

Abstract
The normal human breast comprises an inner layer of luminal epithelial cells and an outer layer of myoepithelial cells separated from the connective tissue stroma by an intact basement membrane. In breast cancer, tumor cells are in direct contact with the surrounding highly activated collagenous stroma, with little or no discernible myoepithelial fence from the original double-layered structure. To understand the evolution of these two scenarios, we took advantage of a three-dimensional hydrated collagen gel approach. The contribution of myoepithelial cells to normal morphogenesis was studied by ablation and rescue experiments, and genes regulated on tumor cell–fibroblast interaction were identified in a tumor environment assay. In normal breast morphogenesis, the ability to correctly polarize sialomucin to the luminal membrane of emerging acini was used as a criterion for apical polarity and functional differentiation. In the assay of breast neoplasia, the consequence of reciprocal tumor cell–fibroblast interaction was addressed morphologically as well as by a differential display approach. Normal breast epithelial cells were purified immunomagnetically and an established cell line, MCF-7, was used as a surrogate tumor cell. With regard to the importance of myoepithelial cells in normal breast epithelial morphogenesis, the collagen gel assay elucidated the following subtleties: In contrast to culturing in basement membrane gels, luminal epithelial cells when cultured alone made structures that were all inversely polarized. This aberrant polarity could be rescued by co-culture with myoepithelial cells. The molecular activity of myoepithelial cells responsible for correct morphogenesis was narrowed down to the laminin-1 component of the basement membrane. As for the consequence of interaction of tumor cells with connective tissue fibroblasts, the assay allowed us to identify a hitherto undescribed gene referred to as EPSTI1. The relevance of the assay-based identification of regulated genes was confirmed in a series of breast carcinomas in which EPSTI1 was highly upregulated compared with normal breast. Few if any of these observations would have been possible on two-dimensional tissue culture plastic.
Keywords: Breast models, Three-dimensional cell culture, Microenvironment, Myoepithelial cell, Luminal epithelial cell, Tumor environment assay, Myofibroblast, EPSTI1
1. Introduction
Three-dimensional cultures of mammary epithelial cells has been pursued extensively for rodent [1-3], canine [4], bovine [5], and human [for review see 6,7] species. Techniques have included the use of reconstituted basement membrane material, hydrated collagen gels, and cellulose sponges with mixtures of extracellular matrix (ECM) components ([1,3,8-10], reviewed in [6]). Here, we focus on some novel aspects of the collagen gel assay as a vehicle in the studies of cellular interactions in the human breast. We concentrated on two intriguing heterotypic cellular interactions in the breast: that of myoepithelial cells with luminal epithelial cells because of the possible tumor-suppressive function of myoepithelial cells, and that of tumor cells with peritumoral fibroblasts because of the apparent tumor-facilitating consequences [11-15]. In both instances, we could model these interactions in hydrated collagen as the matrix of choice. This is because the breast epithelial component is self-supplied with basement membrane from both luminal epithelial and myoepithelial cells, and that the entire gland, including the basement membrane, is an entity suspended in collagenous interstitial stroma. In principle, the same argument is valid for tumor cells except that here the nearest neighbors usually are not the myoepithelial cells but the myofibroblasts [16]. Since normal luminal epithelial cells and invasive tumor cells often share a common lineage affliation, we decided to address the consequences of ablating or adding their respective partner cells, i.e., myoepithelial cells and myofibroblasts, from or to the microenvironment under experimental conditions. Whereas a hallmark of normal luminal epithelial cells is their differentiated polarization of the cellular axis and vectorial secretion, the tumor cells had increased morphological disorganization [16] but no obvious interaction-dependent molecular marker. The latter, thus, had to be defined.
The two sets of three-dimensional collagen-based experimental setups allowed us to draw the following conclusions for normal and tumor cells, respectively: (1) A prominent function of myoepithelial cells is the maintenance of a correct polarity of the luminal epithelial cells and this function requires laminin-1. (2) When peritumoral fibroblasts and invasive tumor cells are in direct contact, metalloproteinases and novel genes are induced. Three-dimensional hydrated collagen gels therefore emerge as an invaluable culture assay for the dissection of cell–cell interactions in breast morphogenesis and neoplasia.
2. Description of methods
2.1. Technical considerations
With regard to the three-dimensional assay for luminal epithelial–myoepithelial interaction we found that valid measures to purify the cells and to prevent culture-induced modifications were indispensable. This was especially true for the myoepithelial cells. In situ, the only proliferating cells are found within the luminal epithelial compartment, and the myoepithelial cells are almost entirely quiescent [17]. In culture, myoepithelial cells rapidly enter the cell cycle and proliferate very fast [18–21]. To date, culture conditions that specifically support only one of the two breast epithelial lineages to purity have not been found. Therefore, measures to establish pure cell populations have had to rely on other technologies. Based on antibodies against lineage-specific cell surface markers, our laboratory and others have succeeded in optimizing methods for immunomagnetic separation of primary luminal epithelial and myoepithelial cells from the mammary gland [14,19,20,22-26]. The separations have used Dynabeads, fluorescence-activated cell sorting (FACS), or the magnetic cell sorting (MACS) system with similar results. Below we focus only on the MACS separation technology, a technology we have used and thus are best familiar with.
In addition to the problems of purification, it is necessary to address culture-induced artifacts in the cellular phenotype. Cultivation of normal human breast epithelial cells was pioneered by the laboratory of Martha Stampfer using a relatively complex serum-containing medium [27]. Later, this medium was replaced by a serum-free medium for long-term cultivation without allowing overgrowth of fibroblasts [28]. Both media have been thoroughly evaluated with regard to their ability to maintain lineage-specific markers in culture [29]. We have used a modification of the serum-free medium in combination with coating of the culture flasks with Vitrogen-100 to obtain moderate expansion of cells with preserved lineage-specific traits [18,30]. We also have devised culture media to expand separated luminal epithelial cells and myoepithelial cells without significant drifting in the phenotype [25]. Thus, the prerequisites for separation and recombination have been fulfilled relatively well, and this is described in detail below.
For tumor cell–fibroblast interactions, a critical point to keep in mind is that not all fibroblasts are created equally. Thus, techniques need to be devised for isolation and characterization of the relevant stromal cells from normal and malignant breast. This topic has been reviewed extensively elsewhere [31,32]. Our initial experimental approach has so far relied on the continuous collection and explantation of biopsies, a relatively time-consuming process. In addition, normal breast biopsies are limited in supply and long-term experiments, or experiments requiring larger quantities of cells, have also been hampered by the diffculty of expanding the cell population. Newly explanted fibroblasts readily convert into myofibroblasts in the presence of serum, but at the same time proliferation ceases along with α-smooth muscle actin induction [33]. To overcome the general built-in technical barrier of using normal primary tissue, methods to extend the life span of explanted cells have been developed recently. Ectopic expression of hTERT, encoding the catalytic subunit of human telomerase, as well as human papillomavirus 16 E6/E7 oncogenes, has been shown to extend the life span of human cells, including fibroblasts [34-36]. Therefore, after having established that myofibroblasts in carcinomas originate primarily from fibroblasts [16], an hTERT-immortalized fibroblast cell line was established, which could substitute for fresh biopsies as a source of myofibroblast partners in the tumor environment assay. Collectively, these technical considerations and improvements have formed the basis for the applications of three-dimensional co-culture assays described in the following sections.
2.2. Functional analysis of the role of myoepithelial cells in breast epithelial polarization
2.2.1. Rationale
The ability to isolate myoepithelial cells from normal breast tissue has enabled us to test their functional role in breast morphogenesis. We had observed previously that while normal breast luminal epithelial cells in reconstituted basement membrane (rBM) form polarized acini with basement membrane, the same cells in collagen gel are incapable of doing so [37]. We therefore chose to test the effect of myoepithelial cells in this assay [14].
2.2.2. Protocol
2.2.2.1. Isolation of myoepithelial cells
Plate epithelial organoids from reduction mammoplasty and allow cells to spread out into monolayer. Trypsinize cells and filter as previously described [25]. Isolate myoepithelial cells by retention of luminal epithelial cells in an immunomagnetic column by two independent separations with anti-sialomucin antibody (carcinoma-associated antigen, clone 115D8, Catalog No. MON 9005, Monosan, The Netherlands). All cell separations should be carried out by use of the miniMACS magnetic cell separation system according to the manufacturer’s instructions (Miltenyi Biotec, GmbH, Gladbach, Germany).
2.2.2.2. Preparation of collagen gels
2.2.2.2.1. Materials
Vitrogen-100 (Cohesion, Palo Alto, CA, USA) collagen gel, chilled to a temperature of 4–6 °C.
Sterile 10× phosphate-buffered saline solution (PBS: (0.2 MNa2HPO4, 1.3 M NaCl, pH 7.4)
0.1 M HCl
0.1 M NaOH
Phenol red or pH paper
2.2.2.2.2. Preparation of collagen solution (2 ml)
Mix 1.6 ml of chilled Vitrogen-100 with 0.2 ml of 10× PBS. Add 0.2 ml of 0.1 MNaOH and mix. Adjust the pH of the solution to 7.4 ± 0.2 by the addition of a few drops of 0.1 MHCl or 0.1 MNaOH. The pH of the solution can be monitored by pH paper or by use of a pH indicator dye such as phenol red. Phenol red can be added to the PBS at a concentration of 0.005 mg/ml.
2.2.2.2.3. Gelation
Suspend 2 × 105 purified luminal epithelial cells with 0.5 ml of collagen solution/well and pipet into a well (24-well dishes, Nunc Roskilde, Denmark). Collagen gelation is initiated by warming the collagen solution to 37 °C. For best results allow a minimum of 60 min for gelation to occur. Add 1 ml of medium (CDM3) to each well and culture cells for 12 days. Change medium three times a week.
All three-dimensional cell cultures should be followed by thorough inspection in an inverted phase contrast microscope and photographed as necessary.
2.2.2.3. Reconstituted basement membrane matrix
Vials of rBM (Matrigel, Collaborative Biomedical Products, Bedford, MA, USA) should be preserved at −80 °C. Thaw vials of rBM on ice for approximately 2 h. Suspend 2 × 105 purified luminal epithelial cells in 0.3 ml of chilled rBM using prechilled pipet tips and pipet into one 24-well dish (Nunc), and allow the gel to solidify for approximately 40 min. Add 1 ml medium and change the medium three times a week.
2.2.2.4. Preparation of gels for cryosectioning
Carefully discard the culture medium and wash the gel twice with prewarmed (approximately 37 °C) PBS. Remove the gel carefully from the culture dish with a rubber policeman/spatula, transfer immediately into ice-cold n-hexane on dry ice (−70 °C), mount the gel in Tissue Freezing Medium (Leica Instruments, GmbH, Heidelberg) prior to sectioning in a cryostat with a thickness setting of 5 or 8 μm for subsequent peroxidase or fluorescence staining, respectively.
2.2.2.5. Evaluation of marker expression
Air-dry sections for 15 min and fix for 5 min in methanol at −20 °C. Preincubate sections with PBS supplemented with 10% normal goat serum. This buffer is also used for dilution of antibodies and for rinsing. Immunoperoxidase staining is performed as previously described using rabbit anti-mouse immunoglobulin as secondary antibody (Z259, DAKO) and PAP complex as tertiary antibody (P850, DAKO) [18]. To visualize the nuclei, counterstain with Mayer’s hematoxylin (Sigma–Aldrich, Albertslund, Denmark) and rinse in running water. For immunofluorescence and double-labeling experiments use isotype-specific antibodies as described [33,38]. To visualize the nuclei combine with counterstaining with either the DNA-specific dye Hoechst 33258 (blue, Bisbenzimide H 33258 fluorochrom, Riedel-de Häen, Germany) or use 1 μg/ml propidium iodide (red, Molecular Probes, Eugene, OR, USA). Rinse sections thoroughly with water, dry, and mount with coverslips by use of Fluoromount-G (Southern Biotechnology) supplemented with 2.5 mg/ml n-propyl gallate (Sigma–Aldrich) as previously described [33,38]. Immunofluorescence can be visualized by either conventional fluorescence microscopy or confocal microscopy.
2.2.2.6. Quantification of polarization
Count all acinar profiles stained with the apical marker sialomucin and counterstain with hematoxylin in three different cryostat sections from at least three different biopsies.
2.2.3. Comments
When embedded into the collagen gel, the purified luminal epithelial cells form acinus-like structures, albeit without any lumen formation [14]. Other studies have reported formation of duct-like, rather than acinus-like, structures in the collagen assay [39,40]. It is likely that the observed difference between our study and those of others is based on the nature of the cell type used. In our work, we have used a highly purified single cell population of luminal epithelial cells whereas others have used either organoids or unseparated mixed populations of cells including myoepithelial cells and stromal cells which facilitate branching morphogenesis most likely through expression of morphogenetic factors such as epimorphin [41]. When sectioned and stained with specific antibodies, we reveal a fundamental difference in the polarization of luminal epithelial cells cultured in collagen compared with our previous reports in the rBM assay [9]. Double staining with sialomucin and epithelium-specific antigen (ESA) or sialomucin and occludin reveals inside-out polarization in collagen and no deposition of BM as compared with correct polarization and appearance of a distinct BM in the rBM assay [14].
2.3. Assay for studying the differences between normal-derived and cancer-derived myoepithelial cells in breast morphogenesis
2.3.1. Rationale
In vivo, luminal epithelial cells are in contact with myoepithelial cells. To decipher the function of myoepithelial cells, we asked whether the inside-out polarity in collagen I gel could be reversed by addition of purified primary myoepithelial cells. Since the rBM could substitute for myoepithelial cells and laminins, and laminins have been reported to be instrumental in different aspects of morphogenesis (for review see [42]), we tested the major laminin isoforms found in the breast epithelial BM, i.e., laminin-1, laminin-5, and laminin-10/11, in the collagen assay. One of the hallmarks of breast cancer progression is the loss of normal tissue architecture including polarity. It has also been postulated that primary breast carcinomas show dramatic increases in the luminal epithelial/myoepithelial ratio and that many invasive breast carcinomas essentially lack myoepithelial cells [43]. This may be so, if the criterion for identification of myoepithelial cells is the expression of a complete differentiation program. However, these studies often have focused exclusively on the presence of an intact BM in general and laminin in particular, both of which are usually absent in invasive breast cancer [44,45]. In contrast, other myoepithelial markers have frequently been reported in breast cancer. Thus, CK14, CK17, and vimentin have been reported to be present in up to 20–33% of invasive breast carcinoma (Fig. 1).
Fig. 1.

Neoplastic myoepithelial cells at the epithelial-stromal junction of a bimodal breast cancer. A cryostat section was double-stained with antibodies against cytokeratin 17 (green) and CAM5.2. (red) (bar, 25 μm).
Similar frequencies of ultrastructurally identified myoepithelial cells have been reported in breast cancers (reviewed in [46]). However, even in cases where myoepithelial markers have been detected, luminal cells remain largely disorganized [47,48]. Based on this information we asked whether cancer-associated myoepithelial cells are deficient in their ability to signal to luminal epithelial cells for polarity, and whether this could be related to their inability to synthesize basement membrane components.
2.3.2. Protocol
2.3.2.1. Preparation of collagen gels
Unless otherwise stated, all steps are the same as in method 2.2.2.
2.3.2.2. Purification of normal- and cancer-derived myoepithelial cells
Normal myoepithelial cells are purified by collection of flow-through of two consequent anti-sialomucin columns. Cancer-derived myoepithelial cells are purified from primary carcinomas using an anti-Thy-1 column (AS0-2, Dianova, GmBH).
2.3.2.3. Addition of myoepithelial cells
Mix 2 × 105 purified normal-derived or cancer-derived myoepithelial cells with 2 × 105 luminal epithelial cells and suspend in collagen solution, allow the gel to solidify as described above.
2.3.2.4. Replacement experiments
Addition of rBM: Add rBM, primarily consisting of laminin-1, to a final concentration of 10% (volume) to the collagen solution prior to gelation.
Addition of laminin: 10 to 100 μg/ml laminin-1 (Sigma–Aldrich), 10 to 100 μg/ml laminin-10/11 (Catalog No. 12163-010; Gibco-BRL, Life Technologies A/S, Vallensbæk, Denmark), and 2.5 μg/ml affnity-purified laminin-5 [49] can be used. Add laminin to the CDM3 medium every time medium is changed.
2.3.3. Comments
In our hands, the myoepithelial cells are able to rescue polarity of luminal epithelial cells in 70% of cases (Fig. 2). The effect of myoepithelial cells appears to be cell type specific since co-cultures of luminal epithelial cells with other breast cells (resident breast fibroblasts) or nonbreast cells (osteosarcoma cells) do not lead to reversal of inside-out polarity. Moreover, in the collagen I gels, myoepithelial cells surround the luminal epithelial cells, resulting in formation of double-layered acini similar to their position in vivo (Fig. 3).
Fig. 2.

Inside-out polarity of luminal epithelial spheres in collagen gel and polarity reversal by myoepithelial cells. Cryostat sections of luminal epithelial: (a) without and (b) with co-cultured myoepithelial cells in collagen gels and stained for sialomucin. Cells were counter-stained with heamotoxylin (bar, 40 μm). Note that in (a), sialomucin is localized to the outside of spheres, where in (b), it is inside the lumen as is the case in the breast.
Fig. 3.
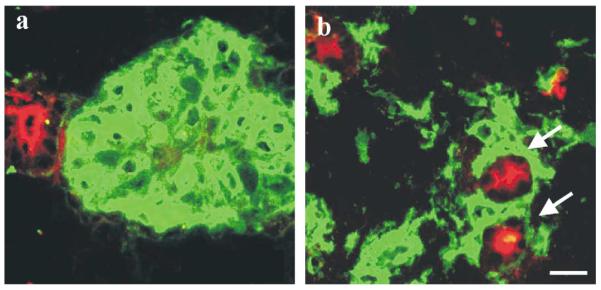
Differential sorting out of myoepithelial cells in reconstituted basement membrane and in collagen gel. Co-cultures of luminal epithelial cells and myoepithelial cells in (a) rBM and (b) collagen gel. Gels were double-stained for thy-1 to demonstrate myoepithelial cells (green) and sialomucin to demonstrate primarily the lumina of acini (red). Note that whereas myoepithelial cells sort out by themselves in rBM, double-layered structures may be formed in the collagen gel (arrows) (bar, 25 μm).
In the replacement assay, both laminin-1 and rBM (10% of final concentration) can reverse polarity of luminal epithelial cells when added to collagen gels, whereas laminin-5 and laminin-10/11 fail to do so. These data indicate that (1) luminal epithelial cells do not make suffcient laminin-1 to allow correct polarity, and (2) normal myoepithelial cells are the source of the polarizing principle through synthesis of laminin-1. The synthesis of basement membrane was further analyzed by immunostaining with monoclonal antibody against the α1-chain of laminin-1 and by reverse transcription polymerase chain reaction (RT-PCR). The only difference between the myoepithelial- and luminal epithelial cells was the lack of laminin α1 chain expression in the latter [14].
Surprisingly, we discovered that 75% of myoepithelial cells derived from tumor tissues completely failed to polarize luminal epithelial cells because they were unable to synthesize laminin-1. To test whether the data obtained with tumor-derived myoepithelial cells in culture agreed with observations in vivo, we stained 12 infiltrating ductal breast carcinomas for laminin-1. Seven carcinomas were completely negative for laminin-1. The other five carcinomas showed only foci of fragmented basement membrane staining, much less pronounced than in normal breast tissue [14]. Double staining for either of the myoepithelial markers maspin, CK17, or keratin-associated BG3C8 and laminin-1 revealed that tumor-associated myoepithelial cells expressed the other markers, but were either negative or weakly positive for laminin-1. Whenever residual normal breast tissue was present in the carcinomas, myoepithelial cells stained strongly for laminin-1 [14].
2.4. The tumor environment assay as a tool for the identification of differentially expressed genes at the tumor cell–stroma interface
2.4.1. Rationale
When breast cancer progresses from in situ carcinoma to invasive carcinoma, fully differentiated myoepithelial cells and the intact basement membrane are often lost. Thus, the major extracellular matrix component to which epithelial cancer cells become exposed is collagen I [50]. Another conspicuous feature of invasive breast carcinomas is the accumulation of myodifferentiated stromal cells, the so-called myofibroblasts, in the immediate vicinity of tumor cells [51-53]. Therefore, it could be argued that to recapitulate critical aspects of the dynamics of tumor morphogenesis in culture, it is essential to at least include collagen I and myofibroblasts even if the tumors constantly remodel their microenvironment [54]. Previously we determined the origin of myofibroblasts in human breast and showed that the major contribution to myofibroblasts come from fibroblasts rather than vascular smooth muscle cells or pericytes ([16,33]; reviewed in [31,32]). By mixing tumor cells with purified stromal cell populations from normal breast in a three-dimensional collagen gel, we were able to recapitulate tumor histology in this assay. More recently, we have used this assay to identify genes that are differentially expressed at the tumor cell–stroma interface. To do this, the gene expression profiles of tumor cells and myofibroblasts cultured separately were compared with that of the cells in co-culture. A critical step in this endeavor is to mix the isolated RNAs from separate cultures in the correct ratio.
2.4.2. Protocol
2.4.2.1. hTERT immortalization of normal breast fibroblasts
Breast fibroblasts are prepared from reduction mammoplasties [55] and transduced with hTERT [56] in the presence of 8 μg/ml polybrene (Sigma–Aldrich). Cells are selected with addition of 0.7 μg/ml puromycin (Life Technologies).
2.4.2.2. Preparation of collagen gels
The materials and procedures are the same as described above (Section 2.2.2.1) The final concentration of collagen may be optimized for the individual cell type used by diluting Vitrogen with sterile MilliQ water prior to adding the other ingredients. Add cells to the gels—one with tumor cells only, one with (myo)fibroblasts only, and one with both tumor cells and (myo)fibroblasts—and transfer each to a six-well plate (Nunc). On gelation, add 5 ml of the same culture medium to each well.
2.4.2.3. Extraction of RNA from cells embedded in three-dimensional collagen gel
After the appropriate culture period, aspirate culture medium, wash twice in pre-warmed PBS (37 °C), and aspirate thoroughly. Add 2.5 ml Trizol to each well, and extract RNA according to the manufacturer’s instructions for cells grown in monolayer. The collagen gels may be somewhat resistant to solubilization, but this can be achieved by additional pipetting and, if necessary, by vortexing after incubation with Trizol.
2.4.2.4. Normalization of RNAs isolated from separate cultures and co-cultures
Prior to mixing the RNAs extracted from cells cultured separately for subsequent comparison with RNA extracted from the co-cultures, the correct mixing ratio must be determined. Select at least two control genes (for instance, GAPDH and TATAbox binding protein) and two lineage-specific markers for tumor cells and fibroblasts, respectively (for instance, cytokeratin 19 for tumor cells and vimentin for fibroblasts; although a number of tumor cells do express vimentin and thus the markers need to be checked for the specific cell type used), and quantify the expression level in the respective gel extracts using real-time PCR [15,57]. Estimate the mixing ratio and mix the RNA from the cells cultured separately in a ratio corresponding to the expression level in the co-culture. The expression profiles in pooled separate cultures can now be compared with that of direct co-culture by a number of techniques, e.g., differential display [15,58], serial analysis of gene expression (SAGE) [59], cDNA arrays [60].
2.4.3. Comments
The comparison of the in situ gene expression profiles of normal breast versus carcinoma is informative, but the carcinomas comprise a number of different cell types in unpredictable ratios. A different option is the direct analysis of gene expression as a consequence of close interaction between selected cancer cells and myofibroblasts in a three-dimensional microenvironment, which recapitulates critical aspects of tumor histology. A prerequisite, however, for isolating truly differentially expressed genes is the mixing of the RNAs isolated from cells cultured separately in a correct ratio as compared with the cells in direct co-culture. We have recently demonstrated that one effect of co-culturing MCF-7 with myofibroblasts is a prominent proliferation of tumor cells as assessed by Ki67 protein expression [61]. For example, if a mixing ratio corresponding to the initial cell number inoculated in the gels were chosen, the contribution from MCF-7 cells would predominate, and a differential expression of the isolated transcripts could most likely not be verified. The present approach has recently led to the identification of a hitherto undescribed gene, epithelial–stromal interaction 1 (breast), EPSTI1. Of note, whereas EPSTI1 was upregulated approximately six times in the co-culture, in the subsequent real-time PCR verification analysis from 14 breast carcinomas and 5 normal specimens, EPSTI1 was upregulated up to 158 times in the carcinomas (range: 5.6- to 158-fold the expression in normal breast) [15]. Thus, the differential expression in the three-dimensional assay may reflect even a more pronounced upregulation in some carcinomas, further signifying the importance of testing gene expression in co-culture systems.
3. Discussion and concluding remarks
From the structural and functional point of view, the ultimate unit in any organ is clearly the organ itself [62]. In the normal breast, an important component of this structural and functional unit is the double-layered acinus composed of luminal and myoepithelial cells embedded into a collagenous stroma. While this is not strictly equivalent to the entire breast, it nevertheless captures an essential unit of function. On the other hand, during breast cancer progression, the characteristic feature of normal breast, i.e., the boundaries that distinguish epithelium and stroma, disappears as the tumor becomes invasive. In the present article, we have described the ability of three-dimensional collagen I cell culture assays to capture and imitate critical aspects of normal and malignant breast tissue architecture through the formation of polarized acini, and by establishment of a tumor–stroma environment. These assays are highly suitable for observing the heterotypic signaling between different cell types within the human breast. Moreover, we have emphasized the utility of the tumor environment assay for the identification of at least one hitherto undescribed gene, EPSTI1, which is upregulated on confrontation of tumor cells with myofibroblasts.
The finding that myoepithelial cells in the normal breast function to maintain correct polarity of the cellular axis of luminal epithelial cells through their ability to synthesize laminin-1 has been strengthened by results from other studies. Thus, it has previously been shown that α1 chain of laminin-1 is associated with polarization of epithelial cells in lung [63] and kidney [64]. Furthermore, laminin-1 was shown to induce milk production in mammary epithelial cells [2,65] and to polarize and induce acinus formation in human submandibular epithelial cells [66,67] and human prostate epithelial cells [67]. Hoffman et al. [67] have shown that a peptide fragment (AG-73) derived from the α1 chain of laminin-1 could induce acinus-like structures in human submandibular gland cells. Bello-DeOcampo et al. [68] compared the ability of matrigel, laminin-1, collagen IV, and fibronectin to induce acinus formation in prostate epithelial cells. They found that rBM and laminin-1 were equally effective in acinus formation, whereas no effect was seen with collagen IV or fibronectin. O’Brien et al. [69] showed that polarity of Madin–Darby canine kidney (MDCK) epithelial cells cultured as cysts in collagen I gel was controlled by the Rac1 GTPase through assembly of laminin into basement membrane. Our recent results with dystroglycan in breast epithelial tumor cells is suggestive of the possibility that this receptor for laminin-1, rather than an integrin, may play an important role in polarity-induced acinus formation [70]. However, Runswick et al. [71] have demonstrated that acinar polarity is governed not only by cell–basement membrane interactions, but also by epithelial–myoepithelial interaction through desmosomes. Desmosomal blocking peptides were shown to disrupt acinus formation and cell positioning of mammary luminal epithelial cells when cultured within rBM [70].
The three-dimensional collagen I assay allows the formation of the double-layered breast acini, a feature that cannot be achieved in the rBM assay because of the intrinsic ability of the laminin component of rBM to allow acinus formation by luminal epithelial cells. Furthermore, the collagen assay allows for identification of aberrant interactions as seen by the inability of tumor-derived/-associated myoepithelial cells to correct polarity of luminal epithelial cells unless they produce laminin-1. This finding may provide a plausible explanation as to why breast carcinomas that show partial myoepithelial differentiation do not necessarily confer a better prognosis for the patients [48]. However, in some breast carcinomas, neoplastic myoepithelial cells are present and appear to be functional. This is the case with the adenoid cystic carcinoma which has a better prognosis and does not metastasize to distant sites [72]. Not surprisingly, in these carcinomas laminin is deposited and the neoplastic luminal epithelial cells polarize correctly. Thus, in grade I carcinoma, which has a favorable prognosis and is often polarized correctly [72], a fragmented BM is present [44,45] and at the ultra-structral level the presence of myoepithelial cells has been reported [73]. In this context, it is tempting to speculate that the presence of myoepithelial cells keeps the genetic alterations within the malignant cells in check via production of basement membrane components and induction of polarity. That overtly malignant breast cells can revert to a “normal” phenotype has been conclusively shown in three-dimensional cultures [74,75]. In vivo, the myoepithelial cells could at least in theory function as both a tumor suppressor and a reverting agent, preventing the conversion of noninvasive tumors to invasive cancers. In the malignant breast, the carcinoma cells of luminal origin outnumber the myoepithelial cells and invade the surrounding collagenous stroma through a disrupted basement membrane. The stromal reaction results in profound cellular and extra-cellular changes commonly referred to as desmoplasia (for review see [31]). The most distinguishing feature of the stromal reaction is the appearance of myofibroblasts, originally described in healing wounds by Gabbiani et al. [76] and later shown in the breast to be derived primarily from resident fibroblasts [16]. Myofibroblasts are absent in normal breast tissue, but are the most abundant stromal cell type in human breast cancer where they constitute up to 80% of the stromal cells and are closely interwoven with the carcinoma cells [53]. Whereas monolayer cultures of breast fibroblasts and defined culture conditions have allowed the identification of a number of novel genes relevant to breast neoplasia [77], this approach has focused mainly on the stromal compartment, i.e., the conversion of fibroblasts to myofibroblasts. The three-dimensional tumor environment assay offers the opportunity to identify genes that may be regulated in either or both compartments on direct tumor cell–myofibroblast interactions and, in addition, perhaps to identify genes that may be particularly relevant for tumor morphogenesis, invasion, and metastasis, all processes that take place in the three-dimensional plane. The three-dimensional co-cultures described here have unraveled new aspects of interactions between different cell types such as the importance of myoepithelium-derived laminin-1 for luminal polarization and identification of hitherto unknown genes expressed as a result of tumor–stromal interactions.
Acknowledgments
We thank Tove Marianne Lund for expert technical assistance, and the Aasted Clinic, the Private Clinic, and the Søllerød Plastic Surgery Clinic are gratefully acknowledged for providing the biopsy material. We also thank the following sources of funding: The Icelandic Research Fund for Graduate Students, Dansk Kræftforskningsfond, The Dagmar Marshalls Fond (to T.G.), The Danish Research Council, The Novo Nordisk Foundation, The Thaysen Foundation, Friis Fonden, The Meyer Foundation, The Danish Cancer Society, The Danish Research Council (to O.W.P. and L.R.-J.), Weimanns Legat (to L.R.-J.), US National Cancer Institute (Grant CA-64786 to M.J.B. and O.W.P.), and US Department of Energy Offce of Biological and Environmental Research (Contract DE-AC03-76SF00098 to M.J.B.).
References
- [1].Emerman JT, Pitelka DR. In Vitro. 1977;13:316–328. doi: 10.1007/BF02616178. [DOI] [PubMed] [Google Scholar]
- [2].Li ML, Aggeler J, Farson DA, Hatier C, Hassell J, Bissell MJ. Proc. Natl. Acad. Sci. USA. 1987;84:136–140. doi: 10.1073/pnas.84.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Barcellos-Hoff MH, Aggeler J, Ram TG, Bissell MJ. Development. 1989;105:223–235. doi: 10.1242/dev.105.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tateyama S, Furukawa H, Yamaguchi R, Nasaka D, Kondo F. Res. Vet. Sci. 1990;49:14–19. [PubMed] [Google Scholar]
- [5].Purup S, Vestergaard M, Sejrsen K. Adv. Exp. Med. Biol. 2000;480:27–43. doi: 10.1007/0-306-46832-8_4. [DOI] [PubMed] [Google Scholar]
- [6].Petersen OW, Rønnov-Jessen L, Bissell MJ. Breast J. 1995;1:22–35. [Google Scholar]
- [7].Schmeichel KL, Weaver VM, Bissell MJ. J. Mamm. Gland Biol. Neoplasia. 1998;3:201–213. doi: 10.1023/a:1018751124382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang J, Guzman R, Richards J, Jentoft V, DeVault MR, Wellings SR, Nandi S. J. Natl. Cancer Inst. 1980;65:337–343. [PubMed] [Google Scholar]
- [9].Petersen OW, Rønnov-Jessen L, Howlett AR, Bissell MJ. Proc. Natl. Acad. Sci. USA. 1992;89:9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Baibakov BA, Chipisheva TA, Guelstein VI, Ermilova VD, Polevaya EB, Vasiliev JM, Margolis LB. In Vitro Cell Dev. Biol. Anim. 1994;30A:490–495. [PubMed] [Google Scholar]
- [11].Sternlicht MD, Kedeshian P, Shao ZM, Safarians S, Barsky SH. Clin. Cancer Res. 1997;3:1949–1958. [PubMed] [Google Scholar]
- [12].Barcellos-Hoff MH, Ravani SA. Cancer Res. 2000;60:1254–1260. [PubMed] [Google Scholar]
- [13].Tlsty TD, Hein PW. Curr. Opin. Genet. Dev. 2001;11:54–59. doi: 10.1016/s0959-437x(00)00156-8. [DOI] [PubMed] [Google Scholar]
- [14].Gudjonsson T, Rønnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. J. Cell Sci. 2002;115:39–50. doi: 10.1242/jcs.115.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nielsen HL, Rønnov-Jessen L, Villadsen R, Petersen OW. Genomics. 2002;79:703–710. doi: 10.1006/geno.2002.6755. [DOI] [PubMed] [Google Scholar]
- [16].Rønnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. J. Clin. Invest. 1995;95:859–873. doi: 10.1172/JCI117736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Joshi K, Smith JA, Perusinghe N, Monoghan P. Am. J. Pathol. 1986;124:199–206. [PMC free article] [PubMed] [Google Scholar]
- [18].Petersen OW, van Deurs B. Cancer Res. 1987;47:856–866. [PubMed] [Google Scholar]
- [19].O’Hare MJ, Ormerod MG, Monaghan P, Lane EB, Gusterson BA. Differentiation. 1991;46:209–221. doi: 10.1111/j.1432-0436.1991.tb00883.x. [DOI] [PubMed] [Google Scholar]
- [20].Clarke C, Titley J, Davies S, O’Hare MJ. Epithelial Cell Biol. 1994;3:38–46. [PubMed] [Google Scholar]
- [21].Lakhani SR, O’Hare MJ. Breast Cancer Res. 2001;3:1–4. doi: 10.1186/bcr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dairkee S, Heid HW. In Vitro Cell Dev. Biol. Anim. 1993;29A:427–432. doi: 10.1007/BF02633993. [DOI] [PubMed] [Google Scholar]
- [23].Gomm JJ, Browne PJ, Coope RC, Liu QY, Buluwela L, Coombes RC. Anal. Biochem. 1995;226:91–99. doi: 10.1006/abio.1995.1196. [DOI] [PubMed] [Google Scholar]
- [24].Stingl J, Eaves CJ, Kuusk U, Emerman JT. Differentiation. 1998;63:201–213. doi: 10.1111/j.1432-0436.1998.00201.x. [DOI] [PubMed] [Google Scholar]
- [25].Péchoux C, Gudjonsson T, Rønnov-Jessen L, Bissell MJ, Petersen OW. Dev. Biol. 1999;206:88–99. doi: 10.1006/dbio.1998.9133. [DOI] [PubMed] [Google Scholar]
- [26].Gudjonsson T, Villadsen R, Nielsen HL, Rønnov-Jessen L, Bissell MJ, Petersen OW. Genes Dev. 2002;16:693–706. doi: 10.1101/gad.952602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Stampfer M, Hallowes RC, Hackett AJ. In Vitro. 1980;16:415–425. doi: 10.1007/BF02618365. [DOI] [PubMed] [Google Scholar]
- [28].Hammond SL, Ham RG, Stampfer MR. Proc. Natl. Acad. Sci. USA. 1984;81:5435–5439. doi: 10.1073/pnas.81.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Taylor-Papadimitriou J, Stampfer M, Bartek J, Lewis A, Boshell M, Lane EB, Leigh IM. J. Cell Sci. 1989;94(Pt 3):403–413. doi: 10.1242/jcs.94.3.403. [DOI] [PubMed] [Google Scholar]
- [30].Petersen OW, van Deurs B. Differentiation. 1988;39:197–215. doi: 10.1111/j.1432-0436.1988.tb00094.x. [DOI] [PubMed] [Google Scholar]
- [31].Rønnov-Jessen L, Petersen OW, Bissell MJ. Physiol. Rev. 1996;76:69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- [32].Rønnov-Jessen L. Breast J. 1996;2:320–339. [Google Scholar]
- [33].Rønnov-Jessen L, Celis JE, van Deurs B, Petersen OW. J. Histochem. Cytochem. 1992;40:475–486. doi: 10.1177/40.4.1552184. [DOI] [PubMed] [Google Scholar]
- [34].Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW. Nat. Genet. 1999;21:115–118. doi: 10.1038/5063. [DOI] [PubMed] [Google Scholar]
- [35].Ouellette MM, Liao M, Herbert BS, Johnson M, Holt SE, Liss HS, Shay JW, Wright WE. J. Biol. Chem. 2000;275:10072–10076. doi: 10.1074/jbc.275.14.10072. [DOI] [PubMed] [Google Scholar]
- [36].O’Hare MJ, Bond J, Clarke C, Takeuchi Y, Atherton AJ, Berry C, Moody J, Silver AR, Davies DC, Alsop AE, Neville AM, Jat PS. Proc. Natl. Acad. Sci. USA. 2001;98:646–651. doi: 10.1073/pnas.98.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Howlett AR, Bissell MJ. Epith. Cell Biol. 1993;2:79–89. [PubMed] [Google Scholar]
- [38].Rønnov-Jessen L, van Deurs B, Nielsen M, Petersen OW. In vitro Cell Dev. Biol. 1992;28A:273–283. doi: 10.1007/BF02634244. [DOI] [PubMed] [Google Scholar]
- [39].Hallowes R, Bone E, Jones W. In: Tissue Culture in Medical Research (II) Richards R, Rajan K, editors. Pergamon Press; Oxford: 1980. [Google Scholar]
- [40].Takahashi K, Kawahara S, Ono T. Jpn. J. Cancer Res. 1990;81:52–57. doi: 10.1111/j.1349-7006.1990.tb02506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hirai Y, Lochter A, Galosy S, Koshida S, Niwa S, Bissell MJ. J. Cell Biol. 1998;140:159–169. doi: 10.1083/jcb.140.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Colognato H, Yurchenco PD. Dev. Dyn. 2000;218:213–234. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- [43].Rudland PS. Cancer Metastasis Rev. 1987;6:55–83. doi: 10.1007/BF00047609. [DOI] [PubMed] [Google Scholar]
- [44].Albrechtsen R, Nielsen M, Wewer U, Engvall E, Ruoslahti E. Cancer Res. 1981;41:5076–5081. [PubMed] [Google Scholar]
- [45].Gusterson BA, Warburton MJ, Mitchell D, Ellison M, Neville AM, Rudland PS. Cancer Res. 1982;42:4763–4770. [PubMed] [Google Scholar]
- [46].Petersen OW, Lind Nielsen H, Gudjonsson T, Villadsen R, Rønnov-Jessen L, Bissell MJ. Breast Cancer Res. 2001;3:213–217. doi: 10.1186/bcr298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wetzels RH, Holland R, van Haelst UJ, Lane EB, Leigh IM, Ramaekers FC. Am J. Pathol. 1989;134:571–579. [PMC free article] [PubMed] [Google Scholar]
- [48].Malzahn K, Mitze M, Thoenes M, Moll R. Virchow’s Arch. 1998;433:119–129. doi: 10.1007/s004280050226. [DOI] [PubMed] [Google Scholar]
- [49].Koshikawa N, Gianelli G, Cirulli V, Miyazaki K, Quaranta V. J. Cell Biol. 2000;148:615–624. doi: 10.1083/jcb.148.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lagace R, Grimaud JA, Schurch W, Seemayer TA. Virchow’s Arch. A. 1985;408:49–59. doi: 10.1007/BF00739962. [DOI] [PubMed] [Google Scholar]
- [51].Skalli O, Ropraz P, Trzeciak A, Benzonana G, Gillessen D, Gabbiani G. J. Cell Biol. 1986;103:2787–2796. doi: 10.1083/jcb.103.6.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tsukada T, McNutt MA, Ross R, Gown AM. Am. J. Pathol. 1987;127:389–402. [PMC free article] [PubMed] [Google Scholar]
- [53].Sappino AP, Skalli O, Jackson B, Schurch W, Gabbiani G. Int. J. Cancer. 1988;41:707–712. doi: 10.1002/ijc.2910410512. [DOI] [PubMed] [Google Scholar]
- [54].Bissell MJ, Radisky DC. Nat. Rev. (Cancer) 2001;1:48–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rønnov-Jessen L, van Deurs B, Celis JE, Petersen OW. Lab. Invest. 1990;63:532–543. [PubMed] [Google Scholar]
- [56].Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- [57].Bustin SA. J. Mol. Endocrinol. 2000;25:169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- [58].Liang P, Pardee A. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- [59].Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- [60].Brown PO, Botstein D. Nat. Genet. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- [61].Petersen OW, Nielsen HL, Gudjonsson T, Villadsen R, Rank F, Niebuhr A, Bissell MJ, Rønnov-Jessen L. Am. J. Pathol. 2003;162:391–402. doi: 10.1016/S0002-9440(10)63834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Bissell MJ, Hall HG. The Mammary Gland: Development, Regulation and Function. 1987. pp. 97–146. [Google Scholar]
- [63].Schuger L, Yurchenco P, Relan NK, Yang Y. Int. J. Dev. Biol. 1998;42:217–220. [PubMed] [Google Scholar]
- [64].Klein G, Langegger M, Timpl R, Ekblom P. Cell. 1988;55:331–341. doi: 10.1016/0092-8674(88)90056-6. [DOI] [PubMed] [Google Scholar]
- [65].Streuli CH, Schmidhauser C, Bailey N, Yurchenco P, Skubitz AP, Roskelley C, Bissell MJ. J. Cell Biol. 1995;129:591–603. doi: 10.1083/jcb.129.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hoffman MP, Kibbey MC, Letterio JJ, Kleinman HK. J. Cell Sci. 1996;109:2013–2021. doi: 10.1242/jcs.109.8.2013. [DOI] [PubMed] [Google Scholar]
- [67].Hoffman MP, Nomizu M, Roque E, Lee S, Jung DW, Yamada Y, Kleinman HK. J. Biol. Chem. 1998;273:28633–28641. doi: 10.1074/jbc.273.44.28633. [DOI] [PubMed] [Google Scholar]
- [68].Bello-DeOcampo D, Kleinman HK, Deocampo ND, Webber MM. Prostate. 2001;46:142–153. doi: 10.1002/1097-0045(20010201)46:2<142::aid-pros1018>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- [69].O’Brien LE, Jou TS, Pollack AL, Zhang Q, Hansen SH, Yurchenco P, Mostov KE. Nat. Cell Biol. 2001;3:831–838. doi: 10.1038/ncb0901-831. [DOI] [PubMed] [Google Scholar]
- [70].Muschler J, Levy D, Boudreau R, Henry M, Campbell K, Bissell MJ. Cancer Res. 2002;62:7102–7109. [PubMed] [Google Scholar]
- [71].Runswick SK, O’Hare MJ, Jones L, Streuli CH, Garrod DR. Nat. Cell Biol. 2001;3:823–830. doi: 10.1038/ncb0901-823. [DOI] [PubMed] [Google Scholar]
- [72].Kasami M, Olson SJ, Simpson JF, Page DL. Histopathology. 1998;32:232–238. doi: 10.1046/j.1365-2559.1998.00383.x. [DOI] [PubMed] [Google Scholar]
- [73].Hayashi Y, Aoki Y, Eto R, Tokuoka S. Acta Pathol. Japon. 1984;34:537–552. doi: 10.1111/j.1440-1827.1984.tb07582.x. [DOI] [PubMed] [Google Scholar]
- [74].Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. J. Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wang F, Hansen RK, Radisky D, Yoneda T, Barcellos-Hoff MH, Petersen OW, Turley EA, Bissell MJ. J. Natl. Cancer Inst. 2002;94:1494–1503. doi: 10.1093/jnci/94.19.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gabbiani G, Ryan GB, Majne G. Experientia. 1971;27:549–550. doi: 10.1007/BF02147594. [DOI] [PubMed] [Google Scholar]
- [77].Rønnov-Jessen L, Villadsen R, Edwards JC, Petersen OW. Am J. Pathol. 2002;161:471–480. doi: 10.1016/s0002-9440(10)64203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]