

Abstract
Intercellular communication is an essential process in stimulating lymphocyte development and in activating and shaping an immune response. B celldevelopment requires cell-to-cell contact with and cytokine production by bone marrow stromal cells. However, this intimate relationship also may be responsible for the transfer of death-inducing molecules to the B cells. 7,12-dimethylbenz[a]anthracene (DMBA), a prototypicalpolycyclic aromatic hydrocarbon, activates caspase-3 in pro/pre-B cells in a bone marrow stromal cell-dependent manner, resulting in apoptosis. Thesestudies were designed to examine the hypothesis that an intrinsic apoptotic pathway is activated by DMBAand that the ultimate death signal is a DMBA metabolitegenerated by the stromal cell sand transferred to the B cell s. Whilea loss of mitochondrial membrane potentialdid not occur in the DMBA/stromal cell -induced pathway, cytochrome c release was stimulated in B cells. Caspase-9 was activated, and formation of the apoptosome was essential to support apoptosis, as demonstrated by suppression of death in Apaf-1fogmutant pro -B cells. Investigation of signaling upstream of the mitochondria demonstrated an essential role for p53. Furthermore, DMBA-3,4-dihydrodiol-1,2-epoxide, a DNA-reactive metabolite of DMBA, wassufficient to upregulate p53, induce caspase-9 cleavage and initiate B cell apoptosis in the absence of stromal cells, suggesting that production of this metabolite by the stromal cellsand transfer to the B cell sis a proximal event in triggering apoptosis. Indeed, we provide evidencethat metabolite transfer from bone marrow stromal cells occurs through membrane exchange, which may represent a novel communication mechanismbetween developing B cells and stromal cells.
Keywords: B cells, stromal cells, apoptosis, DMBA, membrane exchange, p53
Introduction
Polycyclic aromatic hydrocarbons (PAHs)4, such as benzo[a]pyrene (B[a]P) and benz[a]anthracene (BA), are immunosuppressive in rodent models (1–6) and they alter immune responses in humans (7–10). These and other PAHs are capable of binding to and activating the aryl hydrocarbon receptor (AhR)and are produced by a number of industrial processes and by the incomplete combustion of carbon-containing compounds including fossil fuels. Human exposure to environmentally ubiquitous PAHs, such as B[a]P and BA, regularly occurs through ingestion of contaminated food and inhalation of vehicle exhaust or cigarette smoke (11, 12).
7,12-Dimethylbenz[a]anthracence (DMBA) is a highly toxic, methylated derivative of BAthat has been used extensively in model systems designed to elucidate mechanisms of PAH -mediated immunotoxicity and carcinogenicity (13). In rodent models, both B[a]P and DMBA induce a reduction in bone marrow cellularity resulting largely from a massive loss of B cells (5, 6, 14, 15). In an in vitro bone marrow stromal cell/B cell co-culture system, DMBA induces apoptosis of pro-and pre -B cells through apoptosis signaling pathways that resemble those activated during immature B cell clonal deletion (15–19). Interestingly, exposure of B cells alone to PAHsdoes not induce apoptosis (20). B cell death requires contact with AhR-and cytochrome P450 1B1 (CYP1B1)-expressing stromal cells (21, 22).
Bone marrow stromal cells, multipotentcells that are capable of differenti ating into either osteoblasts or adipocytes, are responsible for maintaining the milieu of cytokines and adhesion/interaction/matrix molecules that support B lymphopoiesis (23). In fact, deletion of key molecules that support stromal cell/B cell interactions suppressesB lymphopoiesis (SDF -1/CXC4)(24, 25). Direct contact between stromal cells and B cells not only suppresses spontaneous pre-B cell apoptosis, but also suppressescytokine -and glucocorticoid -induced pre-B cell apoptosis (26–28). Conversely, stromal cell/B cell contact is required for initiation of DMBA-induced pro/pre-B cell apoptosis as treatment of B cells alone with DMBA or with conditioned medium from DMBA-treated stromal cells is not sufficient to induce B cell death (21, 29).
The nature of the death signal transferred from the stromal cellsis unknown. However, we have demonstrated that AhR expression in the stromal cells is required, thatmetabolism of DMBA likely precedestransfer of the death signal to stromal cell -adherent B cells, and that DMBA-induced apoptosis does not result from production of death receptor ligands by stromal cells or by activation of known death receptors on B cells (17, 20, 30). From these results we have hypothesized that the death signal that is transferred between the stromal cells and B cells is highly labile, likely a DMBA metabolite, and that it initiates an intrinsic apoptotic pathway.
The mechanism by which the death signal is transferredfrom the stromal cells to the bone marrow B cellsis somewhat harder to postulate. Communication among cells in the immunesystem largely occurs through production/secretion of cytokines and exchange of membranes (trogocytosis). Cytokines are an unlikely mediator in this case, as we have previously shown thatseveral common death -inducing cytokines and their receptors (e.g. TNF-α, TNF-β, lymphotoxin-β, TNF receptors (TNFR1, TNFR2), Fas, and death receptor 6 (DR6)) are not involved in DMBA-induced death (30). In the absence of evidence for death receptor or cytokine involvement, we hypothesized that a trogocytosis-like mechanism may be at work. Trogocytosis(rapid, contact -dependent, intercellular membrane patch exchange)typically facilitates transfer of antigen following formation of an immunological synapse (31). However, it can occur in antigen-independent cell-cell interactions as well (32).
Analysis of the interactions between stromal cells and B cells during DMBA-induced death signaling may reveal important information on the nature of communication between these bone marrow cell subsets in the presence or absence of environmental chemicals. The studies described herein were designed to examine the hypothesis that an intrinsic apoptotic pathwayis activated by a DMBA metabolite transferred to the B cell via membrane exchange with a stromal cell capable ofmetabolizing DMBA. Accordingly, we defined the intracellular DMBA/stromal cell-induced B celldeath pathway, showing that it is dependent upon cytochrome c release and apoptosome formation and that it is p53-dependent. We determined that a terminal DMBA metabolite, DMBA -3,4-dihydrodiol-1,2-epoxide(DMBA -DE), is sufficient to induce apoptosis in B cells in the absence of stromal cells and showedthat p53 also is critical for DMBA -DE-induced apoptosis. These data all are consistent with transfer of a death-inducing epoxide from the stromal cell to the B cell and the subsequent initiation of the “intrinsic” apoptosis pathway. Finally, we provide evidence that membrane transfer occurs between bone marrow stromal cells and B cells, but not T cells, and suggest that this is the mechanism of transfer of the otherwise labile, death-inducingDMBA metabolite.
Materials and Methods
Materials
The caspase-8-specific antibody was from Axxora (San Diego, CA). The cytochrome c-specific antibody and phenotyping antibodies were from BD Biosciences (Palo Alto, CA). Antibodies specific for cleaved caspases-3 and -9 and cleaved lamin were from Cell Signaling Technology (Beverly, MA). The p53-specific antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Plasmocin was from Invivogen (San Diego, CA). JC-1 was from Molecular Probes (Eugene, OR). Murine rIL-7 was from Research Diagnostics (Flanders, NJ). Caspase inhibitors were from R&D Systems (Minneapolis, MN). DMBA, propidium iodide, Protease Inhibitor Cocktail for Mammalian Cells, and the β-actin-specific antibody were from Sigma Chemical Co. (St. Louis, MO). All other reagents were from Thermo Fisher Scientific (Suanee, GA).
Cell culture
Stromal cell-dependent, CD43+BU -11 cells expressing rearranged cytoplasmic Ig heavy chains (pro/pre-B cells) (15) were co-cultured on cloned BMS2 bone marrow-derived stromal cells (33)(kindly provided by Dr. P. Kincade, Oklahoma Medical Research Foundation). Stocks of BU-11 cells were maintained on BMS2 cell monolayers in an equal mixture of DMEM and RPMI 1640 medium with 5% bovine growth serum (BGS)(Thermo Fisher Scientific, formerly Hyclone), plasmocin, L-glutamine and 2-mercaptoethanol. All cultures were maintained at 37° C in a humidified, 7.5% CO2atmosphere. Cell cultures were determined to be mycoplasma negative by PCR (Mycoplasma Detection Kit; ATCC, Manassas, VA).
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee at Boston University. Primary bone marrow pro-B cell cultures were prepared from wildtype C57BL/6 and B6.129S2-Trp53tm1Tyj/Jor Apaf1fog/J homozygous and heterozygousmice (Jackson Laboratories, Bar Harbor, ME) essentially as described (34). Bone marrow was flushed from the femurs of 4–8 week-old mice. Red blood cells were lysed by incubation in 0.17 M NH4Cl, 10 mM KHCO3, and 1 mM EDTA at 37°C for 5 min. The remaining cells were cultured for 5–7 days in primary B cell medium (RPMI containing 10% FBS, penicillin/streptomycin, L- glutamine, 2-mercaptoethanol, and 16 ng/ml murine rIL-7). This procedure results in a B cell culture in which at least 95% of the cells express CD43 and B220.
B and T lymphocytes were isolated from spleens of C57BL/6 miceby dissociation and use ofSpinSep Mouse B Cell Enrichment and T Cell Enrichment Kits, respectively (StemCell Technologies Inc, Vancouver, BC). To assess purity, aliquots of cells were stained with anti- B220-FITC or FITC-conjugated rat IgG2afor B cells or anti -CD3-FITC or FITC-conjugated rat IgG2bfor T cells, fixed in 0.4% paraformaldehyde, and analyzed on a Becton Dickinson FACScan flow cytometer.
For co-culture experiments, BMS2 cells (2× 104/cm2) were cultured for 24 hr in DMEM containing 5% BGS to form a monolayer which was approximately 75% confluent. BU-11 or primary pro-B cells were added in RPMI containing 5% BGS (or 10% FBS and 16 ng/ml rIL-7 for primary pro-B cells) at a final concentration of 2× 105cells/ml and allowed to associate with the stromal cells overnight. For some experiments, B cells and BMS2 cells were separated by a 3 μm pore size transwell insert. B cell/stromal cell co-cultures were treated with vehicle (DMSO, 0.1% final concentration) or DMBA (1–10 μM)(Sigma) for 2–16 hr. Vehicle, the pan-caspase inhibitor VAD-FMK, the caspase-3 inhibitor DEVD-FMK, or the caspase-6 inhibitor VEID-FMK (15–30 μM) were added to co-cultures 30 min prior to vehicle or DMBA treatment.
For single culture experiments, BU-11 cells (2 × 105cells/ml) were cultured for 24 hr in RPMI containing 5% BGS and 16 ng/ml rIL-7 prior to treatment with vehicle, etoposide (0.1 μg/ml)(Sigma), or DMBA-DE (anti-7,12-dimethylbenz[a]anthracene-3,4-dihydrodiol-1,2-epoxide, 0.001-1μM)(NCI Chemical Carcinogen Reference Standard Repository).
Immunoblotting
B cells were harvested and washed once in cold PBS. For analysis of cleavage of caspases or their substrates, cytoplasmic extracts were prepared as described previously (35). For analysis of cytochrome c release, cytoplasmic fractions from digitonin-permeabilized cells were prepared as described previously (36). Protein concentrations were determined by the Bradford method.
Total proteins (15–50 μg) were resolved on 12%or 15% gels, transferred to a 0.2 μm nitrocellulose membrane, and incubated with primary antibody. Primary antibodies included polyclonal rabbit anti-cleaved caspase-3 (9661), polyclonal rat anti-caspase-8 (ALX-804-447), polyclonal rabbit anti-caspase-9 (9504), polyclonal rabbit anti-p53 (SC-6243), monoclonal mouse anti-cleaved lamin A (2036), and polyclonal rabbit anti-cytochrome c antibody (S2050). Immunoreactive bands were detected using HRP-conjugated secondary antibodies (Biorad, Hercules, CA) followed by ECL. To control for equal protein loading, blots were re-probed with a β-actin-specific antibody (A5441)and analyzed as above.
Apoptosis Analyses
Mitochondrial membrane potential was analyzed by JC-1 and analyzed by flow cytometry as described (37). The percentage of cells with low mitochondrial membrane potential (ΔΨmlow) was determined to be those having an increased green fluorescence with or without a loss of red fluorescence. DNA fragmentation was analyzed by propidium iodide (PI) staining and flow cytometry as described (30, 35). The percentage of cells undergoing apoptosis was determined to be those having a weaker PI fluorescence than cells in the G0/G1 phase of the cell cycle. Caspase activityin cytoplasmic extracts was determined using p-nitroaniline-conjugated substrates and spectrometry, according to the manufacturer’s instructions (Apoalert, Clontech, Palo Alto, CA).
Two-Photon Microscopy
BMS2 cells were plated in DMEM containing 5% BGS on half of a glass bottom 35 mm petri dish with a 14 mm microwell with a number 0 coverglass below the microwell (MatTek Corporation) using a removable barrier placed down the center of the microwell. After incubating for 24 hrs, the barrier was removed. Cultures were treated with 10 μM DMBA for 30 min, washed with PBS to remove free DMBA, incubated for 1 hr, and then washed again with PBS. BU-11 cells were added in RPMI containing 5% BGS and centrifuged onto the BMS2 layer for 10 min at 1500 rpm. Excess non-adhering BU-11 cells were removed by gentle washing, fresh medium was added, and cultures were immediately analyzed by two-photon confocal microscopy for DMBA/metabolite fluorescence.
DMBA Uptake
BMS2 cells were plated in 6 well plates in DMEM containing 5% BGS and allowed to adhere overnight. Cells were treated with vehicle or DMBA (10 μM) for 30 min. Medium was removed, cells were washed with PBS, and new medium was added. Cells were incubated for an additional 5 hr followed by medium removal, washing and replacement. BU-11 cells, in RPMI containing 5% BGS, then were added directly to the BMS2 cells or to1 μm pore size transwell inserts over the BMS2 cells. BU-11 cells were harvested after 18 hr and analyzed immediately for DMBA/metabolite fluorescence on a Dako MoFlo flow cytometer. Only cells in the live B cell gate were analyzed.
DiO Uptake
Vybrant DiO (Invitrogen, Carlsbad, CA) was used essentially as directed by the manufacturer. BMS2 were plated in 12 well plates in DMEM containing 5% BGS and allowed to adhere overnight. Cells were stained with 5 μl/ml Vybrant DiO in culture medium for 20 min at 37° C followed by extensive washing. Medium (DMEM containing 5% BGS) was replaced and BU-11 cells or primary pro-B, splenic B, or splenic T cells from wildtype C57Bl/6 mice were added in RPMI containing 5% BGS. For some experiments, BU-11 cells were added to3 μm pore transwell inserts over the stained BMS2. Lymphocytes were harvested after 16 hr, washed, and fixed in 0.4% paraformaldehyde. In addition, naïve BU-11 cells were resuspended in 16 hr supernatants from wells of DiO-stained BMS2 and incubated at room temperature for 20 min before washing and fixing. Samples were analyzed by flow cytometery. Only cells in the live lymphocyte gate were analyzed.
Statistics
Statistical analyses were performed with Statview (SAS Institute, Cary, NC). Data are presented as means ± standard errors (SE). At least three experiments were performed in each BU-11 protocol. Experiments with primary B and T cells were performed with cells from a minimum of three independently prepared and maintained pools of bone marrow cells or three individual spleens. One-factor ANOVAs were used to analyze the data, with the Dunnett’s or Tukey-Kramer multiple comparisons test to determine significant differences.
Results
The intrinsic pathway is activated in DMBA/stromal cell-induced B cell apoptosis upstream of the caspase cascade
Previous studies demonstrated that, even though caspase-8 is activated in the DMBA/stromal cell-induced apoptosis pathway, production of death-inducing cytokines and triggering of an extrinsic, death-receptor mediated apoptosis pathway isnot the mechanism of DMBA-induced apoptosis (30). These results suggestthat DMBA/stromal cell-induced B cell apoptosis is initiated through the intrinsic pathwayand are consistent with the hypothesis that a DMBA metabolite is the death signal transferred from the stromal cell to the B cell. Therefore, mediators of an intrinsic pathway were evaluated and the potential role for a reactive DMBA metabolite in delivering the death signal to B cells was assessed.
A key feature of the intrinsic apoptotic pathway is mitochondrial outer membrane permeabilization (MOMP). To examine mitochondrial integrity, the non-transformed pro/pre-B cell line BU-11 was treated in co-culture with BMS2 cell monolayers with vehicle (DMSO, 0.1% final concentration) or DMBA (1 μM). Cytosolic proteins recovered from digitonin-permeabilized BU-11 cellswere analyzed for cytochome c by immunoblotting. Cytochrome c release into the cytosol was evident 4–6 hr after DMBA treatment (Fig. 1A), indicating that the B cell mitochondriawere permeabilized.
Fig. 1.

DMBA induces caspase-independent mitochondrial cytochrome c release, but not membrane potential loss, in B cells co-cultured with bone marrow stromal cells. (A) BU-11/BMS2 co-cultures were treated with vehicle (Vh, 0.1% DMSO) or DMBA (1 μM), and BU-11 cells were harvested after 2–8 hr. Cytochrome c release was analyzed by immunoblotting of cytoplasmic extracts from digitonin-permeabilized cells. Data are representative of 3 experiments. (B) BU-11/BMS2 co-cultures were treated with Vh or DMBA (1 μM) or BU-11 cultures were treated with Vh or etoposide (0.1 μg/ml) (inset) for 4–10 hrs. BU-11 cells were analyzed for mitochondrial membrane potential loss by JC-1 staining followed by flow cytometry. Data are are presented as means + SE from at least three experiments. (C) BU-11/BMS2 co-cultures were pre-treated with Vh or the pan-caspase inhibitor VAD-FMK (30 μM) for 30 min prior to treatment with Vh or DMBA (1 μM) for 8 hr. Cytochrome c release was analyzed as above. Data are representative of 3 experiments. *Statistically greater than Vh treated (p<0.05, ANOVA, Tukey-Kramer).
Loss of mitochondrial membrane potential (ΔΨm) often, but not always, occurs in apoptosis mediated by the intrinsic pathway (38, 39). To determine if mitochondrial membrane potential is altered during DMBA-induced apoptosis, BU-11/BMS2 cell co-cultures were treated with vehicle or DMBA, and mitochondrial membrane potential was analyzed in BU-11 cells by JC-1 staining and flow cytometry. BU-11 cells did not undergo significant mitochondrial membrane potential loss at any time point tested (Fig. 1B). Similar results were seen using DiOC6(data not shown). As it is unusual for cells to undergo mitochondria -dependent apoptosis without membrane potential loss, there was a possibility that the JC-1 staining was not sensitive enough to detect changes in mitochondrial membrane potential in BU-11 cells. However, etoposide (0.1 μg/ml), a compound known to induce mitochondrial membrane potential loss (40, 41), induced significant mitochondrial membrane potential loss within 4 hr of treatment that continued to increase through 6 hr (Fig. 1B, inset). These results support the conclusion that MOMPoc curswithout causing a loss of mitochondrial membrane potential in DMBA/stromal cell-induced B cell apoptosis.
Based on the facts that DMBA-induced activation of caspase-8 is caspase-dependent (30) and that cytochrome c release from the mitochondria occurs early in this pathway, it seemed likely that MOMP takes place before the caspase cascade is activated. To test this hypothesis, BU-11/BMS2 cell co-cultures were treated with vehicle or the pan-caspase inhibitor VAD-FMK (30 μM) prior to treatment with vehicle or DMBA. BU-11 cellsthen were harvested and cytoplasmic fractions were analyzed for cytochrome c release from the mitochondria by immunoblotting. Cytochrome c was released from the mitochondria of B cells in DMBA-treated co-cultures, and this release was not blocked by pre-treatment with VAD-FMK (Fig. 1C). Therefore, MOMP appears to precede activation of the caspase cascade in the DMBA/stromal cell-induced apoptosis pathway.
Having determined that mitochondrial permeabilization is likely caspase-independent, the contribution of caspases downstream of cytochrome c release was investigated. BU-11/BMS2 cell co-cultures were treated with vehicle or DMBA, and BU-11 cells were analyzed for caspase-9 activation by immunoblotting for cleaved, active caspase-9 and for caspase-9 catalytic activity. An increase in the formation of active caspase-9 fragments in BU-11 cells occurred 4–6 hr after DMBA treatmentof the co -culture (Fig. 2A). Concomitantly, an increasein caspase -9-like activity was detected 6 hr after DMBA treatment and reached statistical significance at 8 hr (Fig. 2B).
Fig. 2.

The apoptosome is essential for DMBA/stromal cell-induced B cell apoptosis. BU-11/BMS2 co-cultures were treated with Vh or DMBA (1 μM), and BU-11 cells were harvested after 2–10 hr. (A) Total proteins were extracted and analyzed for active caspase-9 fragments and for β-actin by immunoblotting. Data are representative of 4 experiments. (B) Cytosolic proteins were extracted, and caspase-9-like activity was measured using p-nitroaniline-conjugated LEHD substrate. Data were quantified as the average fold increase in caspase-9-like activity relative to the activity in untreated cells and presented as means ±SE from 4 experiments. (C) Primary proB cells isolatedfrom mice either homozygous or heterozygous for the Apaf-1fogmutation, were treated in co-culture with BMS2 with Vh or DMBA (1 μM). Pro-B cells were analyzed for apoptosis by PI staining 16 hr after treatment. The percentage of death measured in naïve cell populationswas subtracted prior to analysis. Data are presented as means + SE from primary pro-B cells prepared from 5–6 individual mice. *Statistically different from Vh (p<0.05, ANOVA, Dunnett’s).* *Statistically greater than all other groups(p<0.05, ANOVA, T ukey-Kramer).
To demonstrate a requirement for caspase-9 activation in the DMBA-induced death pathway, two approaches were taken. First, experiments with LEHD-FMK, a caspase-9 inhibitor, showed that DMBA/stromal cell-induced B cell death was at least partially sensitive to inhibition of caspase-9 (data not shown). Second, primary pro-B cells with mutant Apaf-1 were tested for sensitivity to DMBA/stromal-induced apoptosis. Apaf-1 is an integral component of the apoptosome, and is essential for maximal activation of caspase-9 (42). Primary pro-B cells were generated from the bone marrow of mice hetero-or homozygous for the Apaf-1fogmutation. ProB/BMS2 cell co-cultures were treated with vehicle or DMBA, and pro-B cells were analyzed for apoptosis by hypotonic PI staining and flow cytometry. DMBA induced a significant increase in apoptosis of pro-B cells from mice heterozygous for the Apaf-1fogmutation, but not of pro -B cells from mice homozygous for the Apaf-1fogmutation ( Fig. 2C). These results indicate that MOMP results in assembly of the apoptosome, which results in caspase-9 activation, and is required for DMBA/stromal cell-induced B cell apoptosis.
MOMP can be amplified by caspase-dependent positive feedback loops. Indeed, caspase-8 and Bid arecleaved to their active form in DMBA/stromal cell-induced B cell apoptosis (30). Bid isa BH3-only, direct activator, proapoptotic Bcl-2 family memberthat is cleaved and activated by a number of caspases leading to its translocation to the mitochondria where it stimulates Bax/Bak pore formation (43). Bid can be cleaved directly by caspase-2, caspase-3 and caspase-8 and also may be activated through a caspase-3 to -6 to -8 to Bid loop (44–49). The likely intermediate in the activation of caspase-8 by a caspase-3 feedback mechanism, caspase-6, was activated in B cells following treatment of B cell/stromal cell co-cultures with DMBA (Supplemental Fig. 1A-B). Furthermore, while caspase-6 and -8 activation was largely suppressed by caspase inhibitors, activation of caspase-3 was not (Supplemental Fig. 1C). Thus, the caspase-9 intrinsic pathway likely is amplified by activation of caspases that can cleave and activate Bid, further stimulating MOMP.
The role of p53 in activating the DMBA/stromal cell-induced apoptosis pathway
One of the most common activators of the intrinsic pathway is p53, a tumor suppressor protein that can trigger mitochondrialpermeabilization through transcriptional as well as non -transcriptional mechanisms (50). p53 is a likely candidate for initiation of MOMP in DMBA/stromal cell-induced apoptosis in developing B cells because p53 plays a role in DMBA-induced bone marrow toxicity and immunosupression in vivo(51, 52), and p53 protein levels are increased in the nucleiof BU -11 cells at least at a late time point in this system (53).
To begin to determine how p53 could be acting in DMBA/stromal cell-induced apoptosis, p53 protein expression was examined at early time points. BU-11/BMS2 cell co-cultures were treated with vehicle or DMBA, and BU-11 whole cell lysates were analyzed for p53 expression by immunoblotting. An increase in p53 protein was seen beginning 2–4 hr after DMBA treatment and continuing through 10 hr (Fig. 3A). An apparent decrease in p53 electrophoretic migration is likely due to post-translational modifications, as p53 is known to be modified following cellular stress (54). The early time at which p53 protein levels were increased is consistent with p53 acting upstream of MOMP.
Fig. 3.

Contribution of p53 to DMBA/stromal cell-induced B cell apoptosis. (A) BU-11/BMS2 co-cultures were treated with Vh or DMBA (1 μM), and BU-11 cells were harvested after 2–10 hr. Total proteins were extracted and analyzed for p53 and for β-actin by immunoblotting. Data are representative of 3 experiments. (B, C) Primary pro-B cells isolated from wildtype or p53 mutant mice were treated in co-culture with BMS2 cells with Vh or DMBA (1 μM) for 2–16 hr. (B) Pro-B cells were analyzed for apoptosis by PI staining 16 hr after treatment. The percentage of death measured in naïve cells was subtracted prior to analysis. Data are presented as means + SE from primary pro-B cells prepared from 3–4 individual mice. (C) Total proteins were extracted from pro-B cells and analyzed for cleaved caspase-3 and for β-actin by immunoblotting. A caspase-3 positive control was included on each gel to enable comparison between blots. Data are representative of experiments using primary pro-B cells from 3 individual mice. *Statistically greater than all other treatment groups (p<0.05, ANOVA, Tukey-Kramer). **Statistically less than wildtype DMBA treated (p<0.05, ANOVA, Tukey -Kramer).
To evaluate the requirement for functionalp53 in DMBA-induced apoptosis, primary pro-B cells were generated from wildtype C57Bl/6 mice (controls) and from congenic mice homozygous for a mutation in p53 (Trp53tm1Tyj). Primary pro-B/BMS2 cell co-cultures were treated with vehicle or DMBA, and pro-B cells were analyzed for apoptosis by hypotonic PI staining and flow cytometry and by immunoblotting for cleaved, active caspase-3. Both wildtype and p53 mutant pro-B cells underwent significant apoptosis in response to DMBA treatment as measured by PI staining; however mutant pro-B cells underwent significantly less apoptosis than wildtype pro-B cells at 16 hr (Fig. 3B). An increase in cleaved caspase-3 was detected in extracts from wildtype pro-B cells 4 hrs after DMBA treatment (Fig. 3C). In contrast, primary p53 mutant pro-B cells exhibited a low background level of caspase-3 cleavage, which did not increase over time in response to DMBA (Fig. 3C). These results indicate that p53 is essential for optimal DMBA/stromal cell-induced B cell apoptosis.
DMBA-DE induces p53 expressionand apoptosis in B cells
Results gathered to date, including those described above, indicate that DMBA induces bone marrow stromal cells to produce a death signal that is transferred to developing B cells, initiatinga p53 -dependent intrinsic apoptotic pathway. However, the nature of the stromal cell-derived apoptotic signal that activates the apoptotic pathway in the B cells and the mechanism of its delivery to the B cells remained unclear. A reactive metabolite of DMBA appeared likelyto be the apoptotic signal in this system as DMBA metabolites are known to form DNA adducts (55), which would be predicted to lead to p53 upregulation and mitochondrial permeabilization. In addition, metabolism is known to beimportant in DMBA/stromal cell-induced apoptosis of developing B cells (17, 22).
To assess the ability of a terminal reactive DMBA metabolite to induce developing B cell apoptosis directly, bypassing the need for stromal cells, BU -11 cells cultured in the absence of BMS2 cells were treated with vehicleor a terminal reactive DMBA metabolite, DMBA-3,4-dihydrodiol-1,2-epoxide (DMBA-DE, 1 μM), and apoptosis was assessed by hypotonic PI staining and flow cytometry. A significant increase in apoptosis was observed with DMBA-DE treatment at all time points tested, including the relatively early time point of 4 hr (Fig. 4A). Note that this is considerably earlierthan the point at which BU -11 cell apoptosis is seen in DMBA-treated co-cultures (i.e., 10 hr; (30)). The ability of this terminal reactive metabolite to rapidly induce apoptosis in BU-11 cells while bypassing the need for stromal cells isconsistent with the hypothesis that stromal cells generate and deliver this putative mediator of apoptosis induction to adjacent developing B cells.
Fig. 4.

A terminal metabolite of DMBA induces stromal cell-independent B cell apoptosis. BU-11 cell suspension cultures were treated with Vh or DMBA-DE (1 μM) for the indicated times. (A) BU-11 cells were analyzed for apoptosis by PI staining. The percentage of death measured in naïve cells was subtracted prior to analysis. Data are presented as means + SE from at least 3 experiments. (B) Total proteins were extracted from BU-11 cells and analyzed for p53 and β-actin by immunoblotting. Data are representative of at least 3 experiments. (C) Primary pro-B cells isolated from wildtype orp53 mutant mice were treated with Vh or DMBA-DE (0.01 μM). Pro-B cells were analyzed for apoptosis by PI staining 16 hr after treatment. The percentage of death measured in naïve cells was subtracted prior to analysis. Data are presented as means + SE from primary pro-B cells prepared from 4 individual mice. *Statistically different from Vh (p<0.05, ANOVA, Dunnett’s).
If DMBA-DE induces developing B cell apoptosis in DMBA-treated BU-11/BMS2 cell co-cultures, then it wouldbe predicted that key hallmarks of DMBA -induced apoptosis, such as p53 induction, would be manifest after treatment of BU-11 cells with DMBA-DE. Furthermore, by bypassing the requirement for DMBA metabolism in stromal cells and the subsequent transfer of reactive metabolite to B cells, markers of apoptosis would be expected to be manifest at earlier time pointsthan when treating stromal cell/B cell co -cultures with parent compound (i.e., DMBA). To test these predictions, first BU-11 cells cultured in the absence of stromal cells were treated with vehicle or DMBA-DE and p53 expression was analyzed by immunoblotting. Expression of p53 protein increased within 30 min-1 hr of DMBA-DE treatment (Fig. 4B). Second, primary pro-B cells from wildtype C57Bl/6 and congenic Trp53tm1Tyj mice were treated with vehicle or DMBA-DE (0.01 μM) for 16 hrs, and the B cells were analyzed for apoptosis by hypotonic PI stainingand flow cytometry. DMBA-DE induced significant apoptosis in primary pro-B cells from wildtype mice, but not in pro-B cells from p53 mutant mice (Fig. 4C). A similar resistance to apoptosis was seen with pro-B cells from Apaf-1fogmutant mice ( 46 ±12 % in Apaf-1WT/Mutvs 6 ±6 % in Apaf-1Mut/Mutpro -B cellstreated with 0.1 –0.001 μM DMBA-DE, p<0.05, ANOVA, Tukey-Kramer). The induction of apoptosis with DMBA-DE in a shorter time frame than when treating stromal cell/B cell co-cultures with DMBAand the suppression of apoptosis in p53-and Apaf −1− mutantB cells are consistent with the hypothesis that DMBA-DE initiates the DMBA/stromal cell-induced apoptosis cascade in developing B cells.
Membrane transfer as a mechanism of death signal delivery
To begin to determine the requirements for transfer of the putative effector metabolite from stromal to developing B cells, the requirement for cell-cell contact was evaluated. BU-11 cells either were plated on BMS2 cell monolayers or were cultured in the upper chamber of transwell inserts with the BMS2 cells maintained in the lower chamber. The membranes of these transwells allow free transfer of anything less than or equal to 3 μm in diameter between the upper and lower chambers, which would include various types of vesicles including exosomes (56). These cultures then were treated with vehicle or DMBAfor 16 hr, and apoptosis of BU-11 cells was analyzed by hypotonic PI staining. A significant percentage of the BU-11 cells underwent apoptosis when they were allowed to contact the BMS2 cells directly but not when they were cultured above the BMS2 cells in transwells (Fig. 5A). This result implies that cell-cell contact is required for apoptosis. However, it is formally possible that a DMBA effector metabolite is carried from the BMS2 cells to the BU-11 cells within vesicles and that the failure to observe BU-11 cell apoptosis in the experiment described above reflected the inability of the vesicles to migrate up through the membrane to the BU-11 cells. Therefore, the experiment was repeated with BMS2 cells in the upper chamber and BU-11 cells in the lower chamber. The failure of DMBA to induce apoptosis in this protocol (Fig. 5A) further argues against the possibility of the death signal being delivered by a vesicle and is consistent with the hypothesis that BMS2/BU-11 cell contact is required for delivery of the death signal. Additional studies in which BU-11 cells were treated with DMBA-treated BMS2-derived vesiclessimilarly failed to implicate vesicles in the transfer of the apoptotic signal (data not shown).
Fig. 5.
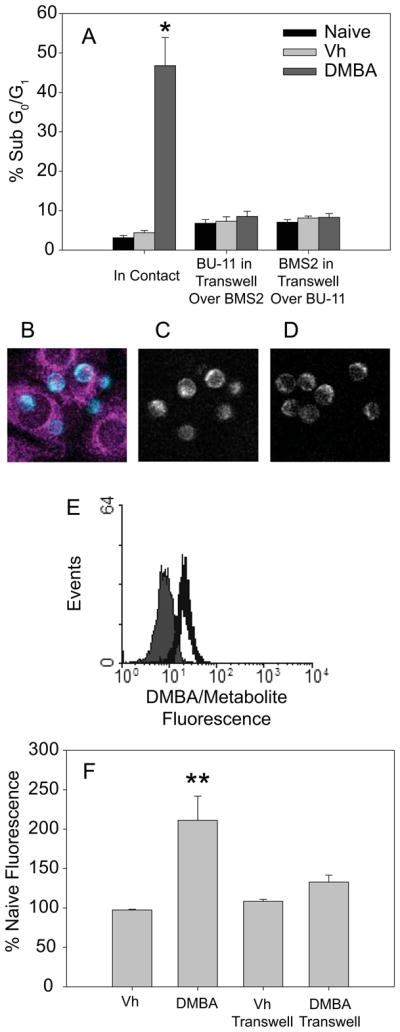
Cell contact is required for DMBA/stromal cell-induced apoptosis, potentially facilitating metabolite transfer from BMS2 to BU-11 cells. (A) BMS2 cells were cultured in plates as usual or in 3 μm pore size transwells. BU-11 cells were added directly to BMS2 monolayers or to transwells suspended over BMS2 monolayers. Co-cultures were treated with Vh or DMBA (1 μM) for 16 hr. BU-11 cells were analyzed for apoptosis by PI staining. Data are presented as means + SE from 3 experiments. (B-D) BMS2 cultures were treated with 10 μM DMBA for 30 min, washed, incubated for 1 hr, then washed again. BU-11 cells then were centrifuged onto the BMS2 cell layer and cultures immediately analyzed by two-photon microscopy for DMBA/metabolite fluorescence transfer. (B) False color image of BU-11 cells in contact with the BMS2 cell monolayer overlayed on the BMS2 cell image. (C) Image of BU-11 cells in contact with the BMS2 cell monolayer. (D) BU-11 cells not in contact with the BMS2 cell monolayer. Representative data from one of three similar experiments are presented. BU-11 cells in contact with BMS2 cells tended to contain a higher level of fluorescence than those no it contact with BMS2 (p<0.06). (E, F) BMS2 cultures were treated with Vh or DMBA (10 μM) for 30 min, washed extensively, incubated for 5 hr, and washed again. BU-11 cells then were added directly to the BMS2 cell layer or to a 1 μm pore size transwell insert above the BMS2 cells. BU-11 cells were analyzed at 18 hr by flow cytometry for fluorescence at 450 nm. (E) Representative fluorescence histogram of BU-11 cells cultured on naïve (filled) or DMBA-treated (open) BMS2 cells. (F) DMBA/metabolite fluorescence expressed as percent of naïve fluorescence. Data are presented as the means + SE from 3 experiments. *Statistically greater than other treatment groups (p<0.05, ANOVA, Tukey-Kramer). **Statistically different from naïve and from DMBA in transwell groups (p < 0.05, ANOVA, Tukey-Kramer).
Taking advantage of the fact that DMBA and its metabolites are weakly fluorescent, transfer of DMBA and/or its metabolites from BMS2 to BU-11 cells was investigated by two-photon microscopy and flow cytometry. It should be noted that DMBA and DMBA-DE are excited and emit fluorescence under identical conditions, so it was not possible to differentiate between DMBA and its metabolitein these experiments (althoug h even if DMBA were to be transferred to the B cells it cannot induce apoptosis). BMS2 cells were grown on one half of a coverglass. The following day, the whole coverglass was treated with DMBA (10 μM) and excess DMBA/metabolites were washed off. BU-11 cells then were centrifuged onto the dish and analysis of adherent BU-11 cells by two-photon microscopy was begun within 15 minutes. BU-11 cells that were in direct contact with BMS2 cells at the time of observation appeared to have a higher fluorescence than BU-11 cells clearly not in contact with BMS2 cells (Fig. 5B-D). Statistical analysis of the fluorescent intensity of BU-11 cells in contact with BMS2 cells as compared with fluorescence of BU-11 cells not in contact with BMS2 cells showed a trend towardsignificance (p=0.06, Student’s T -test).
For a more rigorous quantification of transfer, BU-11 cells were analyzed for DMBA/metabolite uptake by flow cytometry. BMS2 cells were treated with vehicle or DMBA (10 μM) and were washed to remove excess DMBA/metabolites. BU-11 cells were added either directly to the stromal cell monolayer or to the upper chamber of transwells over the BMS2 cells. BU-11 cells that were allowed to contact BMS2 cells directly exhibited a statistically significant increase in fluorescence consistent with DMBA and/or DMBA metabolite uptake(Fig. 5E-F). In contrast, there was no significant increase in fluorescence of BU-11 cells that were separated from BMS2 cells by transwell membranes (Fig. 5E-F). These results suggest that DMBA and/or its metabolites can be transferred from BMS2 cells to BU-11 cells in a contact-dependent manner.
Trogocytosis, acquisition of membrane-anchored antigen by intercellular membrane exchange, is a mechanism commonly used by cells of the immune system, including B cells (57). If lipophilic metabolites of DMBA are transferred from BMS2 cells to BU-11 cells, one mechanismfor that transport may be exchange through the BMS2 cell plasma membrane. To test this hypothesis, BMS2 cells were labeled with the lipophilic plasma membrane dye Vybrant DiO before BU-11 cells were added either directly onto BMS2 cell monolayers or into transwells above the BMS2 cell cultures. After 16 hrs, BU-11 cells then were analyzed for DiO uptake by flow cytometry. In addition, BU-11 cells maintained on unstained BMS2 cells were resuspended in supernatants from DiO-stained BMS2 cells for 20 min before analysis. A large and significant proportion of BU-11 cells took up DiO when they were allowed to contact the stained BMS2 cells directly(Fig. 6A). However, there was no significant uptake of DiO when the cells were separated by a permeable barrier or when BU-11 cells were exposed to supernatants from stained cells (Fig. 6A). If membrane transfer delivers the apoptotic metabolite, then it would be expected that membrane exchange would occur prior to overt apoptosis. To test this prediction, the kinetics of membrane transfer was determined. BMS2 cells were labeled with DiO as described above. BU-11 cells were added to the culture, harvested over time (0.5–2 hr) and analyzed for DiO uptake by flow cytometry. Significant membrane exchange occurred within 2 hrs of contact (Fig. 6B), prior to significant changes in the earliest markers of apoptosis (cytochrome c release; Fig. 1 and increased p53 expression; Fig. 3). These experiments demonstrate, for the first time, the transfer of plasma membrane components from bone marrow stromal cells, on which developing B cells depend for growth and differentiation signals, to associated bone marrow-derived B cells. The results also are consistent with the hypothesis that lipophilic metabolites, embedded in cellmembranes, may be transferred from bone marrow stromal cells to developing B cells.
Fig. 6.

B lineage cells take up membrane lipid from bone marrow stromal cells in a contact-dependent manner. BMS2 cells were left unstained or were labeled with Vybrant DiO plasma membrane dye according to the manufacturer’s protocol. (A) BU-11 cells were added directly to unstained or stained adherent BMS2 cells (Contact) or in a 3 μm pore size transwell insert over the BMS2 cells (Transwell) and harvested after 16 hr. In addition, naïve BU-11 cells were resuspended for 20 min in 16 hr supernatants from unstained or DiO-stained BMS2 cells (Supernatant). DiO uptake was analyzed by flow cytometry in live-gated cells. Inset: The filled gray histogram representsdata from BU -11 cells cultured in contact with unstained BMS2 cells, the open gray lined histogram represents data from BU-11 cells cultured in transwells over DiO-stained BMS2 cells, and the open black lined histogram represents data from BU-11 cells cultured in contact with DiO-stained BMS2 cells. (B) BU-11 cells were added directly to unstained or stained adherent BMS2 cells, harvested after 0.5–2 hrs, and analyzed by flow cytometry. (C, D) Primary pro-B cells, splenic B cells, or splenic T cells from wildtype C57Bl/6 mice were added to unstained or DiO-stained BMS2 cells directly, and lymphocytes were assayed 16 hr later by flow cytometry. (C) Representative histograms of lymphocytes on unstained (filled) or DiO-stained (open) BMS2 cells. (D) Quantification of DiO positive lymphocytes. Data are presented as means + SE from 3–4 experiments or from 3–5 experiments with cells from individual mice. *Statistically different from lymphocytes on unstained BMS2 cell controls (p< 0.05, ANOVA, Tukey-Kramer).
To determine if this membrane component transfer is unique to the BU-11 cell line, primary pro-B cells, splenic B cells or splenic T cells were co-cultured with BMS2 cells and assessed for dye uptakeby flow cytometry. Primary pro-B cells, which undergo apoptosis when treated with DMBA in co-culture with BMS2 cells in a fashion similar to BU-11 cells (30), also behaved similarly to BU-11 cells in this assay, taking up membrane from BMS2 cells (Fig. 6C-D). Interestingly, splenic B cells acquired BMS2 membrane components when they were allowed to contact BMS2 cells, similar to the developing B cells, while splenic T cells didnot acquire appreciable levels of membrane dye (Fig. 6C-D). These results suggest that membrane uptake is not unique to the BU-11 cell line but rather that it is a property of B cells throughout developmentand that transfer of the DMBA -induced apoptotic signal from bone marrow stromal cells to developing B cells may occur through membrane uptake.
Discussion
Previous studies have shown that DMBA induces significant immunosupression (5) by reducing cellularity in the bone marrow (14, 15)as well as by suppressing spleen cell numbers and function (58, 59). The reduced cellularity in bone marrow is caused largely by a loss of B cells (20), and loss of these developing B cells could result in skewing of the B cell repertoire towards apoptosis resistant, auto-reactive clones and/or a diminution in the production of competent mature B cell populations. B cell death requires contact with AhR-and CYP1B1 -expressing stromal cells (21, 22), suggesting that a metabolite of DMBA is the death factor transferred from the stromal cells to the B cells. Here, we addressed the potential for a terminal DMBA metabolite to induce B cell death and investigated the mechanism of transfer of this molecule between its source, the stromal cells, and its target, the B cells.
In order to identify a key component(s) of the DMBA/stromal cell apoptosis pathway that could be used as a point of reference in the study of the DMBA-DE apoptosis pathway, the DMBA/stromal cell pathway was examined. Several key observations support the conclusion that MOMP is the initiating event in DMBA/stromal cell-induced B cell apoptosis. 1) Cytochrome c release from the mitochondria was evident at early time points. 2) The general caspase-inhibitor, Z-VAD-FMK, did not inhibit DMBA-induced cytochrome c release indicating that cytochrome c release occurs upstream of caspase activation. 3) DMBA-induces formation of catalytically active caspase-9. 4) DMBA-induced apoptosis was suppressed in Apaf-1fogprimary pro-B cells, cells that are unable to form apoptosomes required for caspase-9 activation (42, 60). Caspase-8 activation appears to occur as a caspase-dependent downstream event, potentially participating in a positive feedback loop (45, 47, 61).
Unlike many intrinsic pathway-mediated apoptotic programs, mitochondrial membrane potential was not lost in B cells after DMBA treatmentof the co -culture. This is uncommon, but not without precedent (38, 39). In fact, apoptosis induced by a terminal metabolite of B[a]Pin a lung cancer cell line also involves cytochrome c release without ΔΨmloss (62). While the lack of ΔΨmloss in this system is somewhat unusual for an intrinsic apoptotic pathway, the presence or absence of ΔΨmloss may not be important to apoptotic pathways in general. The previously held view that ΔΨml oss mediates apoptosis has been challenged (63–66). Cells from mice lacking cyclophilin D, a protein essential for the mitochondrial permeability transition pore (PTP) complex responsible for ΔΨmloss, do not undergo mitochondrial membrane potential loss but still undergo apoptosis in response to etoposide, x-ray irradiation, staurosporine, and TNF-α (63, 67–69). However, while loss of ΔΨmmay not initiate apoptosis it may enhance it by disruption of the mitochondrial christae thereby enhancing cytochrome c release (66). In light of these data, we conclude that, while the lack ofmitochondrial membrane potential loss in DMBA -induced developing B cell apoptosis is curious, it is functionally irrelevant.
Once identification of an intrinsic apoptosis pathway was established, we investigated potential key protein mediators responsible for initiating MOMP. The pro-apoptotic proteins Bax and Bak are responsible for pore formation leading to MOMP (70). Multiplesignaling pr oteins may trigger the movement of Bax/Bak to the mitochondria to initiate MOMP, including p53 (71, 72), Bid and Bim (43), and JNK (73). We have shown that Bid is cleaved to its active form during DMBA-induced apoptosis (21). However, apoptosis is reduced only ~20% in Bid knockout primary pro-B cells (data not shown), supporting the conclusion that it participates in an amplification loop rather than acts as an initiator of the death pathway. p53 activates apoptosis through two distinct mechanisms: 1) itengages B cl-2 family members at the mitochondrial membrane resulting in neutralization of anti-apoptotic proteins (e.g. Bcl-2) or activation of pro-apoptotic proteins (e.g. Bax), stimulating MOMPto occur, and 2) p53 transcriptionally upregulates pro-apoptotic Bcl-2 family members (e.g. PUMA, Noxa, Bax)(71, 72). Previous studies have shown that DMBA-induced bone marrow toxicity and immunosupression are reduced in the presence of mutant p53 (Trp53tm1Tyj)in vivo (51, 52). Here we demonstrate that p53 expression is upregulated in B cells exposed to DMBA in the co-culture system, and pro-B cells with mutant p53 are largely resistant to DMBA/stromal cell-induced apoptosis. Thus, p53-mediated mitochondrial apoptosis is a likely pathway for DMBA/stromal cell-induced apoptosis.
Treatment of B cells alone with DMBA-DE results in apoptosis coincident with p53 induction, supportingthe hypothesis that th e stromal cell-derived death factor is indeed the terminal DMBA metabolite. Similarly, a terminal metabolite of B[a]P, B[a]P-7,8-dihydrodiol-9,10-epoxide, also can directly induce B cell death (data not shown). Consistent with this hypothesis are results from a previousstudy demonstrating that DMBA/stromal cell -induced apoptosis requires CYP1B1 and that DMBA-diol-epoxide adducts are formed inthe B cells (22). Interestingly, DMBA-DEadducts also form in the stromal cells (22); however, DMBA is not toxic to these cells. Developing B cells are known to be “ultrasensitive” to apoptotic stimuli, and this may result from the fact that these cells express very low levels of the anti-apoptotic protein BCL-2 (74). Furthermore, DMBA-induced death is associated with the down-regulation of NF-κB, a lymphocyte survival factor, another factor that may sensitize B cells to DMBA-induced death, in particular (16).
We hypothesized that thehighly labile and reactive na ture of DMBA-DE would require a specialized delivery mechanism between the stromal and B cells. Indeed, the stromal cell and B cell must be in direct contact for the death factor to be transferred from one cell to the other, resulting in B cell apoptosis. Trogocytosis (i.e. membrane transfer) is a common but underappreciated mechanism of information transfer between cells in the immune system (31). Thus, membrane transfer wasconsidered a possible mechanism for delivery of otherwise labile DMBA-DE to the B cells. Consistent with this hypothesis, DMBA and/or its metabolites and membranes were transferred between the stromal cells and B cells. Interestingly, bone marrow and splenic B cells, but not splenic T cells, efficiently incorporated membranes from bone marrow stromal cells. This may reflect the fact that B cells are capable of performing trogocytosis without support of intracellular signaling or active processes, while T cell trogocytosis is an active, T cell receptor signal-dependent process (57, 75). Typically (although not always (32)) trogocytosis is thought to be triggered by membrane antigen as a mechanism of antigen capture (57). Membrane transfer between stromal cells and bone marrow B cells may represent a more general mechanism of communication.
Results from these studies show, for the first time, that a terminal DMBA metabolite can initiate apoptosis directly in B cells and can initiate an apoptotic pathway that shares features of DMBA/stromal cell apoptosis, consistent with the conclusion that DMBA-DE is the stromal cell- derived death factor that induces B cell death in stromal/B cell co-cultures and, presumably, in vivo. A trogocytosis-like mechanism is likely the way in which the stromal cells transfer such a highly labile and reactive molecule. In addition, these studies provide support for the novel hypothesis that DMBA, via its reactive metabolite, induces a p53-intrinsic apoptosis pathway. Further studies are required to determine themechanisms of p53 activation and mitochondrial interactions in DMBA/stromal-induced B cell apoptosis.
Supplementary Material
Acknowledgments
Technical assistance was provided by the Boston University Medical Campus Flow Cytometry Core Facility.
Footnotes
Funding: National Institutes of Health (RO1ES06086 to DS); Environmental Protection Agency (Science To Achieve Results Graduate Fellowship FP-91651501 to JT); National Institute of Environmental Health Sciences (P42ES007381 to JS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences, the National Institutes of Health, or the Environmental Protection Agency.
Abbreviations: AhR, aryl hydrocarbon receptor; BA, benz[a]anthracene; B[a]P, benzo[a]pyrene; BGS, bovine growth serum; DMBA, 7,12-dimethylbenz[a]anthracene; DMBA-DE, DMBA-3,4-dihydrodiol-1,2-epoxide; FBS, fetal bovine serum: MOMP, mitochondrial outer membrane permeabilization; PAH, polycyclic aromatic hydrocarbon; PI, propidium iodide.
References
- 1.White KL, Jr, Lysy HH, Holsapple MP. Immunosuppression by polycyclic aromatic hydrocarbons: A structure-activity relationship in B6C3F1 and DBA/2 mice. Immunopharmacology. 1985;9:155–164. doi: 10.1016/0162-3109(85)90011-6. [DOI] [PubMed] [Google Scholar]
- 2.Dean JH, Luster MI, Boorman GA, Lauer LD, Luebke RW, Lawson LD. Immunesuppression following exposure of mice to the carcinogen benzo(a)pyrene but not by the noncarcinogenic congener benxo(e)pyrene. Clin Exp Immunol. 1983;52:199. [PMC free article] [PubMed] [Google Scholar]
- 3.Dean J, Ward E, Murray M, Lauer L, House R, Stillman W, Hamilton T, Adams D. Immunosuppression following 7,12-deimthylbenz[a]anthracene exposure in B6C3F1--II. Altered cell-mediated immunity and tumore resistance. Int J Immunopharm. 1986;8:189–198. doi: 10.1016/0192-0561(86)90058-5. [DOI] [PubMed] [Google Scholar]
- 4.Thurmond L, Lauer L, House R, Cook J, Dean J. Immunosuppression following exposure to 7,12-dimethylbenz[a]anthracene (DMBA) in Ah-responsive and Ah-nonresponsive mice. Toxicology and Applied Pharmacology. 1987;91:450–460. doi: 10.1016/0041-008x(87)90066-4. [DOI] [PubMed] [Google Scholar]
- 5.Ward EC, Murray MJ, Lauer LD, House RV, Irons R, Dean JH. Immunosuppression following 7,12-dimethylbenz[a]anthracene exposure in B6C3F1 mice. I. Effect on humoral immunity and host resistance. Toxicol Appl Pharmacol. 1984;75:299–308. doi: 10.1016/0041-008x(84)90212-6. [DOI] [PubMed] [Google Scholar]
- 6.Uno S, Dalton TP, Dragin N, Curran CP, Derkenne S, Miller ML, Shertzer HG, Gonzalez FJ, Nebert DW. Oral benzo[a]pyrene in Cyp1 knockout mouse lines: CYP1A1 important in detoxication, CYP1B1 metabolism required for immune damage independent of total-body burden and clearance rate. Mol Pharmacol. 2006;69:1103–1114. doi: 10.1124/mol.105.021501. [DOI] [PubMed] [Google Scholar]
- 7.Winker N, Tuschl H, Kovac R, Weber E. Immunological investigations in a group of workers exposed to various levels of polycyclic aromatic hydrocarbons. J Appl Toxicol. 1997;17:23–29. doi: 10.1002/(sici)1099-1263(199701)17:1<23::aid-jat387>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 8.Holt PG. Immune and inflammatory function in cigarette smokers. Thorax. 1987;42:241–249. doi: 10.1136/thx.42.4.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol. 2002;2:372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- 10.Szczeklik A, Szczeklik J, Galuszka Z, Musial J, Lolaryzk E, Targosz D. Humoral immunosuppression in men exposed to polycyclic aromatic hydrocarbons and related carcinogens in plluted environments. Environ Health Perpect. 1994;102:302–305. doi: 10.1289/ehp.94102302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waldman JM, Lioy PJ, Greenberg A, Butler JP. Analysis of human exposure to benzo(a)pyrene via inhalation and food ingestion in the Total Human Environmental Exposure Study (THEES) J Expo Anal Environ Epidemiol. 1991;1:193–225. [PubMed] [Google Scholar]
- 12.Phillips DH. Polycyclic aromatic hydrocarbons in the diet. Mutat Res. 1999;443:139–147. doi: 10.1016/s1383-5742(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 13.Smithgall TE, Harvey RG, Penning TM. Oxidation of the trans-3,4-dihydrodiol metabolites of the potent carcinogen 7,12-dimethylbenz(a)anthracene and other benz(a)anthracene derivatives by 3 alpha-hydroxysteroid-dihydrodiol dehydrogenase: effects of methyl substitution on velocity and stereochemical course of trans-dihydrodiol oxidation. Cancer Res. 1988;48:1227–1232. [PubMed] [Google Scholar]
- 14.Heidel SM, MacWilliams PS, Baird WM, Dashwood WM, Buters JT, Gonzalez FJ, Larsen MC, Czuprynski CJ, Jefcoate CR. Cytochrome P4501B1 mediates induction of bone marrow cytotoxicity and preleukemia cells in mice treated with 7,12-dimethylbenz[a]anthracene. Cancer Res. 2000;60:3454–3460. [PubMed] [Google Scholar]
- 15.Yamaguchi K, Matulka RA, Schneider AM, Toselli P, Trombino AF, Yang S, Hafer LJ, Mann KK, Tao XJ, Tilly JL, Near RI, Sherr DH. Induction of preB cell apoptosis by 7,12-dimethylbenz[a]anthracene in long-term primary murine bone marrow cultures. Toxicol Appl Pharmacol. 1997;147:190–203. doi: 10.1006/taap.1997.8263. [DOI] [PubMed] [Google Scholar]
- 16.Mann KK, Doerre S, Schlezinger JJ, Sherr DH, Quadri S. The role of NF-κB as a survival factor in environmental chemical-induced pre-B cell apoptosis. Mol Pharmacol. 2001;59:302–309. doi: 10.1124/mol.59.2.302. [DOI] [PubMed] [Google Scholar]
- 17.Mann K, Matulka R, Hahn M, Trombino A, Lawrence B, Kerkvliet N, Sherr D. The role of polycyclic aromatic hydrocarbon metabolism in dimethylbenz[a]anthracene-induced pre-B lymphocyte apoptosis. Toxicol Appl Pharmacol. 1999;161:10–22. doi: 10.1006/taap.1999.8778. [DOI] [PubMed] [Google Scholar]
- 18.Quadri S, Qadri A, Mann KL, Sherr DH. The bioflavonoid galangin blocks aryl hydrocarbon receptor (AhR) activation and polycyclic aromatic hydrocarbon-induced pre-B cell apoptosis. Mol Pharmacol. 2000;58:515–525. doi: 10.1124/mol.58.3.515. [DOI] [PubMed] [Google Scholar]
- 19.Ryu HY, Mann KK, Schlezinger JJ, Jensen B, Sherr DH. Environmental chemical-induced pro/pre-B cell apoptosis: Analysis of c-Myc, p27Kip1, and p21WAF1 reveals a death pathway distinct from clonal deletion. J Immunol. 2003;170:4897–4904. doi: 10.4049/jimmunol.170.10.4897. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi K, Near RI, Matulka RA, Schneider A, Toselli P, Trombino AF, Sherr DH. Activation of the aryl hydrocarbon receptor/transcription factor and bone marrow stromal cell-dependent preB cell apoptosis. J Immunol. 1997;158:2165–2173. [PubMed] [Google Scholar]
- 21.Allan LL, Mann KK, Matulka RA, Ryu HY, Schlezinger JJ, Sherr DH. Bone marrowstromal -B cell interactions in polycyclic aromatic hydrocarbon-induced pro/pre-B cell apoptosis. Toxicol Sci. 2003;76:357–365. doi: 10.1093/toxsci/kfg239. [DOI] [PubMed] [Google Scholar]
- 22.Heidel SM, Holston K, Buters JT, Gonzalez FJ, Jefcoate CR, Czupyrynski CJ. Bone marrow stromal cell cytochrome P4501B1 is required for pre-B cell apoptosis induced by 7,12-dimethylbenz[a]anthracene. Mol Pharmacol. 1999;56:1317–1323. doi: 10.1124/mol.56.6.1317. [DOI] [PubMed] [Google Scholar]
- 23.Pelayo R, Welner RS, Nagai Y, Kincade PW. Life before the pre-B cell receptor checkpoint: specification and commitment of primitive lymphoid progenitors in adult bone marrow. Semin Immunol. 2006;18:2–11. doi: 10.1016/j.smim.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Ma Q, Jones D, Borghesani PR, Segal RA, Nagasawa T, Kishimoto T, Bronson RT, Springer TA. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4-and SDF -1-deficient mice. Proc Natl Acad Sci U S A. 1998;95:9448–9453. doi: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Egawa T, Kawabata K, Kawamoto H, Amada K, Okamoto R, Fujii N, Kishimoto T, Katsura Y, Nagasawa T. The earliest stages of Bcell development require a chemokine stromal cell-derived factor/pre-B cell growth-stimulating factor. Immunity. 2001;15:323–334. doi: 10.1016/s1074-7613(01)00185-6. [DOI] [PubMed] [Google Scholar]
- 26.Lu L, Osmond DG. Apoptosis during B lymphopoiesis in mouse bone marrow. J Immunol. 1997;158:5136–5145. [PubMed] [Google Scholar]
- 27.Borghesi LA, Smithson G, Kincade PW. Stromal cell modulation of negative regulatory signals that influence apoptosis and proliferation of B lineage lymphocytes. J Immunol. 1997;159:4171–4179. [PubMed] [Google Scholar]
- 28.Manabe A, Murti KG, Coustan-Smith E, Kumagai M, Behm FG, Raimondi SC, Campana D. Adhesion-dependent survival of normal and leukemic human B lymphoblasts on bone marrow stromal cells. Blood. 1994;83:758–766. [PubMed] [Google Scholar]
- 29.Yuroff AS, Jefcoate CR, Czuprynski CJ. Close proximity, but not VLA-4-dependent adherence between pre-B cells and bone marrow stromal cells, is required for DMBA-induced apoptosis of pre-B cells in vitro. Toxicol Lett. 2005;156:253–260. doi: 10.1016/j.toxlet.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 30.Ryu HY, Emberley JK, Schlezinger JJ, Allan LL, Na S, Sherr DH. Environmental chemical-induced bone marrow B cell apoptosis: death receptor-independent activation of a caspase-3 to caspase-8 pathway. Mol Pharmacol. 2005;68:1087–1096. doi: 10.1124/mol.105.014712. [DOI] [PubMed] [Google Scholar]
- 31.Ahmed KA, Munegowda MA, Xie Y, Xiang J. Intercellular trogocytosis plays an important role in modulation of immune responses. Cell Mol Immunol. 2008;5:261–269. doi: 10.1038/cmi.2008.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rechavi O, Goldstein I, Kloog Y. Intercellular exchange of proteins: the immune cell habit of sharing. FEBS Lett. 2009;583:1792–1799. doi: 10.1016/j.febslet.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 33.Pietrangeli C, Hayashi SI, Kincade P. Stromal cell lines which support lymphocyte growth: characterization, sensitivity to radiation and responsiveness to growth factors. Eur J Immunol. 1988;18:863–872. doi: 10.1002/eji.1830180606. [DOI] [PubMed] [Google Scholar]
- 34.Tze LE, Baness EA, Hippen KL, Behrens TW. Ig light chain receptor editing in anergic B cells. J Immunol. 2000;165:6796–6802. doi: 10.4049/jimmunol.165.12.6796. [DOI] [PubMed] [Google Scholar]
- 35.Schlezinger JJ, Emberley JK, Bissonnette SL, Sherr DH. An L-Tyrosine Derivative and PPARγ Agonist, GW7845, Activates a Multifaceted Casp ase Cascade in Bone Marrow B Cells. Toxicol Sci. 2007;98:125–136. doi: 10.1093/toxsci/kfm071. [DOI] [PubMed] [Google Scholar]
- 36.Waterhouse NJ, Steel R, Kluck R, Trapani JA. Assaying cytochrome C translocation during apoptosis. Methods Mol Biol. 2004;284:307–313. doi: 10.1385/1-59259-816-1:307. [DOI] [PubMed] [Google Scholar]
- 37.Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997;411:77–82. doi: 10.1016/s0014-5793(97)00669-8. [DOI] [PubMed] [Google Scholar]
- 38.Shimizu S, Tsujimoto Y. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc Natl Acad Sci U S A. 2000;97:577–582. doi: 10.1073/pnas.97.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD, Green DR. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol. 2001;153:319–328. doi: 10.1083/jcb.153.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Facompre M, Wattez N, Kluza J, Lansiaux A, Bailly C. Relationship between cell cycle changes and variations of the mitochondrial membrane potential induced by etoposide. Mol Cell Biol Res Commun. 2000;4:37–42. doi: 10.1006/mcbr.2000.0251. [DOI] [PubMed] [Google Scholar]
- 41.Lee ST, Hoeflich KP, Wasfy GW, Woodgett JR, Leber B, Andrews DW, Hedley DW, Penn LZ. Bcl-2 targeted to the endoplasmic reticulum can inhibit apoptosis induced by Myc but not etoposide in Rat-1 fibroblasts. Oncogene. 1999;18:3520–3528. doi: 10.1038/sj.onc.1202716. [DOI] [PubMed] [Google Scholar]
- 42.Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES. Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation. J Biol Chem. 1999;274:17941–17945. doi: 10.1074/jbc.274.25.17941. [DOI] [PubMed] [Google Scholar]
- 43.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Molecular cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Zhivotovsky B, Orrenius S. Caspase-2 function in response to DNA damage. Biochem Biophys Res Commun. 2005;331:859–867. doi: 10.1016/j.bbrc.2005.03.191. [DOI] [PubMed] [Google Scholar]
- 45.Cowling V, Downward J. Caspase-6 is the direct activator of caspase-8 in the cytochrome c-induced apoptosis pathway: absolute requirement for removal of caspase-6 prodomain. Cell Death Differ. 2002;9:1046–1056. doi: 10.1038/sj.cdd.4401065. [DOI] [PubMed] [Google Scholar]
- 46.Murphy BM, Creagh EM, Martin SJ. Interchain Proteolysis, in the Absence of a Dimerization Stimulus, Can Initiate Apoptosis-associated Caspase-8 Activation. J Biol Chem. 2004;279:36916–36922. doi: 10.1074/jbc.M402039200. [DOI] [PubMed] [Google Scholar]
- 47.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. The Journal of cell biology. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 49.Slee EA, Keogh SA, Martin SJ. Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: a potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release. Cell Death Differ. 2000;7:556–565. doi: 10.1038/sj.cdd.4400689. [DOI] [PubMed] [Google Scholar]
- 50.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J, Mitchell LA, Lauer FT, Burchiel SW. p53 and ATM/ATR regulate 7,12-dimethylbenz[a]anthracene-induced immunosuppression. Mol Pharmacol. 2008;73:137–146. doi: 10.1124/mol.107.039230. [DOI] [PubMed] [Google Scholar]
- 52.Page TJ, O’Brien S, Holston K, MacWilliams PS, Jefcoate CR, Czuprynski CJ. 7,12-Dimethylbenz[a]anthracene Induced Bone Marrow Toxicity Is p53 Dependent. Toxicol Sci. 2003;2:2. doi: 10.1093/toxsci/kfg115. [DOI] [PubMed] [Google Scholar]
- 53.Ryu HY, Mann KK, Schlezinger JJ, Jensen B, Sherr DH. Environmental chemical-induced pro/pre-B cell apoptosis: analysis of c-Myc, p27Kip1, and p21WAF1 reveals a death pathway distinct from clonal deletion. J Immunol. 2003;170:4897–4904. doi: 10.4049/jimmunol.170.10.4897. [DOI] [PubMed] [Google Scholar]
- 54.Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268:2764–2772. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- 55.Dipple A, Nebzydoski JA. Evidence for the involvement of a diol-epoxide in the binding of 7,12-dimethylbenz(a)anthracene to DNA in cells in culture. Chem Biol Interact. 1978;20:17–26. doi: 10.1016/0009-2797(78)90077-7. [DOI] [PubMed] [Google Scholar]
- 56.Caby MP, Lankar D, Vincendeau-Scherrer C, Raposo G, Bonnerot C. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005;17:879–887. doi: 10.1093/intimm/dxh267. [DOI] [PubMed] [Google Scholar]
- 57.Aucher A, Magdeleine E, Joly E, Hudrisier D. Capture of plasma membrane fragments from target cells by trogocytosis requires signaling in T cells but not in B cells. Blood. 2008;111:5621–5628. doi: 10.1182/blood-2008-01-134155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao J, Lauer FT, Dunaway S, Burchiel SW. Cytochrome P450 1B1 is required for 7,12-dimethylbenz(a)-anthracene (DMBA) induced spleen cell immunotoxicity. Toxicol Sci. 2005;86:68–74. doi: 10.1093/toxsci/kfi176. [DOI] [PubMed] [Google Scholar]
- 59.Gao J, Lauer FT, Mitchell LA, Burchiel SW. Microsomal expoxide hydrolase is required for 7,12-dimethylbenz[a]anthracene (DMBA)-induced immunotoxicity in mice. Toxicol Sci. 2007;98:137–144. doi: 10.1093/toxsci/kfm089. [DOI] [PubMed] [Google Scholar]
- 60.Honarpour N, Gilbert SL, Lahn BT, Wang X, Herz J. Apaf-1 deficiency and neural tube closure defects are found in fog mice. Proc Natl Acad Sci U S A. 2001;98:9683–9687. doi: 10.1073/pnas.171283198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inoue S, Browne G, Melino G, Cohen GM. Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ. 2009;16:1053–1061. doi: 10.1038/cdd.2009.29. [DOI] [PubMed] [Google Scholar]
- 62.Xiao H, Rawal M, Hahm ER, Singh SV. Benzo[a]pyrene-7,8-diol-9,10- epoxide causes caspase-mediated apoptosis in H460 human lung cancer cell line. Cell Cycle. 2007;6:2826–2834. doi: 10.4161/cc.6.22.4891. [DOI] [PubMed] [Google Scholar]
- 63.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans. 2006;34:232–237. doi: 10.1042/BST20060232. [DOI] [PubMed] [Google Scholar]
- 65.Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–840. doi: 10.1007/s10495-006-0525-7. [DOI] [PubMed] [Google Scholar]
- 66.Kinnally KW, Antonsson B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis. 2007;12:857–868. doi: 10.1007/s10495-007-0722-z. [DOI] [PubMed] [Google Scholar]
- 67.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 68.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 69.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 70.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochimica et biophysica acta. 2009;1787:414–420. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chipuk JE, Green DR. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006;13:994–1002. doi: 10.1038/sj.cdd.4401908. [DOI] [PubMed] [Google Scholar]
- 73.Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, Davis RJ. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol. 2002;22:4929–4942. doi: 10.1128/MCB.22.13.4929-4942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Griffiths SD, Goodhead DT, Marsden SJ, Wright EG, Krajewski S, Reed JC, Korsmeyer SJ, Greaves M. Interleukin 7-dependent B lymphocyte precursor cells are ultrasensitive to apoptosis. J Exp Med. 1994;179:1789–1797. doi: 10.1084/jem.179.6.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hudrisier D, Aucher A, Puaux AL, Bordier C, Joly E. Capture of target cell membrane components via trogocytosis is triggered by a selected set of surface molecules on T or B cells. J Immunol. 2007;178:3637–3647. doi: 10.4049/jimmunol.178.6.3637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.