

Abstract
Autophagy is a regulated catabolic process triggered in cells deprived of nutrients or growth factors that govern nutrient uptake. Here we report that autophagy is induced by cetuximab, a therapeutic antibody that blocks EGFR function. Cancer cell treatment with cetuximab triggered autophagosome formation, conversion of microtubule-associated protein 1 light chain 3 from its cytoplasmic to membrane-associated form, and increased acidic vesicular organelle formation. Autophagy occurred when cetuximab inhibited the class I PI3K/Akt/mTOR pathway, but not when it inhibited only the MEK/Erk pathway, and it was accompanied by decreased levels of hypoxia inducible factor-1 alpha (HIF-1α) and Bcl-2. Stable overexpression of a HIF-1α mutant prevented cetuximab-induced autophagy and decrease in Bcl-2 levels. Knockdown of autophagy regulator beclin 1 or cell treatment with autophagy inhibitor 3-methyladenine, a class III PI3K (hVps34) inhibitor, also inhibited cetuximab-induced autophagy. Furthermore, knockdown of beclin 1 or Atg7 or treatment with the lysosome inhibitor chloroquine sensitized cancer cells to cetuximab-induced apoptosis. Mechanistic analysis argued that cetuximab acted by promoting an association between beclin 1 and hVps34, which was inhibited by overexpression of Bcl-2. Our findings suggest that the autophagy protects cancer cells from the pro-apoptotic effects of cetuximab.
Keywords: cetuximab, autophagy, EGFR
Introduction
Macroautophagy (hereafter referred to as autophagy) is a conserved and tightly regulated cellular catabolic process that involves the lysosomal degradation pathway (1, 2). By selectively recycling macromolecules and organelles, autophagy is an integral part of normal cellular function, helping cells survive under starvation conditions to maintain cell growth and the development and homeostasis of organisms (3). When cells lack nutrients or are deprived of growth factors (which govern the uptake of nutrients), autophagy is rapidly induced to fuel the cells’ bioenergetics to prevent cell death. In such circumstances, inhibiting autophagy results in accelerated cell death via apoptosis (4, 5).
Autophagy is morphologically characterized by the appearance of “double-membrane” vacuoles (autophagosomes) in the cytoplasm. The discovery of autophagy-related genes (Atg) and their involvement in the formation of autophagosomes in yeast has greatly increased our knowledge of the molecular basis of autophagy (6). In yeast, all Atg genes have been shown to act downstream of an important serine/threonine kinase, target of rapamycin (TOR) (7). Autophagy is now known to be regulated by at least two pathways —the class I and class III phosphoinositide 3-kinase (PI3K) pathways (2). In mammalian cells, the mammalian TOR (mTOR) complex 1 of the class I PI3K pathway plays an important role upstream of the Atg1 complex, which regulates different steps in autophagosome formation; in contrast, the beclin 1 (Atg6)/hVps34 [mammalian Vps (vacuolar protein sorting) 34 homologue] complex of the class III PI3K pathway is essential for initiating autophagy (8, 9), which is activated by co-factors such as the ultraviolet irradiation resistance-associated tumor suppressor gene (UVRAG) (10) and is inhibited by some interacting proteins, including the B-cell leukemia/lymphoma-2 gene (Bcl-2) and a related protein, Bcl-XL (11, 12).
Autophagy has elicited great interest and attention from cancer biologists and oncologists in recent years because of the increasing evidence indicating that autophagy, in addition to its normal role in cell physiology, is closely linked to both tumorigenesis and cancer cell response to treatments (13, 14). Beclin 1 is commonly deleted in approximately 40% of prostate cancers, 50% of breast cancers, and 75% of ovarian cancers, suggesting it plays a tumor suppressor role in cancer development (8, 9, 15). Monoallelic deletion of beclin 1/Atg6 in mice enhanced tumorigenesis, leading to development of carcinomas of the lung and liver and lymphomas (16). On the other hand, autophagy is detected in cancer cells after various types of treatment, such as anti-estrogens and ionizing radiation (13, 14, 17). Whether the anticancer treatment-induced autophagy leads to cancer cell death or protects cancer cells from the treatments remains controversial, but it appears that the outcome depends highly on the specific context of the cancer cells and treatment types, such as tumor environment and treatment characteristics (2).
The epidermal growth factor receptor (EGFR) is frequently overexpressed in several human cancers of epithelial origin and plays important roles in the development and progression of these cancers. Targeting EGFR with receptor-blocking monoclonal antibodies or small-molecule tyrosine kinase inhibitors has had therapeutic effects, which led to FDA approval for treating several types of human cancer, including head and neck and colorectal cancers. Among several pathways downstream of EGFR that are inhibited pharmacologically by EGFR-targeting agents, downregulation of the hypoxia-inducible factor-1 alpha (HIF-1α) resulting from inhibition of the class-I PI3K/Akt pathway has specifically been suggested to play a major role in mediating the antitumor activity of cetuximab, an EGFR-blocking antibody (18–20). Because the class I PI3K pathway is known to suppress the occurrence of autophagy through mTOR activation, we reasoned that there might be a link between response to cancer cells to EGFR-targeted therapy and induction of autophagy in the cells. To date, no studies have been published reporting whether inhibiting EGFR via therapeutic intervention is linked to occurrence of autophagy in cancer cells and what biological significance the induction of autophagy might have on cancer cells’ response to the treatment.
In this study, we tested our hypothesis by a combination of transmission electron microscopy, fluorescent microscopy, flow cytometry and Western blotting to detect autophagy in several types of cancer cells after cetuximab treatment. We also explored the biochemical mechanisms by which autophagy was induced after cetuximab treatment and the biological significances of the autophagy. Our findings suggest that autophagy occurring after cetuximab treatment likely protects cancer cells against apoptosis resulting from deprivation of growth signaling from the EGFR pathway. Novel approaches that inhibit autophagy may therefore be combined with cetuximab to enhance cancer cells’ therapeutic response.
Materials and Methods
Reagents
Cetuximab was a gift from ImClone Systems Inc. LY294002 was purchased from Calbiochem/EMD Chemicals, Inc.. The antibodies for Western blotting and immunoprecipitation and their relevant sources are as follows: HIF-1α (BD Biosciences Pharmingen); the microtubule-associated protein 1 light chain 3 (LC3) (Novus Biologicals); beclin 1 and Bcl-2 (Santa Cruz Biotechnology); hVps34 (Echelon Biosciences); total and S473-phosphorylated Akt, total and T202/Y204-phosphorylated extracellular signal-activated kinase (Erk), total and S2448-phosphorylated mTOR, total and S371-phosphorylated p70S6K, poly(ADP-ribose) polymerase (PARP), caspase 3, and caspase 7 (Cell Signaling Technology, Inc.). All other chemicals were purchased from Sigma-Aldrich Corp..
Cell lines and cell culture
A431 human vulvar squamous carcinoma cells, DiFi colorectal adenocarcinoma cells, and HCC827 human non-small cell lung cancer cells were described previously (18–24). All cell lines were grown and maintained in Dulbecco’s modified Eagle’s medium or Ham’s F12 medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin and incubated in a humidified atmosphere (95% air and 5% CO2) at 37°C.
cDNA constructs and transfection
cDNA expression constructs containing myristoylated PI3K (Myr-PI3K), myristoylated Akt (Myr-Akt), constitutively active mitogen-activated protein kinase kinase 1 (MEK1) (S217E/S221D), and a HIF-1α oxygen-dependent degradation domain (ODD) deletion mutant (HIF-1α/Δ ODD) were previously described (20, 25). Bcl-2 and Rheb constructs were ordered from Addgene. The GFP-tagged LC3 cDNA expression construct was a gift from Dr. Noboru Mizushima (Tokyo Medical and Dental University, Tokyo, Japan). Transfection of these constructs was performed with Lipofectamine 2000 (Invitrogen).
siRNA and transfection
Two 21-mer oligonucleotide siRNA duplexes targeting beclin 1 (sense sequence: CAGAUACUCUUUUAGACCAtt; antisense sequence: UGGUCUAAAAGAGUAUCUGtg) and Atg7 (sense sequence: GGAACACUGUAUAACACCAtt; antisense sequence: UGGUGUUAUACAGUGUUCCaa), respectively, and a negative control siRNA were acquired from Ambion, Inc.. In pilot experiments, we tested different concentrations of siRNA oligonucleotides with different time intervals of transfection using Lipofectamine 2000. We judged the transfection efficiency based on the uptake of fluorescein isothiocyanate-conjugated oligonucleotides and determined the efficiency of siRNA-mediated protein knockdown by Western blotting analysis.
Immunoprecipitation and Western blotting
Cultured cells were harvested with a rubber scraper and washed twice with cold phosphate-buffered saline (PBS). Cell pellets were lysed and kept on ice for at least 10 min in a buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% Nonidet P-40, 50 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 25 μg/ml leupeptin, and 25 μg/ml aprotinin. The lysates were cleared by centrifugation, and the supernatants were collected. For immunoprecipitation studies, cell lysates were incubated with primary antibodies, and the resulting immune complexes were precipitated with protein A-Sepharose beads (Amersham Biosciences). Whole-cell lysates or immunoprecipitated proteins were then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and subjected to Western blotting with the primary antibodies and horseradish peroxidase (HRP)-labeled secondary antibodies. We visualized the signals with an enhanced chemiluminescence (ECL) detection kit (Amersham Biosciences).
Cell proliferation assay
After desired treatment in cell culture, the cells were incubated in 24-well plates for 2 h at 37°C in a CO2 incubator with 50 μl/well of 10 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and were then lysed with a lysis buffer (500 μl/well) containing 20% SDS in dimethyl formamide/H2O (1:1, v/v; pH 4.7) at 37°C for at least 6 h. We determined the number of surviving cells in each group by measuring the cell lysates’ optical density (OD) at an absorbance wavelength of 570 nm. The OD value in each treatment group was then normalized to that of untreated cells as a percentage of the OD value of the control and plotted against the treatments.
Apoptosis assay
We measured apoptosis by using an enzyme-linked immunosorbent assay (ELISA) kit (Roche Diagnostics Corp.) that quantitatively measures cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) and by Western blot analysis with antibodies that recognize cleaved PARP, caspase 3 or caspase 7 after various treatments, as we previously reported (26, 27).
Transmission electron microscopy
After fixation with a solution containing 3% glutaraldehyde plus 2% paraformaldehyde in 0.1 M PBS (pH 7.3) for 1 h, cell suspension samples were washed and treated with 0.1% buffered osmium tetroxide for 30 min, stained en bloc with 1% Millipore-filtered uranyl acetate, dehydrated in increasing concentrations of ethanol, infiltrated, and embedded in LX-112 medium. Next, the samples were polymerized in a 70oC oven for 2 d. Ultrathin sections were cut with a Leica Ultracut microtome (Leica), stained with uranyl acetate and lead citrate in a Leica EM stainer, and examined with a JEM 1010 transmission electron microscope (JEOL, USA, Inc.) at an accelerating voltage of 80 kV. Digital images were obtained using an AMT imaging system (Advanced Microscopy Techniques Corp.).
Detection and quantification of acidic vesicular organelles(AVOs) with acridine orange vitality staining
We subjected cells to vital staining with acridine orange (1 μg/ml)for 15 min and then examined them under a fluorescence microscope (28). To quantify the amount of AVOs formed, we removed the cells from the culture plate with trypsin-EDTA and analyzed them using a fluorescence-activated cell sorting FACScan flow cytometer and CellQuest software.
Results
Autophagy is induced in cancer cells after cetuximab treatment
Autophagy was induced upon cetuximab treatment in three cancer cell lines examined, including A431 vulvar squamous carcinoma cells, DiFi colorectal adenocarcinoma cells, and HCC827 lung adenocarcinoma cells, all of which are known to be sensitive to cetuximab (Fig. 1). Transmission electron microscopy revealed abundant characteristic autophagosomes in all three cell lines 48 h after cetuximab treatment; in contrast, autophagosomes were scarce in untreated cells (Fig. 1A). We also observed the conversion of the LC3/Atg8 from the cytoplasmic form, LC3-I (18 kDa), to the autophagosomic form, LC3-II (16 kDa), using Western blotting in the same three cell lines shortly after overnight (16-h) exposure to 10 nM or 20 nM cetuximab (Fig. 1B, upper panel). We further confirmed this result quantitatively by determining the percentage of cells with punctate fluorescence after cetuximab treatment or not in the cells transfected with a GFP-tagged LC3 construct (Fig. 1B, lower panel). In all three cell lines, cetuximab-treated cells displayed more punctate fluorescence (LC3-II) than did untreated cells, which showed homogeneous fluorescence (LC3-I) or minimal punctate fluorescence. Finally, after 24–48 h of cetuximab treatment, cells stained with acridine orange dye showed the appearance of AVOs, which is considered a relatively late event in autophagy (Fig. 1C). Fluorescence-activated cell sorting (FACS) analysis showed that the percentage of cells with AVOs was higher for all three cetuximab-treated cell lines than for the corresponding untreated cells. Together, these findings provide strong evidence that cetuximab induced autophagy in cancer cells.
Figure 1.

Cetuximab induces autophagy in cancer cells. A, photographs from transmission electron microscopy showing characteristic autophagosomes (indicated by arrows) in A431, DiFi, and HCC827 cells after 48 h of treatment with 20 nM cetuximab or no treatment. Magnification: 4000× (×50,000 in the highlighted area). N: nucleus. B, upper panel, Western blot showing an increase in LC3-II levels in the same three cell lines after overnight cetuximab treatment. Lower panel, photographs from fluorescence microscopy showing punctate fluorescence (indicated by arrows) of a transfected GFP-tagged LC3 construct in the three cell lines after overnight treatment with 20 nM cetuximab. Cells with punctate GFP were counted and expressed as a percentage of the total number of cells per field. P <0.01 when the percentage of cells with punctate fluorescence was compared between the treated and untreated groups. C, FACS analysis of acridine orange (1 μg/ml) stained cells showing the percentage of AVO-positive cells after treatment with 20 nM cetuximab or no treatment for 24 h or 48 h. The FACS analysis profiles at 48 h are shown. P <0.01 when the percentage of AVO-positive cells in the cetuximab-treated group was compared with that in the untreated control group.
Cetuximab-induced autophagy is mediated through inhibition of the PI3K/Akt/mTOR pathway but not through inhibition of the MEK/Erk pathway
The MEK/Erk and class-I PI3K/Akt/mTOR pathways are two major pathways downstream of EGFR that are inhibited after cetuximab treatment in cancer cells (27, 29, 30). The appearance of the autophagosomic form of LC3 (LC3-II) correlated with the inhibition of Erk, Akt, and mTOR phosphorylation and the decrease in HIF-1α protein after cetuximab treatment in A431 cells (Fig. 2A). However, the increase in LC3-II in the cells was not affected after transient transfection with a constitutively active MEK1 construct (MEKS217E/S221D), which conferred resistance to cetuximab-induced inhibition of Erk phosphorylation (Fig. 2B, left panel). In contrast, the increase was prevented by transient transfection of A431 cells with myristoylated Akt, which was constitutively phosphorylated and thus was insensitive to cetuximab treatment (Fig. 2B, middle panel). Similar results were also found in the cells with experimental elevation of Rheb protein, which increased the basal level of mTOR phosphorylation and counteracted cetuximab-induced inhibition of mTOR phosphorylation (Fig. 2B, right panel), suggesting that inhibition of the Akt/mTOR pathway was required for induction of autophagy after cetuximab. To provide further evidence supporting a conclusion that inhibition of the class I PI3K pathway is causally linked to cetuximab-induced autophagy, we analyzed the autophagic response to cetuximab or a PI3K inhibitor, LY294002, in A431 cells transiently transfected with a constitutively active PI3K (myristoylated PI3K) construct or corresponding control. Both cetuximab and LY294002 increased LC3-II, and decreased Akt phosphorylation in A431 cells transfected with a control (neo) vector but not in the cells transfected with the constitutively active PI3K construct (Fig. 2C). We conducted an additional quantitative experiment showing a statistically significant difference in punctate LC3-GFP protein after cetuximab treatment of A431 cells transfected with the corresponding control vector or myristoylated PI3K or Akt construct (Fig. 2D).
Figure 2.

Constitutive activation of the PI3K/Akt/mTOR pathway, but not constitutive activation of the MEK/Erk pathway, prevents cetuximab-induced autophagy. A, Western blot showing a reverse correlation between an increase in LC3-II and decreases in Erk, Akt, and mTOR phosphorylation and HIF-1α protein by cetuximab. B, left panel, Western blot showing that the expression of a constitutively active MEK1 construct (S217E/S221D) prevented cetuximab-induced inhibition of Erk phosphorylation but failed to prevent cetuximab-induced increase in LC3-II and decrease in HIF-1α. Middle and right panels, Western blot showing that the expression of a constitutively active Akt (Myr-Akt) or Rheb protein prevented cetuximab-induced increase in LC3-II and decrease in HIF-1α. C, Western blot showing that the expression of a constitutively active class-I PI3K construct (Myr-PI3K) prevented cetuximab- or LY294002-induced increase in LC3-II and decrease in HIF-1α. D, photographs from fluorescence microscopy showing the effects of a constitutively active PI3K or Akt construct on cetuximab (20 nM × 16 h)-induced punctate fluorescence (indicated by an arrow) of the transfected GFP-tagged LC3 in A431 cells. Cells with punctate GFP were counted and expressed as a percentage of the total number of cells per field.
Together, these data indicate that the induction of autophagy after cetuximab treatment is mediated through inhibition of the class I PI3K pathway, but not the MEK/Erk pathway.
Downregulation of HIF-1α protein by cetuximab contributes to induction of autophagy
The dependence of cetuximab-induced autophagy on inhibition of the class I PI3K pathway but not on inhibition of the MEK/Erk pathway is reminiscent of our recently findings that downregulation of HIF-1α, which was postulated to play a major role in cetuximab-induced antitumor activity (20), was mediated through cetuximab-induced inhibition of the PI3K/Akt pathway but not through inhibition of the MEK/Erk pathway (18–20). To define the role of HIF-1α downregulation in cetuximab-induced autophagy, we transfected A431 cells with a HIF-1α mutant, HIF-1α/ΔODD, which, unlike the endogenous HIF-1α, is not liable to quick degradation in normoxia, owing to deletion of the ODD (18–20). We examined the effect of expression of HIF-1α/ΔODD on cetuximab-induced autophagy using the same experimental methods in Figure 1. A431 cells overexpressing HIF-1α/ΔODD showed a significant decrease in the numbers of autophagosomes seen under the transmission electron microscope, in the level of LC3 conversion, and in the amount of AVO formation after cetuximab treatment, while none of these changes occurred in the control vector-transfected cells (Fig. 3A–C).
Figure 3.

Overexpression of a HIF-1α mutant prevents cetuximab-induced autophagy A, photographs from transmission electron microscopy showing a marked decrease in the number of characteristic autophagosomes (indicated with an arrow) in HIF-1α/ΔODD-transfected A431 cells compared to the control vector-transfected cells after 48 h of treatment with 20 nM cetuximab. Magnification: 4000× (50,000× in the highlighted area); N: nucleus. B, left panel, Western blot showing that overexpression of HIF-1α/ΔODD construct inhibits the increase in LC3-II level that would have been induced by cetuximab treatment (20 nM × 16 h). Right panel, photographs from fluorescence microscopy pictures showing the effect of overexpression of HIF-1α/ΔODD construct on inhibiting cetuximab (20 nM × 16 h)-induced punctate fluorescence (indicated by an arrow) in A431 cells transfected with a GFP-tagged LC3 construct. The percentage of cells with punctate GFP was plotted against the types of transfection. P <0.01 when the percentage of cells with punctate fluorescence was compared between the treated and untreated groups. C, FACS analysis of acridine orange (1 μg/ml) stained cells showing the effect of overexpression of HIF-1α/ΔODD on lowering the percentage of AVO-positive cells after cetuximab treatment (20 nM × 48 h). FACS analysis profiles at 48 h are shown. P <0.05 when the percentage of AVO-positive cells in the cetuximab-treated group was compared with that in the untreated control group in A431neo cells, but not in A431 HIF-1α/ΔODD cells. D, Western blotting showing the effect of overexpression of HIF-1α/ΔODD construct on inhibiting the increase in LC3-II level after rapamycin treatment (25 μM × 16 h).
Similar to its effect on cetuximab-induced autophagy, overexpression of HIF-1α/ΔODD also strongly prevented autophagy induced by the mTOR inhibitor rapamycin in A431 cells, a result that was accompanied by a marked decrease in the level of endogenous HIF-1α protein (Fig. 3D). Overexpression of HIF-1α/ΔODD inhibited a rapamycin-induced increase in LC3-II level without affecting rapamycin-induced inhibition of mTOR activity, as shown by the decreased phosphorylation of p70S6K, a downstream target of mTOR. This additional result indicates that inhibition of autophagy by the overexpression of HIF-1α/ΔODD is a general phenomenon and supports a novel role of HIF-1α functioning at the downstream of the PI3K/Akt/mTOR pathway, the inhibition of which leads to autophagy.
Cetuximab-induced autophagy involves the beclin 1/hVps34 complex pathway
To gain further insight into the mechanisms of cetuximab-induced autophagy, we examined the effects of knockdown of beclin 1 on cetuximab-induced autophagy (Fig. 4A). Compared with the results in siRNA controls, knockdown of beclin 1 with siRNA prevented the increase in the level of LC3-II by cetuximab. We found similar results after pre-treating the cells with 3-methyalanine (3MA), a class-III PI3K specific inhibitor (Fig. 4B). These findings indicate that the beclin 1/hVps34 pathway is involved in cetuximab-induced autophagy.
Figure 4.
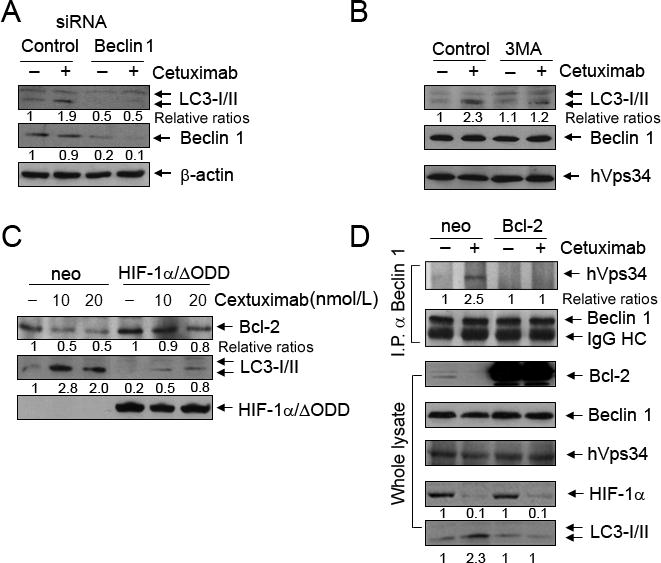
Downregulation of HIF-1α/Bcl-2 and activation of beclin 1/hVps34 complex play roles in cetuximab-induced autophagy. A, Western blot showing that knockdown of beclin 1 with siRNA in A431 cells prevented cetuximab (20 nM × 16 h)-induced increase in LC3-II level, but not in control siRNA-treated cells. The level of β-actin was used as a protein-loading control. B, Western blot showing the effect of treatment with 3MA (20 mM × 16 h) on inhibiting cetuximab (20 nM × 16 h)-induced increase in LC3-II, without affecting the level of hVps34. C, Western blot showing that overexpression of HIF-1α/ΔODD prevented cetuximab (10 and 20 nM × 16 h)-induced decrease in Bcl-2 level and increase in LC3-II level. D, effect of overexpression of Bcl-2 on cetuximab-induced association between beclin 1 and hVps34. Upper panel, Western blot of beclin 1 immunoprecipitates using antibodies against hVps34 or beclin 1 showing that an association between beclin 1 and hVps34 in A431 cells after cetuximab treatment (20 nM × 16h); this association was abolished by the overexpression of a Bcl-2 construct. Lower panel, Western blot of whole cell lysates showing the levels of indicated proteins in Bcl-2-transfected or control vector-transfected A431 cells with and without cetuximab treatment.
Bcl-2, a well known anti-apoptosis protein that is transcriptionally regulated by HIF-1α (31), was recently shown to regulate autophagy by binding to beclin 1 and thereby inhibiting the association between beclin 1 and hVps34, which leads to autophagy (11, 32). We found that cetuximab decreased the level of Bcl-2 protein that occurred along with the induction of autophagy in A431 cells, and that overexpression of HIF-1α/ΔODD prevented cetuximab-induced downregulation of Bcl-2 and autophagy (Fig. 4C). These findings suggest that one of the mechanisms by which autophagy was induced after cetuximab treatment was downregulation of Bcl-2, which released beclin 1 from the Bcl-2/beclin 1 complex, and thereby fostered the association between beclin 1 and hVps34. To test this novel paradigm, we transiently transfected A431 cells with a Bcl-2 construct or corresponding control and then treated the cells with cetuximab or not for 24 h. We examined changes in the association between beclin 1 and hVps34 by Western blotting of beclin 1 immunoprecipitates with antibodies directed against hVps34 and beclin 1, respectively. Cetuximab treatment enhanced the association between beclin 1 and hVps34 in control vector-transfected cells but not in Bcl-2 transfected cells (Fig. 4D). Bcl-2 overexpression inhibited cetuximab-induced increases in the LC3-II level but did not affect cetuximab-induced decreases in the HIF-1α level, indicating that Bcl-2 functions downstream of HIF-1α.
Inhibition of cetuximab-induced autophagy enhances the potential for apoptosis after cetuximab treatment
To determine the biological significance of autophagy on cell growth and survival after cetuximab treatment, we examined the effect of inhibiting autophagy on potential for cetuximab-induced apoptosis. Treatment of A431 cells with cetuximab had only a bottom-line effect on apoptosis induction, as shown by minimal levels of cleavage of PARP, caspase 3 and caspase 7, which serve as markers of apoptosis; however, combining cetuximab with chloroquine, an autophagy-lysosomal inhibitor, or with 3MA substantially increased the cleavage of these proteins (Fig. 5A). The increase in the cleavage of PARP, caspase 3 and caspase 7 was accompanied by a decreased LC3-II level in cells treated with 3MA alone. In chloroquine-treated cells, the level of LC3-II dramatically increased in both cetuximab-untreated or –treated cells, because chloroquine inhibited the lysosomal degradation pathway (33, 34).
Figure 5.

Inhibition of cetuximab-induced autophagy promotes cancer cell responses to cetuximab treatment. A, Western blot showing an increased PARP cleavage by cetuximab (20 nM × 16 h) in A431 cells co-treated with chloroquine (25 μM × 16 h) or 3MA (20 mM × 16 h). The levels of LC3 after the treatments are shown. The level of β-actin was used as protein-loading control. B, Western blotting showing increased PARP, caspase 3 and caspase 7 cleavage by cetuximab (20 nM × 16 h) in A431 cells with knockdown of beclin 1/Atg6 (left panel) or Atg7 (right panel) expression with siRNA. The levels of beclin 1, Atg7 and LC3 are shown. The level of β-actin was used as protein-loading control. C, Quantitative apoptosis ELISA showing that combination treatment of A431 cells with cetuximab (20 nM × 16-h) plus chloroquine (25 μM × 16-h) or 3MA (20 mM × 16-h) induces higher levels of apoptosis than treatment with cetuximab only. D, MTT assay showing enhanced effects of combination treatment with cetuximab (20 nM × 72-h) and indicated concentrations of chloroquine on the survival of A431 cells, compared to treatment with cetuximab or chloroquine alone.
We confirmed the effect of autophagy inhibition by the pharmacological agents (chloroquine and 3MA) on cetuximab-induced apoptosis by using genetic approach through RNA interference. Figure 5B shows that the levels of beclin 1 and Atg7 were lower in beclin 1 siRNA- or Atg7 siRNA-treated A431 cells, respectively, than in the corresponding control siRNA-treated cells, and only the control siRNA-treated cells revealed a cetuximab-induced increase in the level of LC3-II. The level of cleavage of PARP, caspase 3 and caspase 7 after cetuximab treatment was clearly higher in the beclin 1 siRNA- and Atg7 siRNA-treated cells than in the control siRNA-treated cells.
To quantify apoptosis at the cellular level and demonstrate the role of autophagy in protecting cells from cetuximab-induced apoptosis, we compared the levels of histone-associated DNA fragmentation between the cells treated with cetuximab alone and those treated with cetuximab plus chloroquine or 3MA. Compared with the treatment with cetuximab, chloroquine, or 3MA alone, treatment of A431 cells with cetuximab plus chloroquine or 3MA significantly enhanced induction of apoptosis (Fig. 5C). This effect led to substantial decrease in the total number of surviving cells by combination treatment of cetuximab and chloroquine, compared with either treatment alone (Fig. 5D), indicating that inhibition of cetuximab-induced autophagy enhances the potential for apoptosis after cetuximab treatment.
Together with data from the mechanistic studies, we propose a model depicted in Figure 6 showing the novel mechanism by which cetuximab induces autophagy that functions to protect cells from cetuximab-induced apoptosis.
Figure 6.

A model depicting a novel mechanism by which cetuximab induces autophagy. Activation of EGFR/class I PI3K/Akt/mTOR signaling pathway by EGF suppresses autophagy whereas inhibition of the pathway by cetuximab induces autophagy. Cetuximab downregulates HIF-1α, and subsequently decreases Bcl-2, which releases beclin 1 from its association with Bcl-2 to form complex with class III PI3K/hVps34 (directed with a dashed line); the latter complex thereby promotes autophagy. The arrow lines indicate the direction of pathways. The ⊥ symbolizes an inhibitory function by an agent or a molecule. The question mark implies additional mechanisms not explored in current study, by which HIF-1α may suppress autophagy.
Discussion
In this paper, we demonstrated that autophagy was induced in cancer cells after cetuximab treatment, protecting the cells from cetuximab-induced inhibition of cell survival. We show our results from three complementary lines of experiments. First, we offer convincing evidence that autophagy is induced in cancer cells after cetuximab treatment. Second, we explored the mechanisms by which cetuximab induces autophagy, and third, we studied the biological role of autophagy in cancer cells’ response to cetuximab.
Our results, particularly our documentation of the appearance of characteristic autophagosomes on transmission electron microscopy, which is the gold standard for demonstrating autophagy, provide strong evidence that autophagy was induced in cancer cells after cetuximab treatment. The conversion of LC3-I to LC3-II correlates with the extent of autophagosome formation and is widely used as an early marker of autophagy (35). Our quantitative analysis showed that the percentage of punctate GFP-LC3 fluorescence was greater in DiFi and HCC827 cells, in which apoptosis was induced by cetuximab, than in A431 cells, in which apoptosis is usually weak or sometimes not observed after the treatment (19, 20). Furthermore, our quantitative flow cytometric analysis showed that the appearance of AVOs, which is considered a relatively late event in autophagy, occurred earlier and the number of AVOs was greater in DiFi and HCC827 cells than in A431 cells after cetuximab treatment. These findings suggest that the extent of cetuximab-induced autophagy was positively correlated with the extent of cetuximab-induced apoptosis in the different types of cancer cells.
Our studies relevant to the mechanisms of autophagy indicated that both the class I and class III PI3K pathways were involved in cetuximab-induced autophagy, because constitutive activation of the class I PI3K pathway by the expression of constitutively active PI3K or Akt, or Rheb constructs inhibited cetuximab-induced autophagy, as did the inhibition of the class III PI3K pathway by 3MA treatment or silencing beclin 1 or Atg7. In particular, we found that constitutive expression of HIF-1α/ΔODD strongly prevented cetuximab-induced autophagy. This result echoes our recent finding that decrease in HIF-1α through inhibition of protein synthesis by cetuximab plays a major role in cetuximab-mediated antitumor activity (18–20). The requirement of HIF-1α for both cetuximab-mediated antitumor activity and cetuximab-induced autophagy suggests that the two events are intimately linked and share a common pathway to the point at which the two diverge into differential cellular events. Our study suggests a model that a decrease in Bcl-2 resulting from transcription inhibition due to a cetuximab-induced inhibition of HIF-1α protein synthesis diminished a Bcl-2-beclin 1 association, favoring an association between beclin 1 and class III PI3K hVps34, thereby triggering autophagy (11).
A few caveats should be clarified. First, HIF-1α has been previously reported to play an important role in hypoxia-induced autophagy (36–39), which improves cell survival by removing damaged organelles under metabolic stress conditions such as hypoxia (40). Our result showing that HIF-1α overexpression inhibits autophagy seems to contradict its role in hypoxia-induced autophagy. Thus, it is important to point out that hypoxia-induced autophagy requires specific activation of an autophagy-inducing molecule, BNIP3 (another BH3-only Bcl-2 family member similar to beclin 1), which is activated by HIF-1α only in hypoxic cells (39). HIF-1α was originally discovered as a nuclear factor induced under hypoxia (41, 42). It is now known that the level of HIF-1α is regulated by both transcriptional and posttranslational mechanisms. Hypoxia upregulates HIF-1α protein by increasing protein stability across various cell types, whereas cell signaling activated by the PI3K pathway increases HIF-1α protein by increasing protein synthesis in a cell type-dependent manner (43–47). Depending on the types of available co-factors, a HIF-1 heterodimer can transcriptionally activate over 100 genes, including Bcl-2 (31) and BNIP3 (39). Our study was conducted in normoxic conditions, and we found no change in the level of BNIP3 after cetuximab treatment but a decrease in the level of Bcl-2.
A second caveat is that autophagy is known to have dual functions: On one hand, it can serve as a cytoprotective mechanism, allowing tumor cells to survive under conditions of metabolic stress and hypoxia and to escape anticancer treatment-induced cell death; on the other hand, autophagy induced by therapeutic interventions can cause the death of cancer cells that are resistant to apoptosis (48, 49). We found that inhibition of autophagy by a class III PI3K inhibitor, 3MA, or by a lysosomal pathway inhibitor, chloroquine, allowed A431 cells to undergo apoptosis after cetuximab treatment, indicating that autophagy occurring after the treatment protected cells from apoptosis that have occurred in A431 cells after deprivation of EGFR-mediated cell signaling.
The third caveat is that it is noteworthy that our conclusion concerning the role of autophagy in protecting A431 cells from cetuximab-induced apoptosis does not conflict with our finding showing more extensive autophagy in the cells (DiFi and HCC827) prone to apoptosis by cetuximab than in the cells (A431) less sensitive to apoptosis by cetuximab (19), because whether or not cells undergo apoptosis after cetuximab treatment and the extent of apoptosis are intrinsic properties of the cells. As a reactive response to cetuximab-induced apoptosis, autophagy attempts to rescue cells from apoptosis but has limited capability; within a reasonable range, the extent of autophagy is proportional to the extent of apoptosis. Thus, it is expected that inhibition of autophagy in the apoptosis-prone cells should also enhance apoptosis; however, such enhancement may not be readily detectable because of the extent of cetuximab-induced apoptosis in the apoptosis-prone cells.
In our current study, we demonstrated cetuximab-induced autophagy in three cancer cell lines derived from the vulva (A431), colon (DiFi) and lung (HCC827), with most of the mechanistic studies conducted in A431 cells. In our next studies, it will be important to determine whether our findings are also true in additional cancer cell lines, including cancer cells that typically respond to cetuximab mainly by undergoing cell growth arrest rather than cell death via apoptosis. It will be also important to further test the mechanisms identified in current study; that is, in addition to Bcl-2, there are over 100 HIF-1-targeted genes including glucose and lactate transporters and key enzymes for anaerobic glycolysis that are critical for both bioenergetic and biosynthetic metabolism in cancer cells (50). Currently, it is largely unknown how cetuximab-induced autophagy is linked to inhibition of cancer metabolism through downregulation of HIF-1α. Lastly, it will be necessary to determine whether the cetuximab-induced autophagy can be targeted or exploited for therapeutic gains using appropriate in vivo tumor models in nude mice.
In summary, we found that cetuximab induced autophagy that protected cancer cells from the cellular effects of cetuximab treatment; the response was elicited through complex mechanisms involving both inhibition of the class I PI3K/Akt pathway and activation of the class III PI3K pathway. Our data supports a novel model that a decrease in Bcl-2 level resulting from cetuximab-mediated inhibition of class I PI3K/Akt/mTOR/HIF-1α pathway promotes autophagy by releasing beclin 1 from its associations with Bcl-2 to form complex with class III PI3K/hVps34. Further in-depth studies are needed to understand the role of autophagy in EGFR-targeted therapy, which may benefit patients through development of novel combination treatments to improve patients’ response to EGFR-targeted therapy through inhibition of autophagy.
Acknowledgments
Grant supports: NIH grant 5R01CA129036 (to Z.F.), and US DOD CDMRP W81XWH-06-1-0544 (to Z.F.) and NCI cancer center support grant CA016672.
We thank Markeda Wade of the Department of Scientific Publications, The University of Texas M. D. Anderson Cancer Center, for editorial assistance.
Footnotes
Disclosure of Potential Conflict of Interest
The authors have no conflicts of interest to declare concerning the contents of this research article.
References
- 1.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–10. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 3.Yu L, Wan F, Dutta S, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–7. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boya P, Gonzalez-Polo RA, Casares N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Polo RA, Boya P, Pauleau AL, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 6.Klionsky DJ, Cregg JM, Dunn WA, Jr, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–45. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 7.Kamada Y, Sekito T, Ohsumi Y. Autophagy in yeast: a TOR-mediated response to nutrient starvation. Curr Top Microbiol Immunol. 2004;279:73–84. doi: 10.1007/978-3-642-18930-2_5. [DOI] [PubMed] [Google Scholar]
- 8.Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 9.Aita VM, Liang XH, Murty VV, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 10.Liang C, Feng P, Ku B, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 11.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Levine B. Unraveling the role of autophagy in cancer. Autophagy. 2006;2:65–6. doi: 10.4161/auto.2.2.2457. [DOI] [PubMed] [Google Scholar]
- 13.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–34. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 14.Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy. 2006;2:85–90. doi: 10.4161/auto.2.2.2463. [DOI] [PubMed] [Google Scholar]
- 15.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenberg-Lerner A, Kimchi A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis. 2009;14:376–91. doi: 10.1007/s10495-008-0307-5. [DOI] [PubMed] [Google Scholar]
- 18.Luwor RB, Lu Y, Li X, Mendelsohn J, Fan Z. The antiepidermal growth factor receptor monoclonal antibody cetuximab/C225 reduces hypoxia-inducible factor-1 alpha, leading to transcriptional inhibition of vascular endothelial growth factor expression. Oncogene. 2005;24:4433–41. doi: 10.1038/sj.onc.1208625. [DOI] [PubMed] [Google Scholar]
- 19.Lu Y, Liang K, Li X, Fan Z. Responses of cancer cells with wild-type or tyrosine kinase domain-mutated epidermal growth factor receptor (EGFR) to EGFR-targeted therapy are linked to downregulation of hypoxia-inducible factor-1alpha. Mol Cancer. 2007;6:63. doi: 10.1186/1476-4598-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Lu Y, Liang K, Pan T, Mendelsohn J, Fan Z. Requirement of hypoxia-inducible factor-1alpha down-regulation in mediating the antitumor activity of the anti-epidermal growth factor receptor monoclonal antibody cetuximab. Mol Cancer Ther. 2008;7:1207–17. doi: 10.1158/1535-7163.MCT-07-2187. [DOI] [PubMed] [Google Scholar]
- 21.Fan Z, Baselga J, Masui H, Mendelsohn J. Antitumor effect of anti-epidermal growth factor receptor monoclonal antibodies plus cis-diamminedichloroplatinum on well established A431 cell xenografts. Cancer Res. 1993;53:4637–42. [PubMed] [Google Scholar]
- 22.Fan Z, Masui H, Altas I, Mendelsohn J. Blockade of epidermal growth factor receptor function by bivalent and monovalent fragments of 225 anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res. 1993;53:4322–8. [PubMed] [Google Scholar]
- 23.Wu X, Fan Z, Masui H, Rosen N, Mendelsohn J. Apoptosis induced by an anti-epidermal growth factor receptor monoclonal antibody in a human colorectal carcinoma cell line and its delay by insulin. J Clin Invest. 1995;95:1897–905. doi: 10.1172/JCI117871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu B, Fang M, Lu Y, Mendelsohn J, Fan Z. Fibroblast growth factor and insulin-like growth factor differentially modulate the apoptosis and G1 arrest induced by anti-epidermal growth factor receptor monoclonal antibody. Oncogene. 2001;20:1913–22. doi: 10.1038/sj.onc.1204277. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Lu Y, Jin W, Liang K, Mills GB, Fan Z. Autophosphorylation of Akt at threonine 72 and serine 246: a potential mechanism of regulation of Akt kinase activity. J Biol Chem. 2006;281:13837–43. doi: 10.1074/jbc.M602060200. [DOI] [PubMed] [Google Scholar]
- 26.Liu B, Fang M, Schmidt M, Lu Y, Mendelsohn J, Fan Z. Induction of apoptosis and activation of the caspase cascade by anti-EGF receptor monoclonal antibodies in DiFi human colon cancer cells do not involve the c-jun N-terminal kinase activity. Br J Cancer. 2000;82:1991–9. doi: 10.1054/bjoc.2000.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Luwor R, Lu Y, Liang K, Fan Z. Enhancement of antitumor activity of the anti-EGF receptor monoclonal antibody cetuximab/C225 by perifosine in PTEN-deficient cancer cells. Oncogene. 2006;25:525–35. doi: 10.1038/sj.onc.1209075. [DOI] [PubMed] [Google Scholar]
- 28.Takeuchi H, Kondo Y, Fujiwara K, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–46. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 29.Lu Y, Li X, Liang K, et al. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007;67:8240–7. doi: 10.1158/0008-5472.CAN-07-0589. [DOI] [PubMed] [Google Scholar]
- 30.Liang K, Ang KK, Milas L, Hunter N, Fan Z. The epidermal growth factor receptor mediates radioresistance. Int J Radiat Oncol Biol Phys. 2003;57:246–54. doi: 10.1016/s0360-3016(03)00511-x. [DOI] [PubMed] [Google Scholar]
- 31.Carmeliet P, Dor Y, Herbert JM, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–90. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 32.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–88. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura N, Kumamoto T, Kawamura Y, et al. Expression of autophagy-associated genes in skeletal muscle: an experimental model of chloroquine-induced myopathy. Pathobiology. 2007;74:169–76. doi: 10.1159/000103376. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki T, Nakagawa M, Yoshikawa A, et al. The first molecular evidence that autophagy relates rimmed vacuole formation in chloroquine myopathy. J Biochem. 2002;131:647–51. doi: 10.1093/oxfordjournals.jbchem.a003147. [DOI] [PubMed] [Google Scholar]
- 35.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kothari S, Cizeau J, McMillan-Ward E, et al. BNIP3 plays a role in hypoxic cell death in human epithelial cells that is inhibited by growth factors EGF and IGF. Oncogene. 2003;22:4734–44. doi: 10.1038/sj.onc.1206666. [DOI] [PubMed] [Google Scholar]
- 37.Jaattela M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene. 2004;23:2746–56. doi: 10.1038/sj.onc.1207513. [DOI] [PubMed] [Google Scholar]
- 38.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H, Bosch-Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Azad MB, Chen Y, Henson ES, et al. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy. 2008;4:195–204. doi: 10.4161/auto.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–54. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 1997;57:5328–35. [PubMed] [Google Scholar]
- 44.Zelzer E, Levy Y, Kahana C, Shilo BZ, Rubinstein M, Cohen B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1alpha/ARNT. EMBO J. 1998;17:5085–94. doi: 10.1093/emboj/17.17.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–5. [PubMed] [Google Scholar]
- 46.Treins C, Giorgetti-Peraldi S, Murdaca J, Semenza GL, Van Obberghen E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem. 2002;277:27975–81. doi: 10.1074/jbc.M204152200. [DOI] [PubMed] [Google Scholar]
- 47.Minet E, Arnould T, Michel G, et al. ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett. 2000;468:53–8. doi: 10.1016/s0014-5793(00)01181-9. [DOI] [PubMed] [Google Scholar]
- 48.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–5. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoyer-Hansen M, Jaattela M. Autophagy: an emerging target for cancer therapy. Autophagy. 2008;4:574–80. doi: 10.4161/auto.5921. [DOI] [PubMed] [Google Scholar]
- 50.Lum JJ, Bui T, Gruber M, et al. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007;21:1037–49. doi: 10.1101/gad.1529107. [DOI] [PMC free article] [PubMed] [Google Scholar]