2) is a transcription factor that is important in protection against oxidative stress. This study was designed to determine the role of Nrf2 signaling in transcriptional activation of detoxifying and antioxidant genes in an in vivo mouse fetal alcohol syndrome model. Maternal ethanol treatment was found to increase both Nrf2 protein levels and Nrf2-ARE binding in mouse embryos. It also resulted in a moderate increase in the mRNA expression of Nrf2 downstream target detoxifying and antioxidant genes as well as an increase in the expression of antioxidant proteins. Pretreatment with the Nrf2 inducer, 3H-1,2 dithiole-3-thione (D3T), significantly increased Nrf2 protein levels and Nrf2-ARE binding, and strongly induced the mRNA expression of Nrf2 downstream target genes. It also increased the expression of antioxidant proteins and the activities of the antioxidant enzymes. Additionally, D3T pretreatment resulted in a significant decrease in ethanol-induced reactive oxygen species generation and apoptosis in mouse embryos. These results demonstrate that Nrf2 signaling is involved in the induction of antioxidant response in ethanol-exposed embryos. In addition, the potency of D3T in inducing antioxidants as well as in diminishing ethanol-induced apoptosis suggests that further exploration of the antiteratogenic effect of this compound will be fruitful. Antioxid. Redox Signal. 11, 2023–2033.
Introduction
Fetal alcohol spectrum disorders (FASD) are among the most devastating consequences of the widespread use and abuse of alcohol (ethanol). FASD is an umbrella term that includes full-blown fetal alcohol syndrome (FAS) as the severe end of the spectrum of alcohol-induced birth defects. Prenatal ethanol exposure is considered to be the leading known cause of mental retardation in the Western world (1). According to a 1996 Institute of Medicine (IOM) report, the prevalence of FAS in the United States has been estimated at 0.5 to 3.0 cases per 1,000 births, with the full spectrum of ethanol-induced prenatal insult projected to occur several times as frequently (39).
Apoptotic death of selected cell populations is a commonly observed pathogenic feature of ethanol-induced teratogenesis in both in vivo and in vitro model systems (3, 7, 11, 14, 37, 40). Among the proposed mechanisms underlying ethanol-induced apoptosis and malformation is oxidative stress. This is evidenced in studies from our laboratory and others that have shown that (a) antioxidants significantly improve the viability of an ethanol-sensitive cell population, neural crest cells, in vitro (9, 10); (b) ethanol induces superoxide anion generation and lipid peroxidation in cultured mouse embryos (24); (c) superoxide dismutase (SOD) can diminish ethanol-induced cell death and significantly reduce the incidence of neural tube defects (24); and (d) EUK-134, a synthetic SOD and catalase mimetic, can reduce apoptosis and the resulting limb defects in mouse embryos exposed to ethanol in vivo (8).
In addition to using exogenous antioxidants to reduce oxidative stress in ethanol-exposed embryos, a proposed strategy for protecting against oxidative damage entails chemically mediated upregulation of endogenous antioxidants. The current study describes modulation of Nrf2, which is a transcription factor that regulates the induction of detoxifying and antioxidant genes through antioxidant response element (ARE) and, thus, plays a central role in the cellular defense against oxidative stress. Protection mediated by Nrf2 signaling is evidenced by its role in reduction of oxidative damage induced by acute pulmonary injury and hyperoxia (6, 12). Additionally, Nrf2 deficiency enhances the sensitivity of neurons and astrocytes to oxidative stress by reducing the expression of cytoprotective genes (27, 28). Nrf2-deficient mice form higher levels of DNA adducts after exposure to carcinogens (36). A number of studies have also shown that Nrf2 plays an important role in cell survival (27, 28, 30).
Because Nrf2 plays a key role in the transcriptional induction of various antioxidants (21) and an expansive set of antioxidant/detoxification genes that act in synergy to remove ROS is essential for efficient detoxification of ROS, identification of molecules that can induce Nrf2 activation has recently received considerable attention. A wide variety of natural and synthetic small molecules are potent inducers of Nrf2 activity (32). Among these molecules is 3H-1,2 dithiole-3-thione (D3T). D3T is a potent cancer chemopreventive agent that protects against mutation and initiation of neoplasia (34). In addition, activation of the Nrf2 pathway by oral administration of D3T has recently been reported to confer partial protection against MPTP-induced neurotoxicity (4). The protective effects of D3T in animals have been associated with induction of the detoxifying and antioxidant enzymes SOD, catalase and γ-glutamylcysteine synthetase (γ-GCS) (5, 31, 34). Although the exact mechanisms by which D3T activates Nrf2 signaling remain to be defined, it was suggested that the direct interaction between D3T and the sulfhydryl groups of Keap1 can cause dissociation of Keap1 from Nrf2, leading to Nrf2 activation (26). In addition, mitogen-activated protein kinases (MAPKs) have recently been shown to be involved in the activation of Nrf2 by D3T (29).
Although it is clear that Nrf2 is a key transcription factor in regulating a variety of cytoprotective genes in various types of cells and tissues, the potential for this signaling pathway to play a significant protective role relative to teratogenesis by ethanol has not been previously investigated. This study was designed to fill this void by examination of Nrf2-mediated transcriptional activation of detoxifying and antioxidant genes in ethanol-exposed mouse embryos, along with examination of the protective effect of the Nrf2 inducer, D3T. To this end, we have examined the levels of Nrf2 protein and Nrf2-ARE binding activity and have analyzed mRNA and protein expression of Nrf2 downstream target genes as well as the activities of antioxidant enzymes in mouse embryos after exposure to ethanol and D3T in vivo. Additionally, we have determined the potential of D3T to prevent ethanol-induced oxidative stress and apoptosis in mouse embryos. The results of this study demonstrate that Nrf2 signaling is involved in the induction of antioxidants and detoxifying enzymes in ethanol-exposed mouse embryos and that D3T pretreatment can induce antioxidant proteins and phase 2 enzymes and prevent ethanol-induced apoptosis.
Materials and Methods
Animal care and dosing
C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) were mated for 2 h early in the light cycle. The time of vaginal-plug detection was considered 0 days, 0 h of gestation (GD 0:0). Mice were maintained on an ad libitum diet of breeder chow and water. Animals in the experimental groups were administered two intraperitoneal (i.p.) doses of 25.0% (vol/vol) ethanol in lactated Ringer's solution at a dosage of 2.9 g/kg maternal body weight. Control dams were administered lactated Ringer's solution alone. The injections were given 4 h apart, with the first administered at GD 8:0. For D3T pretreatment, pregnant mice were given i.p. injections of either D3T (LKT Laboratories, Inc., St. Paul, MN) at a dosage of 5 mg/kg or its solvent (vegetable oil) on GD 7:6 (i.e., 18 h before they were treated with ethanol or Ringer's solution). For mRNA and protein analysis, pregnant females were killed by cervical dislocation on GD 8:6 (6 h after the first ethanol treatment). For apoptosis assays, pregnant mice were killed on GD 8:12 (12 h after the first ethanol treatment). Abdominal cavities were surgically opened, and the uteri were exposed and removed. Embryos were dissected free of their deciduas in lactated Ringer's solution and then staged by counting the number of somite pairs. Embryos of comparable developmental stage from several litters were pooled for mRNA and protein preparation. Selection of the time points at which analyses were conducted was based on our previous investigations, which demonstrated that significant changes in mRNA expression can be observed by microarray assay in ethanol-exposed mouse embryos as early as 3 h after the first ethanol treatment (18) and that excessive apoptosis can be observed in ethanol-exposed mouse embryos 12–16 h after initial treatment with ethanol (8, 14, 25). All protocols used in this study were approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee.
RNA isolation and reverse-transcription reaction
Total RNA was isolated from whole embryos by using Trizol reagent (Invitrogen, CA) and cleaned with Qiagen RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. RNA quantitation was performed by measuring the absorbance of the RNA sample solutions at 260 nm. The RNA integrity was determined by agarose gel electrophoresis. The RNA samples were treated with DNase-I (Qiagen RNase-free DNase kit). The reverse transcription reaction was carried out in a 100-μl reaction by incubation of 1 μg of RNA with Oligo d(T)16 primers at 48°C for 60 min, by using a commercially available GeneAmp RNA PCR kit (Applied Biosystems, Foster, CA).
Quantitative real-time PCR
Quantitative real-time PCR with SYBR green I detection (Applied Biosystems, Warrington, U.K.) was performed in a MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). Primers listed in Table 1 were designed by using Primer Express Software (Applied Biosystems) and synthesized by Integrated DNA Technologies, Inc (Coralville, IA). Reactions were performed in a total volume of 25 μl, including 12.5 μl 2 × Power SYBR Green PCR Master Mix, 1 μl of 0.4 μM primer, and 2.5 μl of the previously reverse-transcribed cDNA template on 96-well iCycler iQ PCR plates (Bio-Rad). All reactions were carried out at least in duplicate for each sample. The thermocycling profile included 45 cycles consisting of denaturation at 95°C for 15 sec, and annealing and elongation at 60°C for 60 sec. Melting curve and agarose electrophoresis analysis were performed to exclude amplification of nonspecific products. GAPDH was used as a reference for normalization, and relative quantification was performed by using iCycler iQ Optical System Software Version 1 (Bio-Rad).
Table 1.
Sequences of Primers Used for Quantitative Real-Time PCR Analysis of Gene Expression in Mouse Embryos
| Gene | Access. no. | Position | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|---|---|
| Nrf-2 | BC026943 | 1645F-1723R | CGAGATATACGCAGGAGAGGTAAGA | GCTCGACAATGTTCTCCAGCTT |
| SOD1 | NM_011434 | 561F-631R | GTGATTGGGATTGCGCAGTA | TGGTTTGAGGGTAGCAGATGAGT |
| SOD2 | NM_013671 | 193F-263R | TTAACGCGCAGATCATGCA | GGTGGCGTTGAGATTGTTCA |
| SOD3 | NM_011435 | 489F-559R | CATGCAATCTGCAGGGTACAA | AGAACCAAGCCGGTGATCTG |
| CAT | NM_009804 | 1593F-1655R | TGAGAAGCCTAAGAACGCAATTC | CCCTTCGCAGCCATGTG |
| Trx | X77585 | 121F-206R | CCGCGGGAGACAAGCTT | GGAATGGAAGAAGGGCTTGATC |
| Prx-1 | NM_011034 | 393F-471R | GATCCCAAGCGCACCATT | TAATAAAAAGGCCCCTGAAAGAGAT |
| Gpx1 | U13705 | 275F-350R | GAAGAACTTGGGCCATTTGG | TCTCGCCTGGCTCCTGTTT |
| Gpx2 | NM_030677 | 534F-594R | ACCGATCCCAAGCTCATCAT | CAAAGTTCCAGGACACGTCTGA |
| Gpx3 | NM_008161 | 869F-937R | ACAATTGTCCCAGTGTGTGCAT | TGGACCATCCCTGGGTTTC |
| GSR | NM_010344 | 751F-815R | GCTATGCAACATTCGCAGATG | AGCGGTAAACTTTTTCCCATTG |
| NQO-1 | BC004579 | 313F-398R | TATCCTTCCGAGTCATCTCTAGCA | TCTGCAGCTTCCAGCTTCTTG |
| GSTa2 | J03958 | 565F-642R | CGTCCACCTGCTGGAACTTC | GCCTTCAGCAGAGGGAAAGG |
| GSTm2 | J04696 | 714F-780R | GCTCTTACCACGTGCAGCTT | GGCTGGGAAGAGGAAATGGA |
| GSTm3 | J03953 | 292-375 | CACCCGCATACAGCTCATGAT | TTCTCAGGGATGGCCTTCAA |
| GSTp1 | D30687 | 421F-496R | TGGGCATCTGAAGCC1TT TG | GATCTGGTCACCCACGATGAA |
| GAPDH | BC023196 | 165F-355R | AACGACCCCTTCATTGAC | TCCACGACATACTCAGCAC |
Protein preparation and Western blotting
Embryos were washed once in phosphate-buffered saline (PBS) and lysed by incubation for 30 min in RIPA lysis buffer (PBS, 0.5 % sodium deoxycholate, 1% NP-40, 0.1% SDS, 1 mM dithiothreitol) with 1 mM PMSF and protease cocktail inhibitors (Roche, Indianapolis, IN). The samples were then centrifuged at 16,000 g for 10 min at 4°C. The collected supernatants were used for Western blotting. The protein concentration in each sample was measured by using Bio-Rad Protein Assay Reagent (Bio-Rad Laboratories) according to the manufacturer's instructions. Western blots were performed by standard protocols. In brief, total protein (20 μg each lane) was resolved on an SDS–10% polyacrylamide gel and transferred to nitrocellulose membranes. The levels of Nrf2, catalase, and SOD1 and SOD2 proteins were analyzed with the following antibodies: rabbit polyclonal anti-Nrf2 IgG (1:500, Santa Cruz, Santa Cruz, CA), mouse monoclonal anti-catalase antibody (1:500; Sigma, St. Louis, MO), rabbit polyclonal anti-SOD1 IgG (1:1,000; Santa Cruz), and rabbit polyclonal anti-SOD2 IgG (1:500; Santa Cruz), respectively, followed by detection with ECL plus Western-blotting detection reagents (GE Healthcare, Piscataway, NJ). The membranes were scanned on a Bio-Rad Versa Doc Imaging System (model 4000), and the intensity of the protein bands was analyzed by using the Bio-Rad Quantity One software (version 4.5.1).
Electrophoretic mobility shift assay
Nrf2-ARE binding was determined using an electrophoretic mobility shift assay (EMSA). Nuclear extracts were prepared from control and treated mouse embryos by using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology, Rockford, IL) by following the manufacturer's protocol. Binding reactions containing equal amounts of protein (5 μg) and labeled double-strand oligonucleotide probe for mouse glutamate-cysteine ligase (GCLC) ARE (5′-GCACAAAGCGCTGAGTCACGGGGAGG-3′; 5′-GTCCCCGTGACTCAGCGCTTTGTGCG-3′; core ARE sequence underlined) were performed for 20 min at room temperature. Specific binding was confirmed by using 100-fold excess unlabeled ARE oligonucleotide as a specific competitor (Gel Shift Assay Kit; Promega, Madison, WI). The oligonucleotide–protein complex was separated by 4% nondenaturing polyacrylamide gel electrophoresis followed by autoradiography. All gel-shift assays were performed for three sample replicates in each group.
Measurement of antioxidant enzyme activities
Mouse embryos were collected and placed in ice-cold PBS buffer, pH 7.4. The embryos were sonicated, followed by centrifugation at 13,000 g for 10 min at 4°C. The supernatants were collected for measurement of antioxidant enzymes. Total superoxide dismutase (SOD) activity was determined with the SOD Assay Kit from Dojindo Molecular Technologies, Inc. (Gaithersburg, MD) according to the manufacturer's protocol. Catalase activity was measured with an Amplex Red Catalase Assay Kit (Molecular Probes, Eugene, OR) according to the manufacturer's instruction.
Measurement of intracellular ROS generation
Intracellular ROS generation was assayed with the method described by Ozawa et al. (35), with modification. The mouse embryos were washed twice in assay buffer (130 mM KCl, 5 mM MgCl2, 20 mM NaH2PO4, 20 mM Tris-HCl, 30 mM glucose) and homogenized in 100 μl of assay buffer. The homogenate was centrifuged at 10,000 g for 15 min at 4°C, and the supernatant, which was equivalent to 40 μg of protein, was transferred to a 96-well microplate. Assay buffer and 2 μl of 2′,7′-dichlorodihydro-fluoroscein diacetate (DCHFDA; Molecular Probes, Inc., Eugene, OR) (5 mM in DMSO) were added to the microplate to give a total reaction volume of 200 μl. Subsequently, the samples were incubated at 37°C for 15 min. The fluorescence intensity was monitored for 30 min after excitation at 485 nm and emission at 535 nm by using a CytoFlor fluorescence plate reader (Perseptive Biosystems, Foster City, CA). The protein concentration in each sample was measured by using Bio-Rad Protein Assay Reagent (Bio-Rad Laboratories, Inc.), according to the manufacturer's instructions. The ROS level was expressed as relative fluorescence intensity units.
Apoptosis assays
Apoptosis was determined in mouse embryos 12 h after initial maternal ethanol exposure by using two independent assays, caspase-3 activation and Nile blue sulfate vital staining. Caspase-3 activation was analyzed with Western blot. For this assay, protein preparation and Western blot were conducted as described earlier. The activation of caspase-3 was analyzed with rabbit polyclonal against cleaved caspase-3 (Asp 175) (1:500; Cell Signaling Technology, Danvers, MA). The extent of apoptosis was expressed as fold change in caspase-3 activation over control. Caspase-3 activity was determined with fluorimetric assay by using the substrate Ac-DEVD-AMC (Caspase-3 Cellular Activity Assay Kit; Calbiochem, San Diego, CA) according to the manufacturer's instructions. Nile blue sulfate staining was carried out in whole embryos, as described previously (11). In brief, embryos were incubated in a 1:50,000 solution of Nile blue sulfate in Ringer's solution at 37°C for 30 min. Stained specimens were then washed in Ringer's solution and immediately photographed by using an Olympus dissecting photomicroscope. Both experimental and control embryos were processed simultaneously for visual comparison of patterns of cell death. Approximately 10 to 15 embryos for both experimental and control groups were examined.
Statistical analysis
Statistical analyses were performed by using StatView software (SAS Institute Inc., Cary, NC). All data are expressed as mean ± SEM of three separate experiments. Statistical comparisons between groups were analyzed with a one-way ANOVA. Multiple comparison post-tests between groups were conducted by using a Tukey test. Differences between control and treated groups, or between ethanol-treated group and other groups, were considered significant at p < 0.05.
Results
Exposure to ethanol and pretreatment with D3T in vivo result in a significant increase in the level of Nrf2 protein in mouse embryos
To examine the effects of ethanol on Nrf2 signaling in GD 8 mouse embryos, pregnant mice were administered two intraperitoneal (i.p.) doses of 25.0% (vol/vol) ethanol at a dosage of 2.9 g/kg maternal body weight on GD 8. This dose of ethanol produced a peak maternal blood alcohol level (BAL) of ~500 mg/dl 4.5 h after the initial dose. At 6 h after the first dose, the time at which Nrf2 protein level was analyzed, the maternal BAL is ~400 mg/dl (42). Western blot analysis revealed a moderate increase in the level of Nrf2 protein in mouse embryos exposed to ethanol in vivo (p < 0.05) (Fig. 1).
FIG. 1.
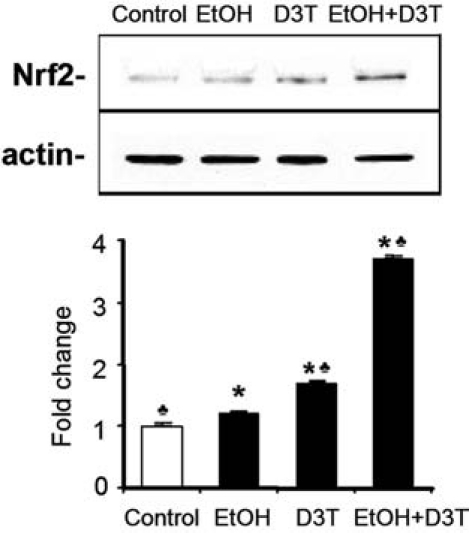
Treatment with ethanol and D3T increases the level of Nrf2 protein in mouse embryos. Western blot was performed to analyze the level of Nrf2 protein in GD 8 mouse embryos 6 h after exposure to vegetable oil or ethanol. Embryo lysates were prepared from whole embryos treated with vegetable oil (Control), 2.9 g/kg ethanol (EtOH), pretreated with 5 mg/kg D3T alone (D3T), or treated with both ethanol and D3T (EtOH+D3T). Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *p < 0.05 vs. control. ♣ p < 0.05 vs. EtOH.
A major focus of the current study was to examine the potential protective property of the Nrf2 inducer, D3T. To this end, the effects of in vivo D3T administration on the expression of Nrf2 and its downstream phase 2 enzymes and antioxidant proteins were determined. Pregnant mice were administered D3T 18 hours before ethanol treatment. Western blot analyses illustrated a 2.8-fold increase in Nrf2 protein in the embryos whose dams had been treated with both ethanol and D3T (Fig. 1).
Exposure to ethanol and pretreatment with D3T in vivo enhance Nrf2-ARE binding activity in mouse embryos
The antioxidant response element (ARE) is a transcriptional regulatory element involved in the activation of genes coding for a number of antioxidant proteins and phase 2 detoxifying enzymes. To further substantiate that treatment with ethanol and D3T can activate Nrf2, we examined the Nrf2-ARE binding activity in mouse embryos exposed to ethanol and D3T. EMSA with an oligonucleotide probe for mouse GCLC ARE showed an increased Nrf2-ARE binding activity in mouse embryos exposed to ethanol for 6 h. Pretreatment with D3T resulted in a significantly greater increase in Nrf2-ARE binding activity in ethanol-exposed mouse embryos than that in mouse embryos in control and ethanol-treated groups (Fig. 2). These data demonstrate that Nrf2 signaling is involved in ethanol-exposed mouse embryos and that it can be induced by Nrf2 inducer in mouse embryos.
FIG. 2.
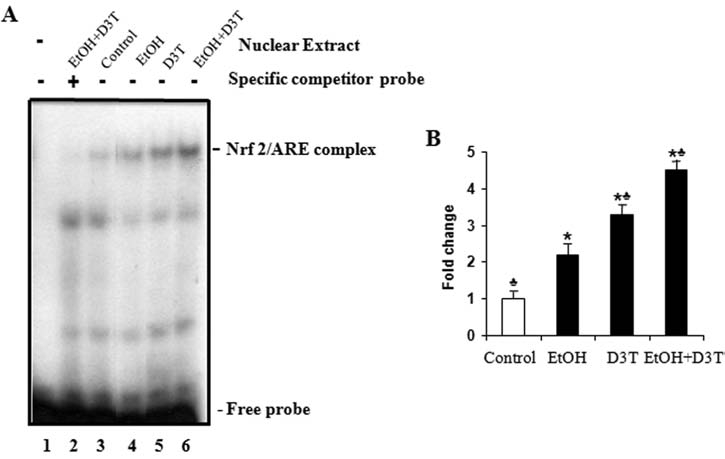
Ethanol and D3T exposure increase the Nrf2-ARE binding activity in mouse embryos. Nrf2-ARE binding was determined by using an EMSA with oligonucleotide probe for GCLC ARE in mouse embryos 6 h after exposure to ethanol. Embryo lysates were prepared from whole embryos treated with vegetable oil (Control), 2.9 g/kg ethanol (EtOH), pretreated with 5 mg/kg D3T alone (D3T), or treated with both ethanol and D3T (EtOH+D3T). (A) A representative gel-shift image. Lane 1, No nuclear extract. Lane 2, Nuclear extracts of embryos treated with ethanol and D3T in the presence of excess specific competitor probe. Lanes 3–6, Nuclear extracts of control and treated mouse embryos in the presence of labeled probe for GCLC ARE. (B) Bars represent the average values of the Nrf2-ARE binding activity of three gel-shift assays. Data represent the mean ± SEM of three separate experiments. *p < 0.05 vs. control. ♣ p < 0.05 vs. EtOH.
Ethanol and D3T exposure induce the expression of Nrf2 downstream antioxidant genes and proteins in mouse embryos
To examine the effects of ethanol on the expression of antioxidant genes that are downstream of Nrf2, mRNA and protein expression were analyzed by using real-time PCR and Western blot, respectively. As indicated by mRNA level, ethanol exposure for 6 h increased the expression of antioxidant genes, including cytoplasmic superoxide dismutase (SOD1), mitochondrial superoxide dismutase (SOD2), extracellular superoxide dismutase (SOD3), catalase (CAT), glutathione reductase (GSR), thioredoxin (TRX), and glutathione peroxidase 1 and 3 (Gpx1 and Gpx3). Except for SOD3, which was increased by threefold, changes in the mRNA level for the remainder of the genes tested in this study were less than onefold over control, indicating that ethanol treatment alone can induce only a moderate increase in antioxidant gene expression (Fig. 3). Western blot analysis also illustrated a moderate increase in the expression of SOD1, SOD2, and catalase protein (Fig. 4), confirming the corresponding changes in major antioxidant proteins that accompany mRNA induction.
FIG. 3.
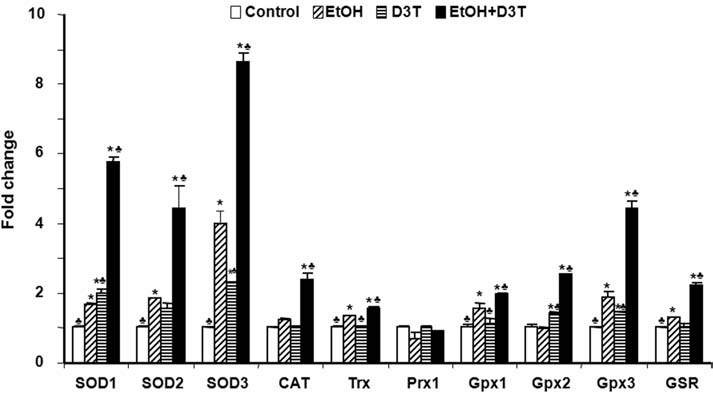
Ethanol and D3T exposure induce the expression of antioxidant genes in mouse embryos. mRNA expression of antioxidant genes was determined by real-time PCR in GD 8 mouse embryos treated with vegetable oil (Control), 2.9 g/kg ethanol (EtOH), pretreated with 5 mg/kg D3T (D3T), or treated with both ethanol and D3T (EtOH+D3T). All data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *p < 0.05 vs. control. ♣ p < 0.05 vs. EtOH.
FIG. 4.
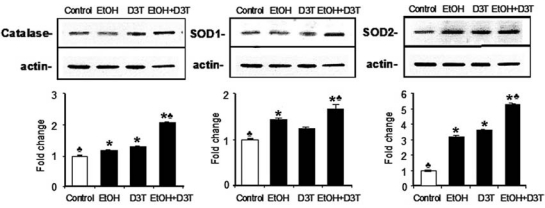
Treatment with ethanol and D3T induces the expression of antioxidant proteins. Western blot was performed to analyze the protein expression of catalase, SOD1, and SOD2 in GD 8 mouse embryos 6 hours after exposure to vegetable oil or ethanol. Embryo lysates were prepared from whole embryos treated with vegetable oil (Control), 2.9 g/kg ethanol (EtOH), pretreated with 5 mg/kg D3T alone (D3T), or treated with both ethanol and D3T (EtOH+D3T). Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *p < 0.05 vs. control. ♣ p < 0.05 vs. EtOH.
Pretreatment with D3T resulted in a significantly greater increase in mRNA and protein expression of antioxidants. Pretreatment with D3T significantly increased the mRNA expression of antioxidant genes, including SOD1, SOD2, SOD3, CAT, Trx, Gpx1, Gpx2, Gpx3, and GSR in ethanol-exposed embryos (p < 0.05). Among these antioxidant genes, the expression of SOD1, SOD2, SOD3, and Gpx3 was increased by four-, three-, seven-, and threefold, respectively, in ethanol-exposed embryos pretreated with D3T as compared with control (Fig. 3). Significant increases in expression of the major antioxidant proteins SOD1, SOD2, and catalase were also observed in ethanol-exposed mouse embryos pretreated with D3T (Fig. 4).
Exposure to ethanol and pretreatment with D3T in vivo result in a significant increase in the activities of antioxidant enzymes in mouse embryos
Nrf2 controls genes encoding phase 2 detoxifying enzymes and antioxidant proteins. The orchestrated induction of these proteins, which is referred to as an Nrf2-mediated defense response, is crucial for cells in counteracting the adverse effects of exogenous insults. To test whether upregulation of Nrf2 by ethanol or D3T can increase the catalytic activity of selected antioxidant enzymes in mouse embryos, the activities of two prototypic antioxidant enzymes, SOD and CAT, were analyzed. As shown in Fig. 5, ethanol exposure for 6 hours resulted in increases in the activities of SOD and CAT. Pretreatment with D3T resulted in a significantly greater increase in the activities of SOD and CAT in ethanol-exposed mouse embryos than the group treated with ethanol alone (Fig. 5), confirming the corresponding changes in the activities of major antioxidant enzymes that accompany mRNA and protein induction.
FIG. 5.
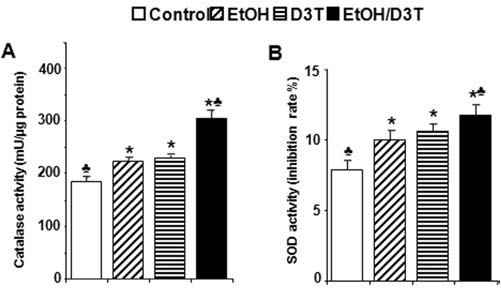
Ethanol and D3T exposure increase the activities of antioxidant enzymes, SOD, and catalase in mouse embryos. Activities of SOD and catalase were determined in GD 8 mouse embryos 6 h after exposure to ethanol. Embryo lysates were prepared from whole embryos treated with vegetable oil (Control), 2.9 g/kg ethanol (EtOH), pretreated with 5 mg/kg D3T alone (D3T), or treated with both ethanol and D3T (EtOH+D3T). Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *p < 0.05 vs. control. ♣ p < 0.05 vs. EtOH.
Ethanol exposure and pretreatment with D3T result in upregulation of genes encoding phase 2 detoxifying enzymes in mouse embryos
Phase 2 detoxifying enzymes are involved in the detoxification of electrophilic chemicals, including carcinogens and cardiovascular toxicants. To explore whether phase 2 enzymes can also be induced by in vivo ethanol exposure in early mouse embryos, mRNA expression of the genes encoding phase 2 enzymes was analyzed by using real-time PCR. As shown in Fig. 6, treatment of early mouse embryos with ethanol for 6 h in vivo increased the mRNA expression of NAD(P) H:quinone oxidoreductase-1(NQO-1) and glutathione S-transferase (GSTa2, GSTm2, GSTm3, and GSTp1). However, similar to ethanol-induced changes in antioxidant gene expression, for most of these genes, the changes in the mRNA level caused by ethanol alone were less than or close to onefold over control. A significantly greater increase in the mRNA levels of the genes encoding phase 2 enzymes was observed in ethanol-exposed embryos pretreated with D3T as compared with both the control group and the groups treated with ethanol alone (p < 0.05) (Fig. 6).
FIG. 6.
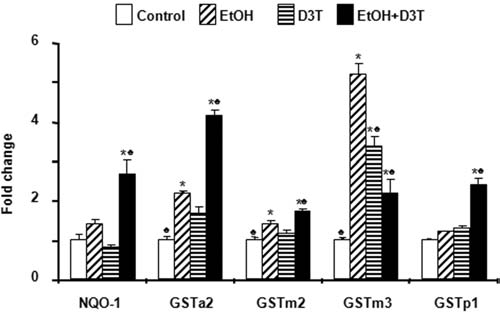
Exposure to ethanol and D3T results in upregulation of genes encoding phase 2 detoxifying enzymes in mouse embryos. mRNA expression of phase 2 detoxifying genes was determined by real-time PCR in GD 8 mouse embryos treated with vegetable oil (Control), 2.9 g/kg ethanol (EtOH), pretreated with 5 mg/kg D3T (D3T), or treated with both ethanol and D3T (EtOH+D3T). Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *p < 0.05 vs. control. ♣ p < 0.05 vs. EtOH.
D3T pretreatment significantly reduces ROS generation in mouse embryos exposed to ethanol in vivo
For analysis of the effects of D3T pretreatment on ROS generation in mouse embryos exposed to ethanol in vivo, a 2′,7′-dichlorodihydro-fluoroscein diacetate (DCHFDA) assay was used. Exposure of mouse embryos to ethanol for 6 h in vivo significantly increased ROS generation, confirming that ethanol can induce ROS generation in early mouse embryos. Pretreatment with D3T significantly reduced ROS generation in mouse embryos exposed to ethanol in vivo (Fig. 7), supporting the premise that chemically induced transcriptional activation of Nrf2 and subsequent induction of detoxifying and antioxidant proteins can reduce ethanol-induced oxidative stress in early mouse embryos.
FIG. 7.
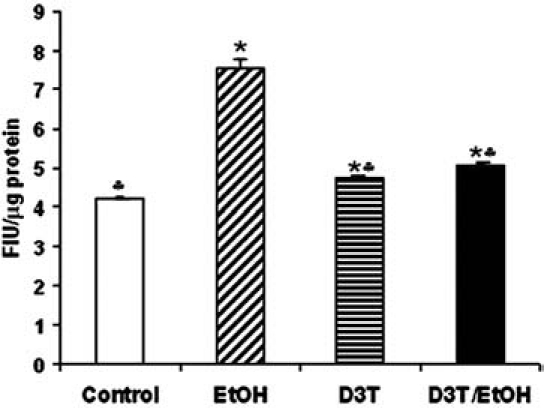
D3T pretreatment significantly reduced ROS generation in mouse embryos exposed to ethanol in vivo. ROS generation was measured in GD 8 mouse embryos 6 h after exposure to vegetable oil (Control), 2.9 g/kg ethanol (EtOH), pretreatment with 5 mg/kg D3T alone (D3T), or treatment with both ethanol and D3T (EtOH+D3T). Data are expressed as relative fluorescence intensity units and represent the mean ± SEM of three separate experiments. *p < 0.05 vs. control. ♣ p < 0.05 vs. EtOH.
D3T pretreatment prevents apoptosis in mouse embryos exposed to ethanol in vivo
To determine the effects of D3T treatment on ethanol-induced apoptosis in early mouse embryos, the effect of D3T on the activation of caspase-3 was examined. As shown in Fig. 8A, in control embryos, caspase-3 exists as procaspase-3 (37 kDa), and little active caspase-3 was observed. In vivo ethanol treatment induced the activation of caspase-3 in mouse embryos, as evidenced by a twofold increase in the expression of its 17-kDa subunit and also resulted in a significant increase in the activity of this protease (Fig. 8B). Pretreatment with D3T resulted in a significant reduction in the activation and activity of caspase-3 in ethanol-exposed mouse embryos as compared with the group treated with ethanol alone.
FIG. 8.
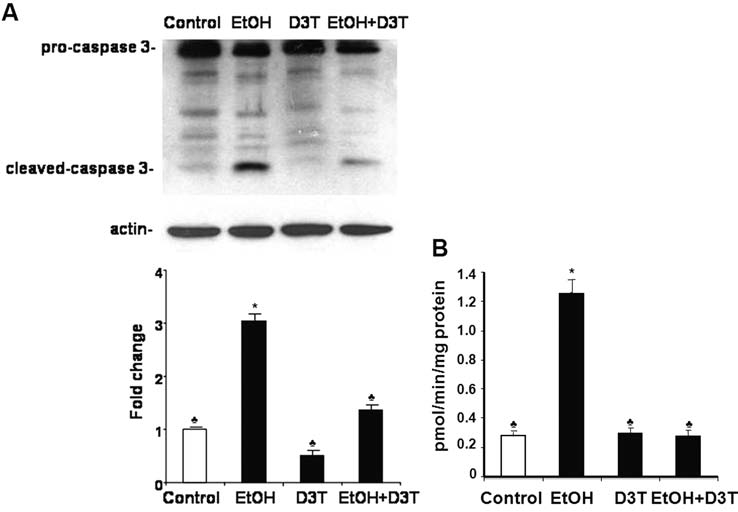
Pretreatment with D3T decreases the activation and activity of caspase-3 in mouse embryos exposed to ethanol in vivo. Western blot analyses and fluorimetric assays were performed to examine the activation (A) and activity (B) of caspase-3, respectively, in GD 8 mouse embryos 12 h after exposure to vegetable oil or ethanol. Embryo lysates were prepared from whole embryos treated with vegetable oil (Control), 2.9 g/kg ethanol (EtOH), pretreated with 5 mg/kg D3T alone (D3T), or treated with both ethanol and D3T (EtOH+D3T). Data are expressed as fold change over control and represent the mean ± SEM of three separate experiments. *p < 0.05 vs. control. ♣ p < 0.05 vs. EtOH.
Although the caspase-3 activation assay allows rapid quantification of apoptosis, it precludes localization of apoptosis in whole embryos. For this, Nile blue sulfate staining was used in conjunction with caspase-3 activation assay to confirm the occurrence and to determine the locations of apoptosis in whole embryos. As expected, compared with the untreated and D3T-treated control embryos, those exposed to ethanol showed excessive Nile blue sulfate staining. The pattern and amount of stain uptake in the ethanol-exposed embryos was typical of that previously reported for this exposure paradigm (14). Pretreatment with D3T dramatically reduced the ethanol-induced increase in Nile blue sulfate staining (Fig. 9), indicating that pretreatment with D3T is, indeed, effective in preventing ethanol-induced apoptosis in mouse embryos.
FIG. 9.
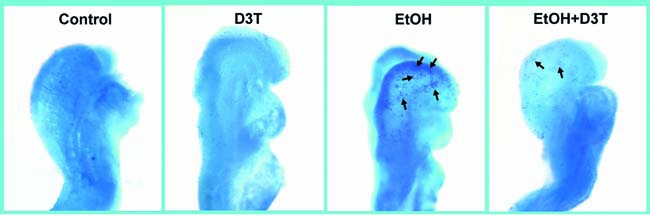
D3T pretreatment reduces ethanol-induced apoptosis. Nile blue sulfate vital staining of comparably staged mouse embryos treated with vegetable oil (Control), pretreated with D3T alone (D3T), treated with ethanol alone (EtOH), or treated with both ethanol and D3T (EtOH+D3T) illustrates excessive stain uptake in mouse embryos exposed to ethanol alone (arrows are directed toward sites of specific uptake in the neural folds and facial region of GD 8 embryos). Pretreatment with D3T results in diminished staining in ethanol-exposed mouse embryos. Shown are representatives of 10 to 15 embryos from several different litters of each group. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Discussion
Utilizing a mouse FASD model, this study provided the first evidence that in vivo ethanol exposure can activate the Nrf2 pathway in early embryos. Activation of Nrf2 results in upregulation of Nrf2 downstream target genes, including those that encode phase 2 detoxifying enzymes and antioxidant proteins such as NQO1, GST, SOD, CAT, TRX, GPx, and GSR. In addition to gene expression changes, Western blot analysis revealed a moderate increase in the expression of Nrf2 and the antioxidant proteins SOD1, SOD2, and catalase, as well as the activities of the antioxidant enzymes, SOD, and catalase in ethanol-exposed mouse embryo. Because exposure of embryos to ethanol results in the generation of ROS, which are known to activate Nrf2 (21), the observed Nrf2 activation was expected. This response is not unique to ethanol-exposed embryos. Similar effects have been observed in cells treated with a numbers of other toxic chemicals, including heavy metals (19, 23), cigarette smoke (22), and arachidonic acid (17). Of particular interest to this study is that an increase in Nrf2 protein has also been observed in livers and hepatocytes of alcohol-fed mice and rats (16).
Most eukaryotic cells possess inherent antioxidant defense mechanisms that include both nonenzymatic neutralization and enzymatic detoxification of oxygen-derived radicals and other reactive species (15). However, early mouse embryos might be expected to have only minimal antioxidant capabilities because the amount of O2 that they metabolize is very low. Indeed, studies by Davis et al. (13) provided evidence that neural crest cells cultured from chick embryos have a deficiency of SOD. The present study showed that Nrf2 is activated in response to ethanol challenge, leading to the transcriptional induction of many antioxidant genes. The teratogenic outcome of ethanol exposure is expected to be determined by the balance between the induced ROS production and the antioxidant capability, as regulated by Nrf2. The fact that excessive apoptosis and dysmorphology result from the exposure regimen used for the present investigation (11, 14) suggests that the ethanol-induced activation of Nrf2 and its downstream antioxidants is an adaptive response that is insufficient to be protective. This is supported by this study's finding that ethanol can induce only a moderate increase in antioxidant and detoxifying gene expression.
One of the promising strategies for protection against oxidative stress that has received considerable attention is supplementation with exogenous antioxidants. We showed previously that exogenous antioxidants, including SOD, catalase, vitamin E, N-acetylcysteine, and EUK-134, a synthetic SOD and catalase mimetic, significantly diminish apoptosis and subsequent dysmorphology in early mouse embryos exposed to ethanol in vitro and in vivo (8–10, 24). Additionally, Spong et al. (38) have shown that NAP, a neuroprotective peptide that has been shown to exhibit antioxidant actions, can decrease ethanol-induced depletion of reduced glutathione and fetal death. However, the protection provided by exogenous antioxidants is not complete. In addition to the fact that teratogenic mechanisms other than oxidative damage may be involved in the teratogenic effects of ethanol, the incomplete protection points to the limitations associated with the use of exogenous antioxidants. The therapeutic efficacy of exogenous antioxidants may be limited by the facts that (a) their systemic delivery might not provide adequate concentrations in the target tissue(s), (b) many antioxidants have very limited passage through a variety of cellular structures and barriers, and (c) limited numbers of exogenous antioxidants can be simultaneously administrated to embryos. In this regard, a more promising strategy for protecting against oxidative injury and ethanol's teratogenesis may be through coordinated upregulation by chemical inducers of a broad spectrum of endogenous antioxidants/phase 2 enzymes in embryonic tissue. As shown by the results of the current study, D3T is effective in this role.
D3T was shown in this study to induce Nrf2 potently in mouse embryos. It significantly increased Nrf2 protein levels and Nrf2-ARE binding activity, and strongly induced the mRNA expression of Nrf2 downstream target genes. Pretreatment with D3T also increased the expression of antioxidant proteins and the activities of antioxidant enzymes, and significantly reduced ROS generation in mouse embryos exposed to ethanol in vivo. In addition, it significantly decreased ethanol-induced activation and activity of caspase-3. Caspase-3 is a key effector in the apoptosis pathway, amplifying the signal from initiator caspases; with its activation signifying full commitment to cellular disassembly (41). The presence of activated caspase-3 has been identified as a marker for active apoptosis (2). Caspase-3 activation has been observed in apoptosis caused by various inducers, including ethanol. For example, ethanol exposure of 7-day postnatal mice has previously been shown to result in caspase-3 activation in a pattern that corresponds to the pattern of apoptotic neurodegeneration (20, 33). The significant decrease in ethanol-induced caspase-3 activation and activity in mouse embryos pretreated with D3T suggests that this compound can reduce ethanol-induced apoptosis in early mouse embryos. This was shown to be the case, as indicated by dramatically reduced Nile blue sulfate staining in ethanol-exposed embryos pretreated with D3T. These results demonstrated that this Nrf2 inducer is indeed effective in preventing oxidative stress and apoptosis in mouse embryos exposed to ethanol in vivo.
In conclusion, this study demonstrates that Nrf2 signaling is involved in the induction of antioxidants and detoxifying enzymes in ethanol-exposed mouse embryos. These findings provide important information regarding the molecular mechanisms of ROS signaling and the endogenous cellular antioxidant defense systems involved in coping with ethanol-induced oxidative stress and cell damage. Although further study regarding the developmental toxicology of D3T is required, the potency of this agent in inducing antioxidant response as well as in preventing ethanol-induced apoptosis suggests that further exploration of its antiteratogenic effect may yield novel strategies for prevention of ethanol-induced birth defects.
Acknowledgments
This work is supported by NIH grants AA017446 (S.-Y.C.), AA013908 (S.-Y.C.), AA11605 (K.K.S), and AA12974 from the National Institute on Alcohol Abuse and Alcoholism.
Abbreviations
ARE, antioxidant response element; CAT, catalase; D3T, 3H-1, 2 dithiole-3-thione; FAS, fetal alcohol syndrome; FASDs, fetal alcohol spectrum disorders; GD, gestational day; Gpx, glutathione peroxidase; GSR, glutathione reductase; GST, glutathione S-transferase; MPTP, 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine; NCCs, neural crest cells; NQO-1, NAD(P)H:quinone oxidoreductase-1; Nrf2, nuclear factor erythroid 2–related factor 2; ROS, reactive oxygen species; SOD1, cytoplasmic superoxide dismutase; SOD2, mitochondrial superoxide dismutase; SOD3, extracellular superoxide dismutase; TRX, thioredoxin; TUNEL, terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling.
References
- 1.Abel EL. Hannigan JH. Maternal risk factors in fetal alcohol syndrome: provocative and permissive influences. Neurotoxicol Teratol. 1995;17:445–462. doi: 10.1016/0892-0362(95)98055-6. [DOI] [PubMed] [Google Scholar]
- 2.Abu-Qare AW. Abou-Donia MB. Biomarkers of apoptosis: release of cytochrome c, activation of caspase-3, induction of 8-hydroxy-2′-deoxyguanosine, increased 3-nitrotyrosine, and alteration of p53 gene. J Toxicol Environ Health B Crit Rev. 2001;4:313–332. doi: 10.1080/109374001301419737. [DOI] [PubMed] [Google Scholar]
- 3.Bonthius DJ. McKim RA. Koele L. Harb H. Kehrberg AH. Mahoney J. Karacay B. Pantazis NJ. Severe alcohol-induced neuronal deficits in the hippocampus and neocortex of neonatal mice genetically deficient for neuronal nitric oxide synthase (nNOS) J Comp Neurol. 2006;499:290–305. doi: 10.1002/cne.21095. [DOI] [PubMed] [Google Scholar]
- 4.Burton NC. Kensler TW. Guilarte TR. In vivo modulation of the parkinsonian phenotype by Nrf2. Neurotoxicology. 2006;27:1094–1100. doi: 10.1016/j.neuro.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 5.Cao Z. Zhu H. Zhang L. Zhao X. Zweier JL. Li Y. Antioxidants and phase 2 enzymes in cardiomyocytes: chemical inducibility and chemoprotection against oxidant and simulated ischemia-reperfusion injury. Exp Biol Med (Maywood) 2006;231:1353–1364. doi: 10.1177/153537020623100809. [DOI] [PubMed] [Google Scholar]
- 6.Chan K. Han XD. Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci U S A. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheema ZF. West JR. Miranda RC. Ethanol induces Fas/Apo [apoptosis]-1 mRNA and cell suicide in the developing cerebral cortex. Alcohol Clin Exp Res. 2000;24:535–543. [PubMed] [Google Scholar]
- 8.Chen SY. Dehart DB. Sulik KK. Protection from ethanol-induced limb malformations by the superoxide dismutase/catalase mimetic, EUK-134. FASEB J. 2004;18:1234–1236. doi: 10.1096/fj.03-0850fje. [DOI] [PubMed] [Google Scholar]
- 9.Chen SY. Sulik KK. Free radicals and ethanol-induced cytotoxicity in neural crest cells. Alcohol Clin Exp Res. 1996;20:1071–1076. doi: 10.1111/j.1530-0277.1996.tb01948.x. [DOI] [PubMed] [Google Scholar]
- 10.Chen SY. Sulik KK. Iron-mediated free radical injury in ethanol-exposed mouse neural crest cells. J Pharmacol Exp Ther. 2000;294:134–140. [PubMed] [Google Scholar]
- 11.Chen SY. Wilkemeyer MF. Sulik KK. Charness ME. Octanol antagonism of ethanol teratogenesis. FASEB J. 2001;15:1649–1651. doi: 10.1096/fj.00-0862fje. [DOI] [PubMed] [Google Scholar]
- 12.Cho HY. Jedlicka AE. Reddy SP. Zhang LY. Kensler TW. Kleeberger SR. Linkage analysis of susceptibility to hyperoxia: Nrf2 is a candidate gene. Am J Respir Cell Mol Biol. 2002;26:42–51. doi: 10.1165/ajrcmb.26.1.4536. [DOI] [PubMed] [Google Scholar]
- 13.Davis WL. Crawford LA. Cooper OJ. Farmer GR. Thomas D. Freeman BL. Generation of radical oxygen species by neural crest cells treated in vitro with isotretinoin and 4-oxoisotretinoin. J Craniofac Genet Dev Biol. 1990;10:295–310. [PubMed] [Google Scholar]
- 14.Dunty WC., Jr Chen SY. Zucker RM. Dehart DB. Sulik KK. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol Clin Exp Res. 2001;25:1523–1535. [PubMed] [Google Scholar]
- 15.Fridovich I. Oxygen: aspects of its toxicity and elements of defense. Curr Eye Res. 1984;3:1–2. doi: 10.3109/02713688408997181. [DOI] [PubMed] [Google Scholar]
- 16.Gong P. Cederbaum AI. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology. 2006;43:144–153. doi: 10.1002/hep.21004. [DOI] [PubMed] [Google Scholar]
- 17.Gong P. Cederbaum AI. Transcription factor Nrf2 protects HepG2 cells against CYP2E1 plus arachidonic acid-dependent toxicity. J Biol Chem. 2006;281:14573–14579. doi: 10.1074/jbc.M600613200. [DOI] [PubMed] [Google Scholar]
- 18.Green ML. Singh AV. Zhang Y. Nemeth KA. Sulik KK. Knudsen TB. Reprogramming of genetic networks during initiation of the fetal alcohol syndrome. Dev Dyn. 2007;236:613–631. doi: 10.1002/dvdy.21048. [DOI] [PubMed] [Google Scholar]
- 19.He X. Lin GX. Chen MG. Zhang J. Ma Q. Protection against chromium (VI)-induced oxidative stress and apoptosis by Nrf2: recruiting Nrf2 into the nucleus and disrupting the nuclear Nrf2/Keap1 association. Toxicol Sci. 2007;98:298–309. doi: 10.1093/toxsci/kfm081. [DOI] [PubMed] [Google Scholar]
- 20.Ieraci A. Herrera DG. Nicotinamide protects against ethanol-induced apoptotic neurodegeneration in the developing mouse brain. PLoS Med. 2006;3:e101. doi: 10.1371/journal.pmed.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kensler TW. Wakabayashi N. Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 22.Knorr-Wittmann C. Hengstermann A. Gebel S. Alam J. Muller T. Characterization of Nrf2 activation and heme oxygenase-1 expression in NIH3T3 cells exposed to aqueous extracts of cigarette smoke. Free Radic Biol Med. 2005;39:1438–1448. doi: 10.1016/j.freeradbiomed.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Korashy HM. El Kadi AO. Transcriptional regulation of the NAD(P)H:quinone oxidoreductase 1 and glutathione S-transferase ya genes by mercury, lead, and copper. Drug Metab Dispos. 2006;34:152–165. doi: 10.1124/dmd.105.005397. [DOI] [PubMed] [Google Scholar]
- 24.Kotch LE. Chen SY. Sulik KK. Ethanol-induced teratogenesis: free radical damage as a possible mechanism. Teratology. 1995;52:128–136. doi: 10.1002/tera.1420520304. [DOI] [PubMed] [Google Scholar]
- 25.Kotch LE. Sulik KK. Experimental fetal alcohol syndrome: proposed pathogenic basis for a variety of associated facial and brain anomalies. Am J Med Genet. 1992;44:168–176. doi: 10.1002/ajmg.1320440210. [DOI] [PubMed] [Google Scholar]
- 26.Kwak MK. Egner PA. Dolan PM. Ramos-Gomez M. Groopman JD. Itoh K. Yamamoto M. Kensler TW. Role of phase 2 enzyme induction in chemoprotection by dithiolethiones. Mutat Res. 2001:480–481. doi: 10.1016/s0027-5107(01)00190-7. 305–315. [DOI] [PubMed] [Google Scholar]
- 27.Lee JM. Calkins MJ. Chan K. Kan YW. Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 28.Lee JM. Shih AY. Murphy TH. Johnson JA. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem. 2003;278:37948–37956. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- 29.Manandhar S. Cho JM. Kim JA. Kensler TW. Kwak MK. Induction of Nrf2-regulated genes by 3H-1, 2-dithiole-3-thione through the ERK signaling pathway in murine keratinocytes. Eur J Pharmacol. 2007;577:17–27. doi: 10.1016/j.ejphar.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 30.Morito N. Yoh K. Itoh K. Hirayama A. Koyama A. Yamamoto M. Takahashi S. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene. 2003;22:9275–9281. doi: 10.1038/sj.onc.1207024. [DOI] [PubMed] [Google Scholar]
- 31.Munday R. Munday CM. Induction of phase II enzymes by 3H-1,2-dithiole-3-thione: dose-response study in rats. Carcinogenesis. 2004;25:1721–1725. doi: 10.1093/carcin/bgh162. [DOI] [PubMed] [Google Scholar]
- 32.Nair S. Li W. Kong AN. Natural dietary anti-cancer chemopreventive compounds: redox-mediated differential signaling mechanisms in cytoprotection of normal cells versus cytotoxicity in tumor cells. Acta Pharmacol Sin. 2007;28:459–472. doi: 10.1111/j.1745-7254.2007.00549.x. [DOI] [PubMed] [Google Scholar]
- 33.Olney JW. Tenkova T. Dikranian K. Muglia LJ. Jermakowicz WJ. D'Sa C. Roth KA. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol Dis. 2002;9:205–219. doi: 10.1006/nbdi.2001.0475. [DOI] [PubMed] [Google Scholar]
- 34.Otieno MA. Kensler TW. Guyton KZ. Chemoprotective 3H-1,2-dithiole-3-thione induces antioxidant genes in vivo. Free Radic Biol Med. 2000;28:944–952. doi: 10.1016/s0891-5849(00)00175-1. [DOI] [PubMed] [Google Scholar]
- 35.Ozawa M. Matsuzuka T. Hirabayashi M. Kanai Y. Redox status of the oviduct and CDC2 activity in 2-cell stage embryos in heat-stressed mice. Biol Reprod. 2004;71:291–296. doi: 10.1095/biolreprod.103.022152. [DOI] [PubMed] [Google Scholar]
- 36.Ramos-Gomez M. Dolan PM. Itoh K. Yamamoto M. Kensler TW. Interactive effects of nrf2 genotype and oltipraz on benzópyrene-DNA adducts and tumor yield in mice. Carcinogenesis. 2003;24:461–467. doi: 10.1093/carcin/24.3.461. [DOI] [PubMed] [Google Scholar]
- 37.Smith SM. Alcohol-induced cell death in the embryo. Alcohol Health Res World. 1997;21:287–295. [PMC free article] [PubMed] [Google Scholar]
- 38.Spong CY. Abebe DT. Gozes I. Brenneman DE. Hill JM. Prevention of fetal demise and growth restriction in a mouse model of fetal alcohol syndrome. J Pharmacol Exp Ther. 2001;297:774–779. [PubMed] [Google Scholar]
- 39.Stratton KR, editor; Howe CJ, editor; Battaglia FC, editor. Fetal alcohol syndrome: diagnosis, epidemiology, prevention, and treatment. Washington, DC: National Academy Press; 1996. [Google Scholar]
- 40.Sulik KK. Cook CS. Webster WS. Teratogens and craniofacial malformations: relationships to cell death. Development. 1988;103:213–217. doi: 10.1242/dev.103.Supplement.213. [DOI] [PubMed] [Google Scholar]
- 41.Thornberry NA. Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 42.Webster WS. Walsh DA. McEwen SE. Lipson AH. Some teratogenic properties of ethanol and acetaldehyde in C57BL/6J mice: implications for the study of the fetal alcohol syndrome. Teratology. 1983;27:231–243. doi: 10.1002/tera.1420270211. [DOI] [PubMed] [Google Scholar]