

Abstract
Follicle-stimulating hormone (FSH), produced by pituitary gonadotrope cells, is required for maturation of ovarian follicles. The FSHβ subunit is the limiting factor for production of mature hormone and provides biological specificity. Activin dramatically induces FSHβ transcription, and the secondary rise in FSH, important for follicular development, is dependent on this induction. Thus, regulation of FSHβ levels by activin is critical for female reproductive fitness. This review discusses activin signaling pathways, transcription factors and FSHβ promoter elements required for activin responsiveness. Since FoxL2, a forkhead transcription factor, was recently shown to be instrumental in relaying activin signaling to the FSHβ promoter, we focus on its role and the inter-relatedness of several key players in activin responsiveness on the FSHβ promoter.
Activin Regulation of FSHβ Gene Expression
FSH is a dimeric glycoprotein hormone composed of a unique β subunit and a common α subunit shared with luteinizing hormone (LH) and thyroid-stimulating hormone. Low FSH levels prevent follicular growth, while high levels are associated with premature ovarian failure [1]. In fact, female mice lacking FSHβ exhibit an arrest in ovarian folliculogenesis, while women with mutations in the FSHβ gene are infertile [2]. The importance of appropriate regulation of FSH levels is also illustrated in FSHβ transgenic mice in which superovulation occurred without ovary depletion when the FSHβ promoter was used to drive expression of the FSHβ gene [3]. Therefore, these studies demonstrate that proper regulation of FSH levels is critical for female fertility.
Transcription of the FSHβ gene is dynamically regulated during the estrous cycle. Changes in FSHβ mRNA levels precede changes in FSH concentration in the circulation, strongly implying that FSHβ transcription is the rate-limiting factor in the production of the mature hormone [4, 5]. Prior to the GnRH-induced ovulatory surge in the afternoon of proestrus, FSHβ mRNA levels increase five fold concomitantly with LHβ. Later, during estrus, transcription of FSHβ again increases by three fold [4, 5]. A secondary increase of FSH also occurs during the human menstrual cycle at the end of the luteal phase and in the beginning of the follicular phase. This secondary FSH rise is necessary for follicular development and is dependent on activin [6, 7]. Notably, in female rats infused with follistatin, an inhibitor of activin, both FSHβ mRNA and FSH levels in the blood can be reduced during the secondary rise [8]. Expression of both intrapituitary follistatin and ovarian inhibin fluctuate during the estrous cycle in opposition to the levels of FSHβ mRNA [8–10], suggesting that bioavailability of activin, through changes in follistatin and/or inhibin levels, is a critical regulatory component of FSHβ synthesis.
Activin is a potent regulator of FSHβ gene expression and was originally identified as a component of ovarian follicular fluid that increased FSHβ synthesis and FSH secretion from pituitary gonadotrope cells [11–13]. Although activin was known for many years to regulate FSH production, it was not until the discovery that FSHβ was synthesized by the gonadotrope-derived LβT2 cell line that the molecular mechanisms of activin induction could begin to be elucidated [14]. In this review, we focus our attention on activin regulation of the FSHβ promoter including recent advances in our understanding of activin-related signal transduction mechanisms. This is quite timely since novel players, not previously associated with activin signaling pathways, have recently come to light.
Activin Signaling via Smad Proteins
Activin signaling in gonadotrope cells through type II receptors (ActRII A/B) and type I receptors (activin receptor-like kinases, ALK 4/7) (Box 1), results in the phosphorylation of receptor-associated Smad proteins, Smad2 and Smad3 (Figure 1) [15–17]. Upon phosphorylation, Smad2/3 bind to Smad4 and translocate into the nucleus of gonadotrope cells [15]. Once in the nucleus, Smad proteins can induce or repress gene expression as a heterodimer or in combination with other transcription factors. Smad3/4 can bind DNA directly through a defined Smad-binding element (SBE) (GTCTAG[N]C) or a Smad half site (GTCT). Smad2 and/or Smad3 have also been shown to interact with transcription factors such as AP-1 [18], FAST-1 [19], FoxO [20], and steroid receptors [21–24].
Box 1. Activin isoforms and activin receptors.
Activin is a dimer of two β subunits. There are multiple β isoforms: βA, βB, βC and βE. βA and βB, which share 65% sequence identity but are differentially expressed, form dimers known to have physiological roles. Thus, activin A is a homodimer of βA subunits and activin B contains two βB subunits. βA and βB can also form heterodimers. Activin A is the predominant isoform expressed in the ovary, while gonadotropes express higher levels of activin B [51]. Activin B is also expressed throughout the pituitary [52] and in cultured pituitary cells [53]. Since activin is present at low levels in the blood, activin action in gonadotrope cells is considered to be predominantly paracrine or autocrine [54]. Both activin A and B can induce FSHβ gene expression in immortalized gonadotrope cells [25]. Activin A binds to the type II receptor A (ActRIIA) on the surface of gonadotrope cells, which in turn results in the recruitment and phosphorylation of the type I receptor, ALK4. Activin B binds with higher affinity to ActRIIB receptors, which selectively recruit and activate ALK7 [55–57]. All of these receptors are expressed in pituitary gonadotrope cells [14, 54]. ALK4 and ALK7 are likely to be functionally interchangeable in gonadotrope cells, since both receptors phosphorylate Smad2 and Smad3 [58], and effects of constitutively active forms of these receptors map to the same regions of the FSHβ promoter [25]. Since the levels of activin receptors remain relatively constant throughout the estrous cycle, modulation of activin signaling probably occurs due to changes in follistatin and/or inhibin levels as well as interactions with other hormone signaling pathways (recently reviewed in [50]).
Figure 1.

Activin signaling regulates transcription of the FSHβ gene. Activin signaling, via ActRIIA/B and ALK4/7 receptors, results in the phosphorylation of Smad2 and 3, which then dimerize with the common Smad4 and translocate into the nucleus of the cell. Activated Smad3, and potentially Smad2, regulate transcription of FSHβ and other specific target genes. The inhibitory Smad7 prevents the phosphorylation of Smad3. Activin signaling also results in activation of the p38 MAPK signaling pathway, potentially via TAK1, however, it is still unclear how p38 relays activin signaling to the FSHβ promoter.
Although it is evident that Smad-dependent signaling is necessary for activin induction of FSHβ in rodents, it is still unclear whether Smads play a role in other mammalian species. The evidence thus far suggests that species-specific differences in the FSHβ promoter result in greater or lesser sensitivity to transcriptional regulation by Smad proteins [25, 26]. Overexpression of Smad7 (an inhibitory Smad) reduces the effects of activin on the murine FSHβ promoter in LβT2 cells [16, 27–29]; however, use of dominant negative (DN) Smads has produced inconsistent results. Overexpression of DN Smad2 reduces activin induction of murine FSHβ mRNA in LβT2 cells [16] and a DN Smad3 lacking the C-terminal tail, reduced activin induction of rat FSHβ [30]. In contrast, a DN Smad3 with mutated phosphorylation sites had no effect on activin induction of the ovine FSHβ gene [31], suggesting a species-specific response to Smad-dependent signaling.
There is also some debate about the contributions of the various Smad proteins to activin induction in gonadotrope cells. Activin induction of murine FSH transcription required Smad4 [32]. Overexpression of Smad3 and Smad4, but not Smad2, induced rat and murine FSHβ gene expression [17, 30, 33]; however, Smad2 overexpression was shown to be low in LβT2 cells [17, 34]. Interestingly, overexpression of a Smad2 isoform lacking exon 3 resulted in FSHβ induction, while replacement of exon 3 of Smad3 with that of Smad2, rendered Smad3 unable to be expressed in transient transfection experiments [17]. In contrast to the rodent promoters, Smad3 overexpression was shown to have a minimal effect on the ovine [31] and human FSHβ promoters [25, 35], which is likely due to their lack of a consensus SBE that is present specifically in the rodent FSHβ promoters. RNAi-mediated knockdown of Smad2 and Smad3 was reported to reduce basal expression of the rodent FSHβ gene [17, 34]. Since LβT2 cells express endogenous activin, it is unclear from these experiments whether basal gene expression is truly affected by Smads. Pretreatment with follistatin could help resolve this question. Several reports demonstrated that knockdown of Smad2 or Smad3 affected responsiveness of the rodent FSHβ gene to activin or a constitutively active form of ALK4 [17, 35]. On the other hand, another report saw no effect of Smad2 knockdown on activin induction of FSHβ [34]. Although Smad2 may be dispensable for activin signaling in gonadotropes, the overall evidence indicates that Smad3 and Smad4 are necessary for induction of the rodent FSHβ promoters. The necessity of Smad proteins for activin induction of other mammalian FSHβ promoters still remains to be investigated.
Smad-Independent Activin Signaling via TAK1 and p38 Kinases
In addition to Smad proteins, activin signaling has been reported to result in the activation of Akt, ERK1/2 and p38 signaling pathways in LβT2 cells and ovine primary pituitary cells [16]. Blockage of Akt signaling using a PI3K inhibitor reduced the amount of Smad2 phosphorylated by activin. However, inhibition of Akt signaling did not alter activin induction of the ovine and murine FSHβ genes suggesting that this pathway is not necessary for activin responsiveness, at least in these species [16]. Inhibition of JNK and ERK1/2 signaling pathways did not appear to alter activin signaling on rodent or ovine FSHβ promoters [16, 29–31]. In contrast, the p38 mitogen-activated protein kinase (MAPK) appears to play a role in activin induction of FSHβ gene expression, since an inhibitor of p38 MAPK reduced FSHβ mRNA expression. We, and others, have shown that treatment of LβT2 cells with inhibitors of p38 reduced activin signaling on ovine and murine FSHβ promoters [28, 29, 31], although one report did not see an effect [16]. Interestingly, the TAK1 inhibitor, 5Z-7-Oxoaeanol, has been reported to block activin induction of ovine FSHβ gene expression [31]. Overexpression of TAK1 along with its binding proteins, TAB2/TAB3, resulted in the induction of the ovine FSHβ promoter to equivalent levels as activin. A DN-TAK1 also reduced activin induction [31]. These data indicate that the TAK1/p38 signaling pathway may also play an important role in activin responsiveness in gonadotrope cells.
Defining Activin Responsive Regions on the FSHβ Promoter
Activin responsive regions on the FSHβ promoter were originally characterized as putative SBEs although only one of these elements in the rodent promoter has been shown to directly bind Smad proteins [30, 33, 35]. Since the other elements do not bind Smads directly or bind them with low affinity, the focus shifted to defining what other transcription factors bind to these elements and whether these factors can interact with Smads to tether them to the promoter. To fully define these elements, it is important to determine whether they are necessary and sufficient for activin induction of FSHβ gene expression using cis mutations in the FSHβ promoter and tandem copies of the element linked to a heterologous promoter, respectively.
Smad Regulation of FSHβ Gene Expression via the -267 SBE
A single consensus SBE at –267/–260 of the murine and –266/–259 of the rat FSHβ promoter has been extensively characterized (Figure 2) [30, 33, 35]. Mutation of this element diminishes the response to activin [30], although the response is not completely blocked, suggesting the presence of additional regulatory elements on the rodent promoter that are responsive to activin signaling. The overall evidence indicates that Smad2/3/4 bind to this consensus element. The DNA binding domains of recombinant Smad3/4 were reported to bind the –267 site [35]. Antibodies to Smad2/3/4 also alter complex formation on this region of the promoter in gel-shift assays using LβT2 nuclear extracts, suggesting their presence in the complexes [30, 33, 35]. Magnetic separation of proteins bound to a biotinylated FSHβ oligonucleotide and DNA precipitation experiments also demonstrated binding of Smad2/3/4 to this element [30, 35]. Interestingly, this SBE is present only in rodent FSHβ promoters, indicating that it may be involved in the species-specific sensitivity to Smad proteins. In support of this idea, addition of this element to the human FSHβ promoter increased its responsiveness to activin and Smad3 overexpression [35]. It is possible that the consensus SBE in the murine and rat promoters plays a species-specific role in the rapid regulation of FSHβ synthesis necessary in the short rodent estrous cycle.
Figure 2.

FSHβ promoter elements important for activin responsiveness. Numerous studies have identified elements in the mouse, rat, sheep, pig, and human FSHβ promoters that are critical for activin induction. Since all of the elements have been examined in the murine promoter, it was used as a basis for comparison. The number above each element corresponds to the number in the original publication, while the number next to the sequence indicates the position of the 5′ nucleotide that is presented. Mutated residues are underlined and the relevant publications are indicated in brackets. The –350 site binds FoxL2 in the murine but not the human promoter; other species have not yet been examined. The –267 SBE is present only in the rodent promoters. The murine –208 and corresponding human –223 FoxL2 sites play a critical role in activin responsiveness; other species have not yet been examined. The binding elements in the more proximal region of the FSHβ promoter are not as clearly defined. The forkhead (FH) consensus is aligned beneath the corresponding sites in this region to help identify potential FoxL2 binding elements.
Pbx/Prep and Ptx Regulation of FSHβ Gene Expression via the –120 and –54 Elements
In silico scanning of the ovine FSHβ promoter revealed many putative SBEs [14]. Mutation of a putative Smad half site at –134 in the ovine and the homologous –120 in the murine proximal promoter resulted in a substantial decrease in activin responsiveness (Figure 2) [36]. The TALE homeodomain proteins Pbx1 and Prep1 were found to bind to this element in vitro and in vivo [36]. EMSA demonstrated that Pbx1 and Prep1 recognized a CTGTCTATCCAA element encompassing the putative Smad half site on the ovine promoter. Chromatin immunoprecipitation experiments showed that both proteins were recruited to the murine proximal FSHβ promoter. Pbx1 and Prep1 were also shown to interact with Smad2/3/4, and a Smad4 antibody disrupted a higher-order complex containing Pbx and Prep observed in EMSA [36], while antibody to Smad 2/3 caused a supershift [27], suggesting that Smad proteins can be recruited to the proximal FSHβ promoter indirectly via protein-protein interactions with Pbx/Prep. Corpuz et al. found that the –267 consensus SBE, that binds Smads directly, and the –120 Pbx/Prep site, that tethers Smads, were each sufficient to convey activin responsiveness when linked in tandem to a heterologous promoter [25]. In contrast, tandem copies of a Smad half site were not sufficient for induction by activin, indicating that additional high-affinity interactions than those provided by a Smad half site are required [25]. In addition to Pbx/Prep, Smads have also been shown to interact with Ptx [33, 37, 38]. Ptx binds to a paired-like homeodomain-binding site at –54 of the rat and murine FSHβ promoters (–65 of the human promoter) (Figure 2) [38, 39] to coordinate basal expression and activin induction. Thus, these studies provide evidence that activin signaling to the FSHβ promoter involves the transcription factors, Pbx/Prep and Ptx, which tether Smad proteins to the promoter.
FoxL2 Regulation of FSHβ Gene Expression via Sites at –350, –208, –153, and –106
Mutation of an additional putative Smad half site (–167 and –153 in the ovine and murine promoters, respectively) resulted in a substantial decrease in activin responsiveness (Figure 2) [36]. Smad regulation of this element was suggested by experiments showing that FSHβ induction by Smad3 overexpression was reduced with mutations in the murine –153 element [25, 27]. However, many attempts to demonstrate Smad binding directly to this site were unsuccessful, making it unclear whether this site represented a bona fide SBE or an element that binds additional transcription factors which may coordinate with Smad proteins to elicit an activin response.
Further analysis of the FSHβ promoter demonstrated that FoxL2, a member of the forkhead family of transcription factors, was essential for activin responsiveness. FoxL2 was previously reported to be expressed in gonadotrope cells and to regulate transcription of the follistatin and GnRH receptor genes [40–42]. FoxL2-mediated induction of the follistatin and GnRH receptor genes was dependent on adjacent Smad sites, and FoxL2 functioned in complex with Smad3. Interestingly, Blepharophimosis Ptosis Epicanthus Inversus Syndrome (BPES) is an autosomal dominant disorder which is characterized by distinctive eyelid abnormalities that result from a mutation in the FoxL2 gene. Two clinical subtypes have been described, and type I is associated with premature ovarian failure [43].
Our recent studies have shown that FoxL2 binds the –153 element in the murine FSHβ promoter and the homologous –164 element in the human promoter (Figure 2) [25]. The homologous porcine –164 site was also reported to bind FoxL2 with a much higher affinity than the murine or human promoters due to a single base-pair substitution [44]. Mutation of the putative Smad half site (AGAC) in this element disrupts FoxL2 binding, indicating that these bases play an important role in the FoxL2 binding element. In addition to the proximal –153 site, the murine and human FSHβ promoters contain other FoxL2 binding sites. Elements at –350 and –208 in the murine promoter, and the equivalent –223 site in the human promoter, bind FoxL2 (Figure 2) [25]. These upstream sites bind FoxL2 with higher affinity, and, interestingly, lack an overlapping Smad half-site sequence. Furthermore, in contrast to the follistatin gene, an adjacent Smad half-site at -355 does not contribute to FoxL2 binding or activin induction of the murine FSHβ gene. Taken together, these results indicate that FoxL2 function is not solely to recruit Smads to the promoter.
Somewhat surprisingly, the FoxL2 elements were not sufficient to convey activin responsiveness on their own. However, these elements were necessary for the response since cis mutation of any of these elements rendered the FSHβ promoter unresponsive to activin, even in the presence of intact SBEs. Induction of the murine promoter by Smad3 overexpression was also diminished by mutations in the FoxL2 sites, implying protein-protein interactions between FoxL2 and Smad proteins. Although the human promoter is not responsive to Smad overexpression, it still requires the FoxL2 sites for induction by activin [25], suggesting that activin directly activates FoxL2.
Analysis of the murine FSHβ promoter also revealed two other activin responsive regions at –139 and –106 (Figure 2) [27] that were originally identified as a steroid hormone response element [24] and an AP-1 element [45]. FoxL2 was reported by Corpuz et al [25] to bind to an extended element (Figure 2) compared to the previously reported 7-base-pair forkhead element [46], which is in agreement with FoxL2 binding as a dimer [44]. In this case, the –153 element would extend to –138 and may partially explain the involvement of the previously identified –139 hormone response element in activin signaling. Interestingly, the AP-1 binding site at –120 in the ovine promoter was shown to mediate GnRH response in cell culture model systems [45, 47], but was reported to be dispensable for activin responsiveness. In contrast, the corresponding -106 element in the murine promoter was reported to play a role in activin induction, although it was not established which proteins mediated this effect [27]. More recently, Lamba et al. identified the –106 site as a FoxL2 binding site though different base pairs were mutated [44]. Since the FoxL2 binding site in the reverse orientation in the murine promoter (CTAAACAA) is adjacent to the –120 site (discussed previously as a Pbx binding element), it is possible that specific cis-mutations interfere with the binding of FoxL2 and/or Pbx/Prep to this region. The role of the –106 element in GnRH signaling may be due to the necessity for endogenous activin in GnRH responsiveness on the FSHβ gene [14, 29].
Runx Regulation of Murine FSHβ Gene Expression via an Element at –159
Su et al. demonstrated that disruption of a sequence juxtaposed to the –167 activin-responsive site within the ovine FSHβ promoter caused severe dysregulation of basal expression and transcriptional regulation by activin in LβT2 cells and in vivo [48]. In silico analysis indicated that this sequence could be a putative binding site for the Runx family of transcription factors. Recently, we have demonstrated that Runx1, 2 and 3 are expressed in gonadotrope cells and that overexpression of Runx proteins blocks activin induction of the murine, ovine and human FSHβ promoters [49], indicating that repression by Runx is conserved among these mammalian species. We also identified a Runx cis-regulatory element at –159 in the murine FSHβ promoter next to the –153 site critical for activin responsiveness (Figure 2). Furthermore, we showed that the –159 site was necessary for Runx2 binding to the FSHβ promoter and essential for Runx2 repression of activin induction [49]. The modulation of activin responsiveness by Runx proteins highlights how FSHβ expression levels are tightly regulated in gonadotrope cells and provides an additional candidate for negative feedback of activin action. Given the complexity of TGFβ family signaling pathways characterized in a variety of cell types, it is probable that many additional regulators of activin signal transduction in gonadotrope cells await discovery.
Summary
As this review discusses, activin signaling in pituitary gonadotrope cells involves an impressive array of transcription factors in the induction of FSHβ transcription in mammalian species (Figure 3). It is also evident that comprehension of activin signal transduction to the FSHβ promoter is still at a relatively rudimentary level. Many important players have been identified, but how they coordinate with each other to elicit a transcriptional response remains to be determined (Box 2). For activin induction of rodent FSHβ promoters, Smad proteins play a critical role in relaying the activin signal through direct actions on the DNA, as well as being recruited to the promoter via interactions with other proteins such as Pbx/Prep and potentially Ptx and FoxL2 (Figure 3). FoxL2 binding to the FSHβ promoter is also essential for activin responsiveness. For the human or ovine genes, it is still not clear whether Smad proteins are required for activin signaling. Moreover, the full panoply of factors and cofactors that provide positive or negative feedback to the activin signal await further investigation. Activin has been recently reported to interact synergistically with both GnRH and steroid hormone signaling pathways to upregulate FSHβ gene expression (recently reviewed in [50]). In addition, a negative regulator of activin induction, Runx was recently described (Figure 3) [49]. However, further research is needed to comprehend how activin signaling integrates into the complex network of hormonal signaling pathways activated in the gonadotrope cell during the estrous cycle. Such research may lead to new treatments for infertility or subfertility that can result from malfunction in FSH production as well as the development of novel contraceptive methods.
Figure 3.
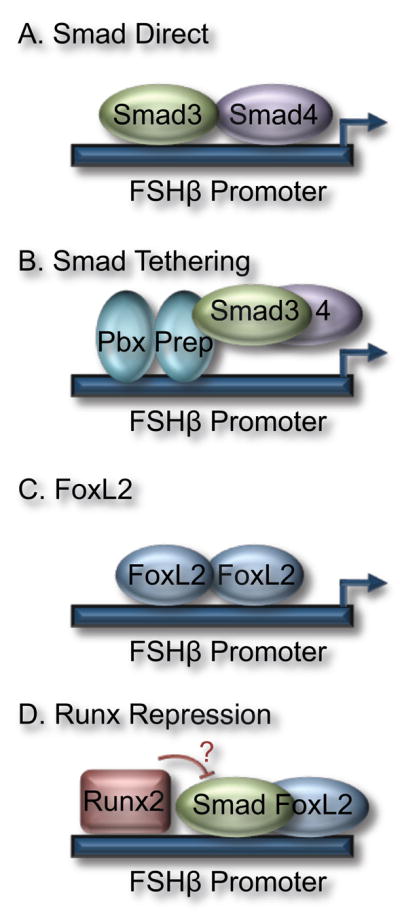
Different mechanisms for modulation of activin responsiveness on the FSHβ promoter. (a) Smad3 and 4 bind directly to the FSHβ promoter. Direct Smad binding has been demonstrated on rodent FSHβ promoters via a consensus SBE. (b) Smad3/4 can also interact with other transcription factors such as Pbx/Prep and be recruited to the FSHβ promoter via tethering. Although Ptx and FoxL2 can interact with Smads, it remains to be determined whether they tether Smads to the FSHβ promoter. (c) FoxL2 binds directly to the FSHβ promoter in several mammalian species, and FoxL2 binding elements are critical for activin induction. It is unknown whether FoxL2 is regulated directly by activin signaling pathways and if FoxL2 acts in a Smad-independent manner in the pituitary, as in the ovary. (d) Runx proteins diminish induction of the FSHβ gene, potentially through interactions with Smad3 or other regulators of activin induction such as FoxL2.
Box 2. Outstanding Questions.
Are Smads necessary for activin responsiveness on the human and ovine FSHβ promoters?
How does activin signaling via the p38 MAPK pathway regulate FSHβ gene expression?
How do Smad proteins co-operate with Pbx/Prep and Ptx to mediate FSHβ transcription?
Does activin signaling directly activate FoxL2, e.g., via phosphorylation?
Since FoxL2 elements are necessary, but not sufficient, for activin responsiveness, does FoxL2 require Smads or other transcription factors to induce FSHβ gene expression?
Acknowledgments
We thank Kellie Breen and Scott Kelley for critical reading of the manuscript and editorial comments. This work was supported by NICHD/NIH through a cooperative agreement (U54 HD012303) as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research (P.L.M.). D.C. was supported by NIH grants R03 HD054595, R21 HD058752 and R01 HD057549. P.L.M. was supported by NIH grants R01 HD020377 and R01 DK044838. V.G.T. was supported by NIH grant K01 DK080467.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chand AL, et al. Inhibin and premature ovarian failure. Hum Reprod Update. 2010;16:39–50. doi: 10.1093/humupd/dmp031. [DOI] [PubMed] [Google Scholar]
- 2.Kumar TR, et al. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15:201–204. doi: 10.1038/ng0297-201. [DOI] [PubMed] [Google Scholar]
- 3.Su P, et al. Conditional induction of ovulation in mice. Biol Reprod. 2005;73:681–687. doi: 10.1095/biolreprod.104.039164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ortolano GA, et al. Follicle-stimulating hormone beta subunit messenger ribonucleic acid concentrations during the rat estrous cycle. Endocrinology. 1988;123:2946–2948. doi: 10.1210/endo-123-6-2946. [DOI] [PubMed] [Google Scholar]
- 5.Halvorson LM, et al. Dynamic regulation of pituitary follistatin messenger ribonucleic acids during the rat estrous cycle. Endocrinology. 1994;134:1247–1253. doi: 10.1210/endo.134.3.8119165. [DOI] [PubMed] [Google Scholar]
- 6.DePaolo LV, et al. Suppression of pituitary secretion of follicle-stimulating hormone by porcine follicular fluid during pro-oestrus and oestrus in the rat: effects on gonadotrophin and steroid secretion, follicular development and ovulation during the following cycle. J Endocrinol. 1979;83:355–368. doi: 10.1677/joe.0.0830355. [DOI] [PubMed] [Google Scholar]
- 7.Hoak DC, Schwartz NB. Blockade of recruitment of ovarian follicles by suppression of the secondary surge of follicle-stimulating hormone with porcine follicular field. Proc Natl Acad Sci USA. 1980;77:4953–4956. doi: 10.1073/pnas.77.8.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Besecke LM, et al. Pituitary follistatin regulates activin-mediated production of follicle-stimulating hormone during the rat estrous cycle. Endocrinology. 1997;138:2841–2848. doi: 10.1210/endo.138.7.5279. [DOI] [PubMed] [Google Scholar]
- 9.Besecke LM, et al. Gonadotropin-releasing hormone regulates follicle-stimulating hormone-beta gene expression through an activin/follistatin autocrine or paracrine loop. Endocrinology. 1996;137:3667–3673. doi: 10.1210/endo.137.9.8756531. [DOI] [PubMed] [Google Scholar]
- 10.Woodruff TK, et al. Inhibin A and inhibin B are inversely correlated to follicle-stimulating hormone, yet are discordant during the follicular phase of the rat estrous cycle, and inhibin A is expressed in a sexually dimorphic manner. Endocrinology. 1996;137:5463–5467. doi: 10.1210/endo.137.12.8940372. [DOI] [PubMed] [Google Scholar]
- 11.Ling N, et al. Pituitary FSH is released by a heterodimer of the β-subunits from the two forms of inhibin. Nature. 1986;321:779–782. doi: 10.1038/321779a0. [DOI] [PubMed] [Google Scholar]
- 12.Weiss J, et al. Perifusion of rat pituitary cells with gonadotropin-releasing hormone, activin, and inhibin reveals distinct effects on gonadotropin gene expression and secretion. Endocrinology. 1993;132:2307–2311. doi: 10.1210/endo.132.6.8504735. [DOI] [PubMed] [Google Scholar]
- 13.Weiss J, et al. Homologous desensitization of gonadotropin-releasing hormone (GnRH)-stimulated luteinizing hormone secretion in vitro occurs within the duration of an endogenous GnRH pulse. Endocrinology. 1995;136:138–143. doi: 10.1210/endo.136.1.7828524. [DOI] [PubMed] [Google Scholar]
- 14.Pernasetti F, et al. Cell-specific transcriptional regulation of FSHβ by activin and GnRH in the LβT2 pituitary gonadotrope cell model. Endocrinology. 2001;142:2284–2295. doi: 10.1210/endo.142.6.8185. [DOI] [PubMed] [Google Scholar]
- 15.Norwitz ER, et al. Activin A augments GnRH-mediated transcriptional activation of the mouse GnRH receptor gene. Endocrinology. 2002;143:985–997. doi: 10.1210/endo.143.3.8663. [DOI] [PubMed] [Google Scholar]
- 16.Dupont J, et al. Activin signaling pathways in ovine pituitary and LbetaT2 gonadotrope cells. Biol Reprod. 2003;68:1877–1887. doi: 10.1095/biolreprod.102.012005. [DOI] [PubMed] [Google Scholar]
- 17.Bernard DJ. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone beta subunit in mouse gonadotrope cells. Mol Endocrinol. 2004;18:606–623. doi: 10.1210/me.2003-0264. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, et al. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature. 1998;394:909–913. doi: 10.1038/29814. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, et al. A transcriptional partner for MAD proteins in TGF-beta signalling. Nature. 1996;383:691–696. doi: 10.1038/383691a0. [DOI] [PubMed] [Google Scholar]
- 20.Seoane J, et al. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 21.Matsuda T, et al. Cross-talk between transforming growth factor-beta and estrogen receptor signaling through Smad3. J Biol Chem. 2001;276:42908–42914. doi: 10.1074/jbc.M105316200. [DOI] [PubMed] [Google Scholar]
- 22.Song CZ, et al. Glucocorticoid receptor inhibits transforming growth factor-beta signaling by directly targeting the transcriptional activation function of Smad3. Proc Natl Acad Sci USA. 1999;96:11776–11781. doi: 10.1073/pnas.96.21.11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang HY, et al. From transforming growth factor-beta signaling to androgen action: identification of Smad3 as an androgen receptor coregulator in prostate cancer cells. Proc Natl Acad Sci USA. 2001;98:3018–3023. doi: 10.1073/pnas.061305498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thackray VG, et al. Androgens, progestins and glucocorticoids induce follicle-stimulating hormone β-subunit gene expression at the level of the gonadotrope. Mol Endocrinol. 2006;20:2062–2079. doi: 10.1210/me.2005-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corpuz PS, et al. FoxL2 is required for activin induction of the mouse and human follicle-stimulating hormone β-subunit genes. Mol Endocrinol. 2010;24:1037–1051. doi: 10.1210/me.2009-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, et al. Activator protein-1 and smad proteins synergistically regulate human follicle-stimulating hormone beta-promoter activity. Endocrinology. 2008;149:5577–5591. doi: 10.1210/en.2008-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGillivray SM, et al. Activin and glucocorticoids synergistically activate follicle-stimulating hormone β-subunit gene expression in the immortalized LβT2 gonadotrope cell line. Endocrinology. 2007;148:762–773. doi: 10.1210/en.2006-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thackray VG, Mellon PL. Synergistic induction of follicle-stimulating hormone beta-subunit gene expression by gonadal steroid hormone receptors and smad proteins. Endocrinology. 2008;149:1091–1102. doi: 10.1210/en.2007-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coss D, et al. p38 mitogen-activated kinase is critical for synergistic induction of the FSH beta gene by gonadotropin-releasing hormone and activin through augmentation of c-Fos induction and Smad phosphorylation. Mol Endocrinol. 2007;21:3071–3086. doi: 10.1210/me.2007-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gregory SJ, et al. Synergy between activin A and gonadotropin-releasing hormone in transcriptional activation of the rat follicle-stimulating hormone-beta gene. Mol Endocrinol. 2005;19:237–254. doi: 10.1210/me.2003-0473. [DOI] [PubMed] [Google Scholar]
- 31.Safwat N, et al. Transforming growth factor beta activated kinase1 (TAK1) is a key mediator of ovine follicle stimulating hormone beta subunit expression. Endocrinology. 2005;146:4814–4824. doi: 10.1210/en.2005-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, et al. Activin A induction of FSH{beta} subunit transcription requires SMAD4 in immortalized gonadotropes. J Mol Endocrinol. 2010;44:349–362. doi: 10.1677/JME-09-0142. [DOI] [PubMed] [Google Scholar]
- 33.Suszko MI, et al. Regulation of the rat follicle-stimulating hormone beta-subunit promoter by activin. Mol Endocrinol. 2003;17:318–332. doi: 10.1210/me.2002-0081. [DOI] [PubMed] [Google Scholar]
- 34.Suszko MI, et al. Smad3 mediates activin-induced transcription of follicle-stimulating hormone beta-subunit gene. Mol Endocrinol. 2005;19:1849–1858. doi: 10.1210/me.2004-0475. [DOI] [PubMed] [Google Scholar]
- 35.Lamba P, et al. Acute regulation of murine follicle-stimulating hormone beta subunit transcription by activin A. J Mol Endocrinol. 2006;36:201–220. doi: 10.1677/jme.1.01961. [DOI] [PubMed] [Google Scholar]
- 36.Bailey JS, et al. Activin regulation of the follicle-stimulating hormone β-subunit gene involves Smads and the TALE homeodomain proteins Pbx1 and Prep1. Mol Endocrinol. 2004;18:1158–1170. doi: 10.1210/me.2003-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suszko MI, et al. Smad3 and Pitx2 cooperate in stimulation of FSHbeta gene transcription. Mol Cell Endocrinol. 2008;281:27–36. doi: 10.1016/j.mce.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 38.Lamba P, et al. Paired-like homeodomain transcription factors 1 and 2 regulate follicle-stimulating hormone beta-subunit transcription through a conserved cis-element. Endocrinology. 2008;149:3095–3108. doi: 10.1210/en.2007-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zakaria MM, et al. Pituitary homeobox 1 activates the rat FSHbeta (rFSHbeta) gene through both direct and indirect interactions with the rFSHbeta gene promoter. Mol Endocrinol. 2002;16:1840–1852. doi: 10.1210/me.2002-0088. [DOI] [PubMed] [Google Scholar]
- 40.Blount AL, et al. FoxL2 and Smad3 coordinately regulate follistatin gene transcription. J Biol Chem. 2009;284:7631–7645. doi: 10.1074/jbc.M806676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellsworth BS, et al. The gonadotropin releasing hormone (GnRH) receptor activating sequence (GRAS) is a composite regulatory element that interacts with multiple classes of transcription factors including Smads, AP-1 and a forkhead DNA binding protein. Mol Cell Endocrinol. 2003;206:93–111. doi: 10.1016/s0303-7207(03)00235-1. [DOI] [PubMed] [Google Scholar]
- 42.Ellsworth BS, et al. FOXL2 in the pituitary: molecular, genetic, and developmental analysis. Mol Endocrinol. 2006;20:2796–2805. doi: 10.1210/me.2005-0303. [DOI] [PubMed] [Google Scholar]
- 43.Crisponi L, et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001;27:159–166. doi: 10.1038/84781. [DOI] [PubMed] [Google Scholar]
- 44.Lamba P, et al. A novel role for the forkhead transcription factor FOXL2 in activin A-regulated follicle-stimulating hormone beta subunit transcription. Mol Endocrinol. 2009;23:1001–1013. doi: 10.1210/me.2008-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strahl BD, et al. Two proximal activating protein-1-binding sites are sufficient to stimulate transcription of the ovine follicle-stimulating hormone-beta gene. Endocrinology. 1997;138:2621–2631. doi: 10.1210/endo.138.6.5205. [DOI] [PubMed] [Google Scholar]
- 46.Benayoun BA, et al. The identification and characterization of a FOXL2 response element provides insights into the pathogenesis of mutant alleles. Hum Mol Genet. 2008;17:3118–3127. doi: 10.1093/hmg/ddn209. [DOI] [PubMed] [Google Scholar]
- 47.Huang HJ, et al. The promoter for the ovine follicle-stimulating hormone-beta gene (FSHbeta) confers FSHbeta-like expression on luciferase in transgenic mice: regulatory studies in vivo and in vitro. Endocrinology. 2001;142:2260–2266. doi: 10.1210/endo.142.6.8202. [DOI] [PubMed] [Google Scholar]
- 48.Su P, et al. Expression and regulation of the beta-subunit of ovine follicle-stimulating hormone relies heavily on a promoter sequence likely to bind Smad-associated proteins. Endocrinology. 2007;148:4500–4508. doi: 10.1210/en.2006-1635. [DOI] [PubMed] [Google Scholar]
- 49.Breen KM, et al. Runt-related transcription factors impair activin indicution of the folicle-stimulating hormone beta-subunit gene. Endocrinology. 2010;151:2669–2680. doi: 10.1210/en.2009-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thackray VG, et al. Hormones in synergy: Regulation of the pituitary gonadotropin genes. Mol Cell Endocrinol. 2010;314:192–203. doi: 10.1016/j.mce.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uccella S, et al. Localization of inhibin/activin subunits in normal pituitary and in pituitary adenomas. Pituitary. 2000;3:131–139. doi: 10.1023/a:1011431123208. [DOI] [PubMed] [Google Scholar]
- 52.Roberts V, et al. Production and regulation of inhibin subunits in pituitary gonadotropes. Endocrinology. 1989;124:552–554. doi: 10.1210/endo-124-1-552. [DOI] [PubMed] [Google Scholar]
- 53.Bilezikjian LM, et al. Characterization and the regulation of inhibin/activin subunit proteins of cultured rat anterior pituitary cells. Endocrinology. 1993;133:2545–2553. doi: 10.1210/endo.133.6.8243276. [DOI] [PubMed] [Google Scholar]
- 54.Thompson TB, et al. Beta A versus beta B: is it merely a matter of expression? Mol Cell Endocrinol. 2004;225:9–17. doi: 10.1016/j.mce.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 55.Attisano L, et al. Activation of signalling by the activin receptor complex. Mol Cell Biol. 1996;16:1066–1073. doi: 10.1128/mcb.16.3.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bilezikjian LM, et al. Autocrine/paracrine regulation of pituitary function by activin, inhibin and follistatin. Mol Cell Endocrinol. 2004;225:29–36. doi: 10.1016/j.mce.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 57.Bilezikjian LM, et al. Pituitary actions of ligands of the TGF-beta family: activins and inhibins. Reproduction. 2006;132:207–215. doi: 10.1530/rep.1.01073. [DOI] [PubMed] [Google Scholar]
- 58.Bernard DJ, et al. Activin B can signal through both ALK4 and ALK7 in gonadotrope cells. Reprod Biol Endocrinol. 2006;4:52. doi: 10.1186/1477-7827-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]