

Abstract
Background
Brain alterations in structure and function have been identified in people with risk factors for sporadic type Alzheimer’s disease (AD), suggesting that alterations can be detected decades before AD diagnosis. While the effect of Apolipoprotein E (ApoE) ε4 on the brain is well studied, less is known about the effect of family history of AD. We examined the main effects of family history and ApoE ε4 on brain integrity, in addition to assessing possible additive effects of these two risk factors.
Methods
Diffusion tensor imaging was performed in 136 middle-aged asymptomatic participants stratified on family history and ApoE ε4. Mean diffusivity and fractional anisotropy (FA) were entered in factorial analyses to test the effect of AD risk on microstructural brain integrity. We performed a post hoc analysis of the three principle diffusivities (λ1, λ2, λ3) to provide potential additional insight on underlying tissue differences.
Results
Parental family history of AD was associated with lower FA in regions of the brain known to be affected by AD, including cingulum, corpus callosum, tapetum, uncinate fasciculus, hippocampus, and adjacent white matter. Contrary to previous reports there was no main effect of ApoE ε4; however, there was an additive effect of family history and ApoE ε4 where family history positive participants who were also ApoE ε4 carriers had the lowest FA compared to the other groups.
Conclusions
The data indicate that unknown risk factors contained in family history are associated with changes in microstructural brain integrity in areas of the brain known be affected by AD. Importantly, the results provide further evidence that AD pathology may be detected prior to cognitive changes, perhaps decades before disease onset.
Keywords: Alzheimer’s disease, family history, ApoE ε4, diffusion tensor imaging, MRI, white matter
1. Introduction
A greater understanding of preclinical brain changes that occur in Alzheimer’s disease (AD) may facilitate earlier identification and treatment of the disease. One approach to understanding early alterations is to study individuals who are disease-free, but at increased risk for AD due to presence of one or more risk factors for disease, such as apolipoprotein (ApoE) ε4, or family history of AD.
ApoE ε4 is a major risk factor for late-onset or sporadic AD. The ε4 allele frequency is approximately 15% in the general population but approximately 40% in patients with AD. There is a significant load effect of ε4, and presence of this risk factor appears to be associated with earlier development of AD (1). Compared to non-carriers, cognitively normal ε4 carriers show differences in brain structure (2, 3) functional activation (4), glucose metabolism (5), perfusion (6), and anisotropic diffusion (7, 8) [see Cherbuin et al. for a systematic review (9)]. ApoE ε4 is not a determinant of AD development, but instead interacts with other genetic and environmental factors to define AD risk.
Parental family history of AD is another risk factor associated with increased odds of developing dementia (10-12). The presence of family history together with ApoE ε4 increases cumulative lifetime risk for AD. For first degree relatives of AD patients, possessing a 3/3 genotype is associated with a life time risk for AD of 29.2%. The life time risk goes up to 46.1% for first degree relatives with a 3/4 genotype, and up to 61.4% for those with a 4/4 genotype (13). In addition, family history of AD has been shown to act alone or in concert with ApoE ε4 to modulate magnetic resonance blood oxygenation level dependent (BOLD) signal during metacognitive (14) and memory tasks (15-18), alter resting perfusion as measured with arterial spin labeling MRI (17), and alter cerebral metabolism rate of glucose (CMRglc) as measured with positron emission tomography (19, 20).
In this study, diffusion tensor imaging (DTI) was used to assess brain microstructure in a cohort of asymptomatic middle aged persons with increased risk for AD either by way of family history or ApoE ε4 genotype, or both factors, compared to controls. DTI facilitates the probing of tissue microstructure through its sensitivity to Brownian motion of water molecules (21, 22) and is sensitive to white matter integrity which is hypothesized to be compromised at an early stage of AD development (23); although how early they become detectable in the preclinical stage is presently unknown. Laboratory work in mice suggests that white matter alterations may be detected even prior to the appearance of tau or beta amyloid pathology (24)
Previous studies have not dissociated the effect of family history and ApoE ε4 on brain white matter. We sought to examine the effect of these two variables using diffusion tensor imaging, employing fractional anisotropy (FA), mean diffusivity (MD) and three principle diffusivities to characterize potential white matter alterations associated with risk. Several studies have evaluated white matter alterations in AD and mild cognitive impairment (MCI) (25-32); however, it is possible that the preclinical picture of AD is different from the disease state. We therefore performed an exploratory voxel-wise analysis of family history and ApoE ε4 to evaluate white matter in a potentially preclinical stage of AD, when no cognitive abnormalities are yet present.
Finally, evidence suggests that maternal origin of family history risk is associated with a greater effect on the brain than paternal origin (20). Accordingly, we also determined the extent to which origin of family history is reflected in differential white matter alterations.
2. Methods
2.1 Participants
One hundred and thirty-six cognitively normal participants (mean age 56.99 ± 5.35 SD years; mean education 16.44 ± 2.21 SD years; 45 men, 91 women) participated in the study. Sixty-three participants did not have a known family history of AD (self-report) and were recruited using advertisements posted in the newspaper, and through an invitation sent to a university campus mailing list. Seventy-three participants had at least one parent with AD (FH+) and were recruited from the Wisconsin Registry for Alzheimer’s Prevention (33) a longitudinal registry of cognitively normal adults who have at least one parent with sporadic AD. To verify the diagnosis of AD in the parent, parental medical records (and autopsy reports, available and verified by a pathologist as AD in ten cases) were reviewed by a multidisciplinary diagnostic consensus panel. Frequently, the clinical work-up and diagnosis in the parent were conducted through a network of statewide clinics affiliated with the Wisconsin Alzheimer’s Institute, all using similar diagnostic procedures. A diagnosis of AD was made using CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) criteria (34). There were no families included with known autosomal dominant mutations. All participants underwent ApoE genotyping using polymerase chain reaction sequencing. Within the FH+ group, thirty-five (48.6%) participants were ε4 positive and the remaining thirty-eight (51.4%) were ε4 negative (five 4/4, twenty-eight 3/4, twenty-eight 3/3, two 2/4, and ten 2/3). A group of 63 participants with no family history of AD (FH-) were recruited from the community and matched to the demographic characteristics of the FH+ sample. Absence of first-degree family history of AD was determined through self-report of the participant in response to a detailed medical history questionnaire. To be included in the FH- group, both parents had to survive to at least age 70 and not carry a diagnosis of dementia or exhibit frank symptoms of dementia of any kind. Twenty-one (33.3%) FH-participants were ε4 positive and forty-two (66.7%) were ε4 negative (two 4/4, nineteen 3/4, thirty-seven 3/3, two 2/3, and two 2/2). Demographics for the four sub-groups are presented in Table 1.
Table 1.
Demographic and Neuropsychological Test Scores by Group Membership
| FH+ε4+ (N, Mean, SD) |
FH+ε4-(N, Mean, SD) |
FH-ε4+(N, Mean, SD) |
FH-ε4-(N, Mean, SD) |
P | |
|---|---|---|---|---|---|
| AGE | 35, 57.5, 5.7 | 38, 57.4, 5.4 | 21, 55.4, 5.7 | 42, 58.4, 4.2 | .07 |
|
| |||||
| EDUCATION | 35, 16.3, 2.5 | 38, 16.1, 2.7 | 21, 16.9, 1.9 | 42, 16.7, 2.3 | .46 |
|
| |||||
| SEX (M/F) | 12/23 | 14/24 | 5/16 | 14/28 | n/a |
|
| |||||
| MMSE | 35, 29.5, 0.8 | 38, 29.4, 0.9 | 21, 29.5, 0.7 | 40, 29.4, 1 | .86 |
|
| |||||
| WRAT Reading | 35, 51.6, 3.5 | 38, 52.9, 3.7 | 21, 54.3, 2.7 | 42, 52.9, 4.2 | .07 |
|
| |||||
| DIGIT SPAN | 35, 17.7, 4.7 | 38, 19.0, 3.9 | 21,19.7, 4.1 | 42, 18.7, 4.2 | .39 |
|
| |||||
| TRAILS A | 35, 28.9, 8.2 | 38, 27.7, 7.9 | 21, 24.2, 6.5 | 42, 29.3, 9.3 | .12 |
|
| |||||
| TRAILS B | 35, 67.8, 23.4 | 38, 57.3, 17.9 | 21, 53.7, 14.9 | 42, 63.9, 31.3 | .10 |
|
| |||||
| COWAT | 35, 43.7, 11.9 | 38 44.2, 9.9 | 21, 47.6, 11.1 | 42, 46.6, 13.6 | .53 |
|
| |||||
| RAVLT TOTAL | 35M, 50.9, 8.0 | 38, 51.6, 9 | 21, 53.3, 7.2 | 42, 50.3, 6.5 | .51 |
|
| |||||
| BVMT TOT | 35, 24.1, 5.5 | 38, 26.9, 4.4 | 21 30.5, 4.1 | 40, 26.8, 5.2 | .00 |
|
| |||||
| RAVLT DR | 35, 9.6, 2.7 | 38, 10.1, 2.6 | 21 10.5, 2.8 | 42, 10.2, 2.5 | .62 |
|
| |||||
| BVMT DR | 35, 9.5, 2.0 | 38, 10.4, 1.7 | 21, 11.2, 1.3 | 40, 10.8, 1.4 | .00 |
|
| |||||
| CES-D | 33, 6.4, 8.4 | 38, 4.2, 3.6 | 21, 7.1, 6.9 | 40, 5.1, 4.2 | .22 |
Note: All reported scores above are raw scores. Abbreviations: MMSE (Mini Mental State Exam); WRAT (Wide Range Achievement Test); TRAILS (Trails Making Tests) COWAT (Controlled Oral Word Association Test); RAVLT (Rey Auditory Verbal learning Test); BVMT (Brief Visual Memory Test), DR (delayed recall); CES-D (Center for Epidemiologic Studies Depression Scale).
Participants’ consent was obtained under a protocol approved by the University of Wisconsin Health Sciences Institutional Review Board. Exclusions included MRI scanner incompatibility, history of major psychiatric disease (e.g., schizophrenia, bipolar disorder, substance abuse, major depression) or major medical conditions (e.g., history of neurological disorders including prior head trauma with loss of consciousness, cancer requiring chemotherapy or radiation, insulin dependent diabetes, and untreated hypertension), abnormal cognitive testing (more than one standard deviation below the mean on any test) or abnormal structural MRI indicating disease or a condition that would affect normalization procedures (e.g. tumor or hydrocephalus). From a total of 166 scans, seven scans were excluded due to signal drop out or artifact, one was discarded due to incomplete coverage of the brain; one scan was dropped due to differing acquisition parameters, and 21 participants were excluded due to abnormal cognitive testing. In these latter 21 cases, participants were first identified by a cognitive score that was more than one standard deviation below the mean on any test. Once identified, all of the participant’s scores were reviewed by a neuropsychologist to confirm below normal cognitive function before exclusion from the analysis.
2.2 Neuropsychological testing
Participants underwent neuropsychological evaluation which included the following tests: Folstein Mini Mental State Exam (35), Digit Span from the Wechsler Adult Intelligence Scale-III (36), Wide Range Achievement Test-III, Reading Test, Rey Auditory Verbal Learning Test (37), Brief Visual Memory Test, Trail Making Test A & B (38), and Controlled Oral Word Association Test (39).
2. 3 Magnetic resonance imaging
Participants were imaged on a General Electric 3.0 Tesla SIGNA (Waukesha, WI) MRI system with a quadrature birdcage head coil. The diffusion tensor imaging parameters have been published previously (40). Briefly, DTI was performed using a cardiac-gated, diffusion-weighted, spin-echo, single-shot, echo planar imaging pulse sequence, in twelve encoding directions. Three averages were acquired and the cerebrum was covered using 39 contiguous 3-mm thick axial slices. The acquired voxel size of 2 × 2 × 3 mm was interpolated to 0.9375 mm isotropic dimensions (256 ×256 in-plane image matrix). High order shimming was performed prior to the DTI acquisition to optimize the homogeneity of the magnetic field across the brain and to minimize EPI distortions. A B0 field map was obtained using a pair of non-EPI gradient echo images at two echo times to address residual spatial distortions from B0 inhomogeneities.
2. 4 Diffusion tensor image processing
Image distortions in the DTI data caused by eddy currents were corrected using a 2D affine co-registration function, align linear, in the Automated Image Registration (AIR) software package (http://www.bishopw.loni.ucla.edu/AIR5/). Non-linear image distortion from static field (B0) inhomogeneities was corrected using the B0 field map and methods implemented in the Prelude (Phase Region Expanding Labeller for Unwrapping Discrete Estimates) and Fugue (FMRIB’s Utility for Geometrically Unwarping EPIs) tools from the FSL software suite (41). Next, three-dimensional maps of the diffusion tensor and derived measures, MD, FA, and the three principle diffusivities λ1, λ2, λ3, were calculated. MD is an index of isotropic diffusion of water molecules and FA is a derived measure of diffusion anisotropy or directional diffusion. Less reported though perhaps more specific to underlying pathology, are the three principle diffusivities composing the diffusion tensor, namely eigenvalue1 (λ1) eigenvalue2 (λ2) and eigenvalue3 (λ3). The principle diffusivity, λ1, has been referred to as parallel or axial diffusion, and decreases in this value have been associated with axonal damage (42), and Wallerian degeneration (43). The intermediate (λ2) and smallest diffusivity (λ3) are more closely related to loss of myelin, and either singly or together (λ2+ λ3/2) are referred to as perpendicular diffusivity. Higher perpendicular diffusivity has been associated with Wallerian degeneration (43), but it more typically associated with myelin alterations such as dysmyelination (44), and demyelination (45).
FA maps were normalized via 12-parameter affine transformation and nonlinear deformation using Statistical Parametric Mapping software (SPM5 available at http://www.fil.ion.ucl.ac.uk/spm) to a custom FA template comprised of an average of 121 FA maps acquired from healthy participants who matched the demographic composition of the present study sample (102 of the participants included in the template were part of the present study sample). Spatial normalization parameters were derived from the normalization of each individual’s FA image, and then applied to their FA, MD, λ1, λ2, and λ3 maps. All of the normalized DTI maps were inspected for accurate normalization using the “check registration” function in SPM and selecting specific fiber tracts for comparison, including the corpus callosum and superior longitudinal fasciculus. Following the generation of our results via SPM, the regions where significant results were obtained were again inspected in every subject to ensure against errors in registration. The normalized images were smoothed using a 10-mm isotropic Gaussian kernel.
2. 5 Statistical analysis
We expected that FH+ participants and ε4+ participants would show alterations in tissue microstructure in AD-vulnerable brain regions; however, as little is known about the preclinical picture of AD, we performed an exploratory analysis that would allow us to detect alterations in less well-defined regions. We performed a 2×2 ANCOVA in SPM5, where the two factors were FH and ε4. Age, education, and gender were included as covariates because of the known or potential effects they may have on brain structure. As it was possible that structural effects would be subtle in this non-impaired group of participants, analyses were thresholded at p<.005 uncorrected. In order to exclude clusters of a very small size and increase the anatomical plausibility of our results, we employed a cluster size threshold of 20 contiguous voxels. Both FA and MD were dependent variables in two separate ANCOVAs. In order to further characterize the main FA and MD results, we evaluated the three principle diffusivities (λ1, λ2, and λ3) in the regions where the main effects of FA and MD were found, using the main effects as a mask. These analyses were thresholded at p<.05. The analysis of family history was conducted using a one-way ANOVA comparing four groups that were divided based on family history detail: maternal family history (FHm), paternal family history (FHp), maternal and paternal family history (FHmp), and negative family history (FH-). The Statistical Package for the Social Sciences (SPSS 15.0, Chicago) was used for the analysis of behavioral data.
3. Results
3. 1 Behavioral
As shown in Table 1, there were no significant differences between groups on age or education. With regard to neuropsychological function, there was a significant difference between groups on the Brief Visual Memory Test (BVMT) total score F(3,130) = 7.3, p < .001, and BVMT delayed recall F(3,130) = 5.7, p < .001. BVMT total score was highest in the FH-ε4+ group (m = 30.5), followed by FH+ε4- (m = 26.9), then FH-ε4- (m = 26.8), and finally FH+ε4+ (m = 24.1). Delayed recall was highest in FH-ε4+ (m = 11.2), followed by FH-ε4- (m = 10.8), then FH+ε4- (m = 10.4), and FH+ε4+ (m = 9.5). There was no significant group difference on any of the other neuropsychological tests.
3. 2 Imaging
Main effect of family history
As hypothesized, increased risk for AD was associated with differences in white matter anisotropy. Specifically, positive family history was associated with lower fractional anisotropy in several brain regions (shown in Figure 1). A factorial ANOVA where factor 1 was Family History (FH+, FH-) and factor 2 was ApoE ε4 status (ε4+, ε4-) indicated a main effect of FH in bilateral anterior corona radiata, left uncinate fasciculus/inferior fronto-occipital fasciculus, left superior corona radiata, left superior longitudinal fasciculus, left tapetum, bilateral posterior corona radiata, small portions of the body of the corpus callosum, right posterior cingulum, and bilateral hippocampus and adjacent white matter posterior to the hippocampus. Although there was no main effect of FH on mean diffusivity, FH was associated with higher perpendicular diffusivity (λ2, and λ3) in the same regions where we found an FA effect (p<.05). At p< .05, there was no relationship between FH and parallel diffusion (λ1).
Figure 1.
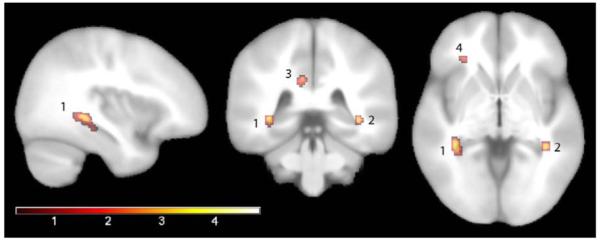
Main effect of family history
Microstructural differences were detected in participants with a parental family history of AD compared to controls. These participants had lower fractional anisotropy in right posterior hippocampus (region 1), left posterior hippocampus (region 2), right posterior cingulum bundle (region 3), left anterior corona radiata and right anterior corona radiata (region 4), left uncinate fasciculus/inferior fronto-occipital fasciculus, left superior corona radiata, left superior longitudinal fasciculus, bilateral posterior corona radiata, and small portions of the body of the corpus callosum. Analyses of FA were adjusted for age, sex, and education. Brain sections are displayed in radiological orientation and the color bar shows the magnitude of the t statistic. Results are shown at p<.005 uncorrected.
Main effect of ApoE ε4
There was no main effect of ApoE ε4 on FA, MD, or on any of the principle diffusivities.
Weighted effect of risk
In order to assess the extent to which risk factors are additive, weighted contrast vectors were assigned in SPM5, where the greatest negative weight was assigned to the group possessing both risk factors, FH+ε4+ (-1), followed by FH+ε4- (-.5), then FH-ε4+ (.5), and finally the highest positive weight to the group with neither risk factor, FH-ε4- (1). As hypothesized, risk followed a step-wise progression where possessing both risk factors was associated with the lowest FA, followed by possessing FH only, then ε4 only, and finally, the highest FA was found in the no risk factor group. This effect was present in left anterior corona radiata, bilateral uncinate fasciculus/inferior fronto-occipital fasciculus, bilateral superior corona radiata, left superior longitudinal fasciculus, left tapetum, bilateral posterior corona radiata, right external capsule, right posterior cingulum bundle, and bilateral hippocampus and adjacent white matter posterior to the hippocampus. Weighted contrast vectors assigned in the analysis of MD, and the principle diffusivities did not show any regions where this pattern was significant.
Maternal family history
There was no significant age or education difference between the four groups. Using data extracted from the maximum t value voxel in the right sided white matter cluster posterior to the hippocampus where we previously found a main effect of family history and an additive effect of FH and ApoE, (36, -39, -3), we performed a one-way ANOVA where hippocampal FA was the dependent variable and group (FH-, FHm, FHp, FHmp) was the independent variable. Controlling for age, sex, and education, there was a significant effect of group, F(3, 132) = 3.4, p < .05. Follow-up t-tests indicated that fractional anisotropy was significantly lower in the maternal family history group (m = 3.08, SD = .36) compared to the control group (m = 3.25, SD = .31), t(111) = 2.7, p < .01. There was no significant difference between FHp (3.11, SD = .43) or FHmp (M = 3.04, SD = .17) compared to control, nor were they significantly different from each other or FHm. Figure 2 illustrates the group means for hippocampal FA in the four groups.
Figure 2.

Maternal family history
The maternal family history group (n = 46) had significantly lower fractional anisotropy in right sided white matter posterior to the hippocampus compared to participants who were FH and ε4 negative (n = 63). There was no significant difference between any of the other groups. The fractional anisotropy values were adjusted for age, sex, and education.
4. Discussion
In the present study, parental family history was associated with altered brain microstructure manifested as lower FA measured by DTI. Furthermore, FA was lower with increased risk load. Specifically, possessing both positive family history and ApoE ε4 was associated with the lowest FA compared to possessing only one or no risk factors. Affected brain regions were those that are also affected in AD, including the corpus callosum (26, 46-49), the cingulum (27, 50, 51), and medial temporal gray and white matter (29, 52). Our findings in the vicinity of the hippocampus proper and white matter adjacent to the hippocampus are similar to previous findings associated with ApoE ε4 (8). To fully understand FA measurements in gray matter, multiexponential modeling of slow and fast diffusion components is needed (53); however, FA measurements in the hippocampus are plausible. The hippocampus has an organized cytoarchitecture that restricts water diffusion in such a way as to cause anisotropic diffusion (54), and hippocampal differences in diffusion have also been reported in MCI (55) and AD (29).
Participants with family history of AD in the present study also showed lower FA in left tapetum. The tapetum is a tract with fibers running between posterior corpus callosum and medial temporal lobe, and is thought to form a connection between the medial temporal lobe and the contralateral temporal cortex. Degeneration of the tapetum that is secondary to temporal lobe abnormality has been observed in temporal lobe epileptics (56). It is possible that preclinical AD involves a similar course of degeneration, with early AD pathology affecting the medial temporal lobe, followed by degeneration of cross-connecting fibers. This is supported by several studies in AD indicating that posterior corpus callosum is affected to a greater degree than the anterior (genu) portion of the corpus callosum (26, 46-49). A difference was also found in the left uncinate fasciculus, a tract connecting anterior temporal lobe with frontal orbital cortex. Alzheimer’s patients show lower anisotropy in the uncinate compared to control (27, 57), and amnestic MCI is associated with lower FA on the left, with uncinate FA also being correlated with episodic memory in aMCI (58).
In general, regions where we found an effect of family history were clustered on the left side of the brain. Our hippocampal results were bilateral, but we found a slightly larger cluster on the left. Several of the microstructural findings in people with increased risk for AD due to MCI are asymmetrical, with several results favoring greater left medial temporal lobe alterations (29, 59, 60), which may reflect a unique pattern of AD development. It is also possible that the changes are bilateral but that limitations in imaging or statistical analysis fail to reveal bilateral alterations. This is somewhat supported by our observation that raising the statistical threshold in our analysis yielded greater unilateral effects.
Only a few studies have examined the effect of risk for AD on DTI measures in a cognitively normal population. Using transverse relaxation rate (R2), a measure that—based on its sensitivity to small changes in tissue water content—is sensitive to myelin breakdown, Bartzokis et al. have demonstrated an effect of ApoE ε4 on the trajectory of aging (61). In cognitively normal adults aged 55 to 75 years, ApoE ε4 resulted in a steeper age-related decline in myelin compared to controls. In a later study, Bartzokis et al. also demonstrated that ApoE ε4 associated myelin alterations are related to cognitive processing speed (62).
The effect of ApoE ε4 in cognitively normal populations has also been investigated using DTI. Honea et al. (63) examined the effect of ApoE ε4 on FA in older adults (over the age of 60 years) and found that ε4 was associated with lower FA in the left parahippocampal gyrus white matter. Similarly, Nierenberg et al. (7), compared ApoE ε4 carriers to non-carriers, and found that carriers exhibited lower FA in parahippocampal white matter. In the present study, there was no main effect of ApoE ε4, however, our participant sample was substantially younger than that of Nierenberg et al. (7) and Honea et al. (63). In another study, Persson et al. (8) found that ApoE ε4 was associated with decreased FA in left hippocampus in a location similar to our finding. However, when ApoE status was taken into account in our sample, we found that decreased FA was more strongly related to family history rather than ApoE ε4, and that ApoE ε4 was additive.
The lack of main effect of ApoE ε4 was an interesting finding of the present study, and suggests that previous findings associated with ApoE ε4 may in fact have been due to family history of AD (except in cases where the contribution of family history was controlled). It is well known that the ε4 allele is overrepresented in family history positive cohorts (33), which could make it difficult to interpret the effects of ε4, if there is something embedded in family history that exerts an effect beyond ε4 as suggested by the present results. The mechanism by which family history of AD bestows increased risk for the disease is ambiguous. Unlike ApoE ε4, there is no clear biomarker associated with family history, and presumably the effects of this risk factor are mediated through an as of yet unknown gene or genes. Although it is surprising that there was no main effect of ApoE ε4, family history by itself and in combination with ε4 is known to increase risk for the development of AD, and exert effects that suggest a unique role in AD pathogenesis. Asymptomatic middle age persons with a family history have elevated plasma A-beta (64) and neurocognitive changes consistent with early AD (65) that are independent of ApoE genotype. Once AD has developed, presence of both family history and ApoE ε4 together are associated with an identifiable cognitive phenotype of AD, namely an amnestic presentation (66). Family history of AD is also associated with a deficit in odor identification that is independent of ApoE genotype. Anosmia is known to be a marker of incipient AD, and the single study of this phenomenon in siblings of AD patients revealed a similar pattern to ours, indicating that even though ApoE is not an independent predictor of deficit, it is additive: siblings of AD patients who also carried the ApoE ε4 genotype showed the greatest deficit. Finally, the potential existence of novel genetic markers embodied in family history is supported by neuroimaging studies that have stratified by family history and ApoE genotype. Family history affects the magnitude of MRI perfusion measurements and BOLD signals (14-18) as well as PET measurements of CMRglc (19, 20).
Further complicating the family history effect are data indicating that the effect of family history is multi faceted. Evidence supports a higher rate of maternal inheritance of AD (67), compared to paternal inheritance, and maternal family history is associated with a lower CMRglc in AD susceptible brain regions compared to paternal family history and absence of family history (19, 20). Furthermore, having both maternal and paternal family history confers a higher risk of developing AD compared to having only one parent with the disease, or no parental family history (12). Consistent with these studies, our analysis indicated that maternal family history is associated with significantly lower hippocampal FA compared to absence of family history. Although it was not a significant effect, there was a trend toward lower hippocampal FA in the small group of participants who had two parents diagnosed with AD. Parental family history of AD is an understudied risk factor that clearly needs increased attention, given the results of this study and others (14-20, 65, 68, 69).
Although AD has historically been considered a disease of gray matter, white matter clearly undergoes an alteration in the disease course. Post mortem assessment of white matter in AD has revealed several pathological differences compared to control, such as denudation of the ventricular ependyma, gliosis, and loss of myelinated axons in the deep white matter (70). Biochemical analysis of post mortem white matter in AD patients has shown increased quantities of Aβ40 and Aβ42, less total myelin protein, decreased white matter cholesterol, and increased total fatty acid content (71). Interestingly, the pattern of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates the development of myelin (72), an observation that suggests that break down of white matter is a key event in the development of AD pathology (23). Our findings of altered white matter integrity in a largely middle-aged cohort lend support to the hypothesis that white matter change is an early event in the development of AD pathology.
Several underlying pathologies may be responsible for the diffusion differences observed in the present study. Future studies, preferably those employing biomarkers or additional modalities of imaging, will be needed to evaluate the contributions of inflammation (73-76), amyloid deposition (77, 78), or tau pathology (79), to alterations in white matter. Vascular health is associated with white matter integrity, and future work will also be needed to ascertain the contribution of vascular risk factors to white matter alterations and AD risk. Arterial spin labeling perfusion methods in combination with DTI hold promise for this avenue of work.
Although we found alterations in radial diffusion suggesting differences in myelin, as opposed to axonal changes, further work will be needed to assess whether preclinical AD involves primary myelin degradation (80), or white matter loss that is secondary to neuronal or axonal degradation. Finally, it should be noted that the interpretation of differences in FA and the eigenvalues is limited. Only a handful of studies have been carried out in humans that relate DTI measures to underlying anatomy and pathology in humans using ex vivo brains (81-84). In particular, attempts to differentiate between axonal loss, Wallerian degeneration, and demyelization by analyzing the eigenvalues of the tensor are largely based animal studies not human brains.
A few additional limitations of the present work should be acknowledged. First, our study adopted a voxel-wise approach of analysis. This approach has been widely embraced by many investigators, benefits from automation, provides the capability of quickly analyzing large quantities of brain images, allows for the evaluation of brain regions that may not have been considered a priori, and appears to produce results that largely concur with region of interest (ROI) based methods (27, 85). However, it is limited by spatial normalization methods and has been challenged as a result (86). In the present study, images were inspected visually for errors in registration, and a field mapping procedure was used to overcome susceptibility-related distortion, however, we cannot completely rule out normalization differences as the source of our results. Furthermore—due to large number of statistical comparisons—voxel-wise results are usually evaluated using stringent criteria for significance. This can result in artificially high correlations in highly localized brain regions (87). In the present study, we employed a threshold that was less stringent. Our study population consisted of largely middle-aged adults who are increased risk for AD, rather than a patient population, hence we expected that structural effects would likely be subtle. Still, it should be recognized that employing a threshold that is not corrected for multiple comparison brings with it the risk of a type 1 error and that significant results must consequently be regarded with caution.
The results of our study may also be limited by the resolution of our DTI scan. The resolution of typical DTI (3 mm thick sections in the present study) means that the technique is prone to partial volume effects. Depending on the location of acquired voxels over brain regions where we found significant effects, it is possible that the FA measures could be influenced by the inclusion of cerebrospinal fluid (CSF). This is particularly true for our hippocampal cluster, which was located in the posterior part of this structure. Future studies with smaller voxel sizes are needed to contribute toward more accurate measurements in structures that are proximal to CSF.
T2-wieghted imaging (with or without suppression of CSF) can provide complementary information to DTI. Abnormal white matter appears hyperintense on T2-weighted scans and in healthy adults, these lesions are most likely attributable to ischemia. T2-weighted scans can be used for quantifying total lesion burden, or for comparing regional distribution of lesion burden between groups. We did not examine white matter lesions in this study; thus, we can not conclude whether our results obtained with DTI were due to differences in the distribution of ischemic white matter damage between groups. The mechanisms that underlie DTI measured white matter alterations in preclinical AD require further study.
Additional limitations of the present study include the fact the number of people in the group of participants who had both a mother and a father with AD was small, limiting our analysis of family history load. Although these results are suggestive, they require replication in additional study samples. The sample was on average highly educated, largely Caucasian, from higher socioeconomic strata, and self-selected based on a shared interest in memory research with a specific focus on family history of AD, limiting the study’s generalizability. Additional studies are needed to further evaluate the unique interplay between family history and ApoE ε4 risk factors in the pathogenesis of AD.
Acknowledgements
The authors gratefully acknowledge the assistance of Donald McLaren, Brent Thiel, Britta Jabbar, Michele Fitzgerald, and Erik Kastman. In addition, we would like to acknowledge the kind support of researchers and staff at the Waisman Center, University of Wisconsin–Madison, where MR imaging took place. Finally, we thank our volunteers for their participation in this research.
Funding
This work was supported by the National Institutes of Health [AG021155 to S.C.J., MH62015 to A.L.A., and R01 AG27161 to M.A.S]; a Merit Review Grant from the Department of Veterans Affairs [S.C.J.], and by the facilities and resources at the William S. Middleton Memorial Veterans Hospital.
Footnotes
Disclosure: The authors report no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 2.Lind J, Larsson A, Persson J, et al. Reduced hippocampal volume in non-demented carriers of the apolipoprotein E epsilon4: relation to chronological age and recognition memory. Neurosci Lett. 2006;396:23–27. doi: 10.1016/j.neulet.2005.11.070. [DOI] [PubMed] [Google Scholar]
- 3.Wishart HA, Saykin AJ, McAllister TW, et al. Regional brain atrophy in cognitively intact adults with a single APOE epsilon4 allele. Neurology. 2006;67:1221–1224. doi: 10.1212/01.wnl.0000238079.00472.3a. [DOI] [PubMed] [Google Scholar]
- 4.Bookheimer SY, Strojwas MH, Cohen MS, et al. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reiman EM, Caselli RJ, Yun LS, et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 6.Fleisher AS, Bondi MW, Taylor C, Perry M, Liu TT, Thal LJ, Buxton RB, Dale AM. IC-P-043 Functional perfusion MRI shows decreased hippocampal signal with normal aging in ApoE4 carriers. Alzheimer’s and Dementia. 2006;2:S671–S672. [Google Scholar]
- 7.Nierenberg J, Pomara N, Hoptman MJ, Sidtis JJ, Ardekani BA, Lim KO. Abnormal white matter integrity in healthy apolipoprotein E epsilon4 carriers. Neuroreport. 2005;16:1369–1372. doi: 10.1097/01.wnr.0000174058.49521.16. [DOI] [PubMed] [Google Scholar]
- 8.Persson J, Lind J, Larsson A, et al. Altered brain white matter integrity in healthy carriers of the APOE epsilon4 allele: a risk for AD? Neurology. 2006;66:1029–1033. doi: 10.1212/01.wnl.0000204180.25361.48. [DOI] [PubMed] [Google Scholar]
- 9.Cherbuin N, Leach LS, Christensen H, Anstey KJ. Neuroimaging and APOE genotype: a systematic qualitative review. Dement Geriatr Cogn Disord. 2007;24:348–362. doi: 10.1159/000109150. [DOI] [PubMed] [Google Scholar]
- 10.Mayeux R, Sano M, Chen J, Tatemichi T, Stern Y. Risk of dementia in first-degree relatives of patients with Alzheimer’s disease and related disorders. Arch Neurol. 1991;48:269–273. doi: 10.1001/archneur.1991.00530150037014. [DOI] [PubMed] [Google Scholar]
- 11.Green RC, Cupples LA, Go R, et al. Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA. 2002;287:329–336. doi: 10.1001/jama.287.3.329. [DOI] [PubMed] [Google Scholar]
- 12.Jayadev S, Steinbart EJ, Chi YY, Kukull WA, Schellenberg GD, Bird TD. Conjugal Alzheimer disease: risk in children when both parents have Alzheimer disease. Arch Neurol. 2008;65:373–378. doi: 10.1001/archneurol.2007.61. [DOI] [PubMed] [Google Scholar]
- 13.Martinez M, Campion D, Brice A, et al. Apolipoprotein E epsilon4 allele and familial aggregation of Alzheimer disease. Arch Neurol. 1998;55:810–816. doi: 10.1001/archneur.55.6.810. [DOI] [PubMed] [Google Scholar]
- 14.Johnson SC, Ries ML, Hess TM, et al. Effect of Alzheimer disease risk on brain function during self-appraisal in healthy middle-aged adults. Arch Gen Psychiatry. 2007;64:1163–1171. doi: 10.1001/archpsyc.64.10.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson SC, Schmitz TW, Trivedi MA, et al. The influence of Alzheimer disease family history and apolipoprotein E epsilon4 on mesial temporal lobe activation. J Neurosci. 2006;26:6069–6076. doi: 10.1523/JNEUROSCI.0959-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu G, McLaren DG, Ries ML, et al. The influence of parental history of Alzheimer’s disease and apolipoprotein E {varepsilon}4 on the BOLD signal during recognition memory. Brain. 2008 doi: 10.1093/brain/awn254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleisher AS, Podraza KM, Bangen KJ, et al. Cerebral perfusion and oxygenation differences in Alzheimer’s disease risk. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bassett SS, Yousem DM, Cristinzio C, et al. Familial risk for Alzheimer’s disease alters fMRI activation patterns. Brain. 2006;129:1229–1239. doi: 10.1093/brain/awl089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mosconi L, Mistur R, Switalski R, et al. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology. 2008 doi: 10.1212/01.wnl.0000333247.51383.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosconi L, Brys M, Switalski R, et al. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci U S A. 2007;104:19067–19072. doi: 10.1073/pnas.0705036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basser PJ. Inferring microstructural features and the physiological state of tissues from diffusion-weighted images. NMR Biomed. 1995;8:333–344. doi: 10.1002/nbm.1940080707. [DOI] [PubMed] [Google Scholar]
- 22.Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B. 1996;111:209–219. doi: 10.1006/jmrb.1996.0086. [DOI] [PubMed] [Google Scholar]
- 23.Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging. 2004;25:5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. author reply 49-62. [DOI] [PubMed] [Google Scholar]
- 24.Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME, Bowers WJ. Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia. 2009;57:54–65. doi: 10.1002/glia.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salat DH, Tuch DS, van der Kouwe AJ, et al. White matter pathology isolates the hippocampal formation in Alzheimer’s disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duan JH, Wang HQ, Xu J, et al. White matter damage of patients with Alzheimer’s disease correlated with the decreased cognitive function. Surg Radiol Anat. 2006;28:150–156. doi: 10.1007/s00276-006-0111-2. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Schuff N, Du AT, et al. White matter damage in frontotemporal dementia and Alzheimer’s disease measured by diffusion MRI. Brain. 2009 doi: 10.1093/brain/awp071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Medina D, deToledo-Morrell L, Urresta F, et al. White matter changes in mild cognitive impairment and AD: A diffusion tensor imaging study. Neurobiology of Aging. 2006;27:663–672. doi: 10.1016/j.neurobiolaging.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 29.Fellgiebel A, Wille P, Muller MJ, et al. Ultrastructural hippocampal and white matter alterations in mild cognitive impairment: a diffusion tensor imaging study. Dement Geriatr Cogn Disord. 2004;18:101–108. doi: 10.1159/000077817. [DOI] [PubMed] [Google Scholar]
- 30.Rose SE, Janke AL, Chalk JB. Gray and white matter changes in Alzheimer’s disease: a diffusion tensor imaging study. J Magn Reson Imaging. 2008;27:20–26. doi: 10.1002/jmri.21231. [DOI] [PubMed] [Google Scholar]
- 31.Rose SE, McMahon KL, Janke AL, et al. Diffusion indices on magnetic resonance imaging and neuropsychological performance in amnestic mild cognitive impairment 10.1136/jnnp.2005.074336. J Neurol Neurosurg Psychiatry. 2006;77:1122–1128. doi: 10.1136/jnnp.2005.074336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fellgiebel A, Muller MJ, Wille P, et al. Color-coded diffusion-tensor-imaging of posterior cingulate fiber tracts in mild cognitive impairment. Neurobiol Aging. 2005;26:1193–1198. doi: 10.1016/j.neurobiolaging.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Sager MA, Hermann B, La Rue A. Middle-aged children of persons with Alzheimer’s disease: APOE genotypes and cognitive function in the Wisconsin Registry for Alzheimer’s Prevention. J Geriatr Psychiatry Neurol. 2005;18:245–249. doi: 10.1177/0891988705281882. [DOI] [PubMed] [Google Scholar]
- 34.Morris JC, Heyman A, Mohs RC, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology. 1989;39:1159–1165. doi: 10.1212/wnl.39.9.1159. [DOI] [PubMed] [Google Scholar]
- 35.Folstein MF, Folstein SE, McHugh PR. “Mini Mental State”: a practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatry Research. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 36.Wechsler D. Wechsler Adult Intelligence Scale-Third Edition. Psych Corp.; New York: 1997. [Google Scholar]
- 37.Spreen O, Strauss E. A Compendium of Neuropsychological Tests. Oxford; New York: 1998. [Google Scholar]
- 38.Reitan RM, Wolfson D. The Halstead-Reitan Neuropsychological Test Battery: Theory and clinical interpretation. 2 ed. Neuropsychology Press; Tucson: 1993. [Google Scholar]
- 39.Benton AL. Neuropsychological assessment. Annual Review of Psychology. 1994;45:1–23. doi: 10.1146/annurev.ps.45.020194.000245. [DOI] [PubMed] [Google Scholar]
- 40.Bendlin BB, Ries ML, Lazar M, et al. Longitudinal changes in patients with traumatic brain injury assessed with diffusion-tensor and volumetric imaging. Neuroimage. 2008;42:503–514. doi: 10.1016/j.neuroimage.2008.04.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23(Suppl 1):S208–219. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 42.Song SK, Sun SW, Ju WK, Lin SJ, Cross AH, Neufeld AH. Diffusion tensor imaging detects and differentiates axon and myelin degeneration in mouse optic nerve after retinal ischemia. Neuroimage. 2003;20:1714–1722. doi: 10.1016/j.neuroimage.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 43.Pierpaoli C, Barnett A, Pajevic S, et al. Water diffusion changes in Wallerian degeneration and their dependence on white matter architecture. Neuroimage. 2001;13:1174–1185. doi: 10.1006/nimg.2001.0765. [DOI] [PubMed] [Google Scholar]
- 44.Song SK, Sun SW, Ramsbottom MJ, Chang C, Russell J, Cross AH. Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. Neuroimage. 2002;17:1429–1436. doi: 10.1006/nimg.2002.1267. [DOI] [PubMed] [Google Scholar]
- 45.Song SK, Yoshino J, Le TQ, et al. Demyelination increases radial diffusivity in corpus callosum of mouse brain. Neuroimage. 2005;26:132–140. doi: 10.1016/j.neuroimage.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 46.Naggara O, Oppenheim C, Rieu D, et al. Diffusion tensor imaging in early Alzheimer’s disease. Psychiatry Res. 2006;146:243–249. doi: 10.1016/j.pscychresns.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Schuff N, Jahng GH, et al. Diffusion tensor imaging of cingulum fibers in mild cognitive impairment and Alzheimer disease. Neurology. 2007;68:13–19. doi: 10.1212/01.wnl.0000250326.77323.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kavcic V, Ni H, Zhu T, Zhong J, Duffy CJ. White matter integrity linked to functional impairments in aging and early Alzheimer’s disease. Alzheimers Dement. 2008;4:381–389. doi: 10.1016/j.jalz.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugihara S, Kinoshita T, Matsusue E, Fujii S, Ogawa T. Usefulness of diffusion tensor imaging of white matter in Alzheimer disease and vascular dementia. Acta Radiol. 2004;45:658–663. doi: 10.1080/02841850410008388. [DOI] [PubMed] [Google Scholar]
- 50.Choo IH, Lee DY, Oh JS, et al. Posterior cingulate cortex atrophy and regional cingulum disruption in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 51.Rose SE, Chen F, Chalk JB, et al. Loss of connectivity in Alzheimer’s disease: an evaluation of white matter tract integrity with colour coded MR diffusion tensor imaging. J Neurol Neurosurg Psychiatry. 2000;69:528–530. doi: 10.1136/jnnp.69.4.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kantarci K, Jack CR, Jr., Xu YC, et al. Mild cognitive impairment and Alzheimer disease: regional diffusivity of water. Radiology. 2001;219:101–107. doi: 10.1148/radiology.219.1.r01ap14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ronen I, Kim KH, Garwood M, Ugurbil K, Kim DS. Conventional DTI vs. slow and fast diffusion tensors in cat visual cortex. Magn Reson Med. 2003;49:785–790. doi: 10.1002/mrm.10431. [DOI] [PubMed] [Google Scholar]
- 54.Shepherd TM, Ozarslan E, Yachnis AT, King MA, Blackband SJ. Diffusion tensor microscopy indicates the cytoarchitectural basis for diffusion anisotropy in the human hippocampus. AJNR Am J Neuroradiol. 2007;28:958–964. [PMC free article] [PubMed] [Google Scholar]
- 55.Cho H, Yang DW, Shon YM, et al. Abnormal integrity of corticocortical tracts in mild cognitive impairment: a diffusion tensor imaging study. J Korean Med Sci. 2008;23:477–483. doi: 10.3346/jkms.2008.23.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim H, Piao Z, Liu P, Bingaman W, Diehl B. Secondary white matter degeneration of the corpus callosum in patients with intractable temporal lobe epilepsy: a diffusion tensor imaging study. Epilepsy Res. 2008;81:136–142. doi: 10.1016/j.eplepsyres.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 57.Taoka T, Iwasaki S, Sakamoto M, et al. Diffusion anisotropy and diffusivity of white matter tracts within the temporal stem in Alzheimer disease: evaluation of the “tract of interest” by diffusion tensor tractography. AJNR Am J Neuroradiol. 2006;27:1040–1045. [PMC free article] [PubMed] [Google Scholar]
- 58.Fujie S, Namiki C, Nishi H, et al. The role of the uncinate fasciculus in memory and emotional recognition in amnestic mild cognitive impairment. Dement Geriatr Cogn Disord. 2008;26:432–439. doi: 10.1159/000165381. [DOI] [PubMed] [Google Scholar]
- 59.Chua TC, Wen W, Chen X, et al. Diffusion tensor imaging of the posterior cingulate is a useful biomarker of mild cognitive impairment. Am J Geriatr Psychiatry. 2009;17:602–613. doi: 10.1097/JGP.0b013e3181a76e0b. [DOI] [PubMed] [Google Scholar]
- 60.Wang L, Goldstein FC, Veledar E, et al. Alterations in cortical thickness and white matter integrity in mild cognitive impairment measured by whole-brain cortical thickness mapping and diffusion tensor imaging. AJNR Am J Neuroradiol. 2009;30:893–899. doi: 10.3174/ajnr.A1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bartzokis G, Lu PH, Geschwind DH, Edwards N, Mintz J, Cummings JL. Apolipoprotein E genotype and age-related myelin breakdown in healthy individuals: implications for cognitive decline and dementia. Arch Gen Psychiatry. 2006;63:63–72. doi: 10.1001/archpsyc.63.1.63. [DOI] [PubMed] [Google Scholar]
- 62.Bartzokis G, Lu PH, Geschwind DH, et al. Apolipoprotein E affects both myelin breakdown and cognition: implications for age-related trajectories of decline into dementia. Biol Psychiatry. 2007;62:1380–1387. doi: 10.1016/j.biopsych.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 63.Honea RA, Vidoni E, Harsha A, Burns JM. Impact of APOE on the Healthy Aging Brain: A Voxel-Based MRI and DTI Study. J Alzheimers Dis. 2009 doi: 10.3233/JAD-2009-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ertekin-Taner N, Younkin LH, Yager DM, et al. Plasma amyloid beta protein is elevated in late-onset Alzheimer disease families. Neurology. 2008;70:596–606. doi: 10.1212/01.WNL.0000278386.00035.21. [DOI] [PubMed] [Google Scholar]
- 65.La Rue A, Hermann B, Jones JE, Johnson S, Asthana S, Sager MA. Effect of parental family history of Alzheimer’s disease on serial position profiles. Alzheimers Dement. 2008;4:285–290. doi: 10.1016/j.jalz.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Snowden JS, Stopford CL, Julien CL, et al. Cognitive phenotypes in Alzheimer’s disease and genetic risk. Cortex. 2007;43:835–845. doi: 10.1016/s0010-9452(08)70683-x. [DOI] [PubMed] [Google Scholar]
- 67.Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC. Increased risk of dementia in mothers of Alzheimer’s disease cases: evidence for maternal inheritance. Neurology. 1996;47:254–256. doi: 10.1212/wnl.47.1.254. [DOI] [PubMed] [Google Scholar]
- 68.La Rue A, R OH, Matsuyama SS, Jarvik LF. Cognitive changes in young-old adults: effect of family history of dementia. J Clin Exp Neuropsychol. 1995;17:65–70. doi: 10.1080/13803399508406582. [DOI] [PubMed] [Google Scholar]
- 69.Hom J, Turner MB, Risser R, Bonte FJ, Tintner R. Cognitive deficits in asymptomatic first-degree relatives of Alzheimer’s disease patients. J Clin Exp Neuropsychol. 1994;16:568–576. doi: 10.1080/01688639408402668. [DOI] [PubMed] [Google Scholar]
- 70.Scheltens P, Barkhof F, Leys D, Wolters EC, Ravid R, Kamphorst W. Histopathologic correlates of white matter changes on MRI in Alzheimer’s disease and normal aging. Neurology. 1995;45:883–888. doi: 10.1212/wnl.45.5.883. [DOI] [PubMed] [Google Scholar]
- 71.Roher AE, Weiss N, Kokjohn TA, et al. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry. 2002;41:11080–11090. doi: 10.1021/bi026173d. [DOI] [PubMed] [Google Scholar]
- 72.Braak H, Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol. 1996;92:197–201. doi: 10.1007/s004010050508. [DOI] [PubMed] [Google Scholar]
- 73.Rogers J, Webster S, Lue LF, et al. Inflammation and Alzheimer’s disease pathogenesis. Neurobiol Aging. 1996;17:681–686. doi: 10.1016/0197-4580(96)00115-7. [DOI] [PubMed] [Google Scholar]
- 74.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McGeer EG, McGeer PL. Inflammatory processes in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:741–749. doi: 10.1016/S0278-5846(03)00124-6. [DOI] [PubMed] [Google Scholar]
- 76.McGeer PL, McGeer EG. Local neuroinflammation and the progression of Alzheimer’s disease. J Neurovirol. 2002;8:529–538. doi: 10.1080/13550280290100969. [DOI] [PubMed] [Google Scholar]
- 77.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 78.Roher AE, Kuo YM, Esh C, et al. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Mol Med. 2003;9:112–122. [PMC free article] [PubMed] [Google Scholar]
- 79.Stenset V, Bjornerud A, Fjell AM, et al. Cingulum fiber diffusivity and CSF T-tau in patients with subjective and mild cognitive impairment. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 80.Bartzokis G, Sultzer D, Lu PH, Nuechterlein KH, Mintz J, Cummings JL. Heterogeneous age-related breakdown of white matter structural integrity: implications for cortical “disconnection” in aging and Alzheimer’s disease. Neurobiol Aging. 2004;25:843–851. doi: 10.1016/j.neurobiolaging.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 81.Schmierer K, Wheeler-Kingshott CA, Boulby PA, et al. Diffusion tensor imaging of post mortem multiple sclerosis brain. Neuroimage. 2007;35:467–477. doi: 10.1016/j.neuroimage.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gouw AA, Seewann A, Vrenken H, et al. Heterogeneity of white matter hyperintensities in Alzheimer’s disease: post-mortem quantitative MRI and neuropathology. Brain. 2008;131:3286–3298. doi: 10.1093/brain/awn265. [DOI] [PubMed] [Google Scholar]
- 83.Larsson EM, Englund E, Sjobeck M, Latt J, Brockstedt S. MRI with diffusion tensor imaging post-mortem at 3.0 T in a patient with frontotemporal dementia. Dement Geriatr Cogn Disord. 2004;17:316–319. doi: 10.1159/000077162. [DOI] [PubMed] [Google Scholar]
- 84.Englund E, Sjobeck M, Brockstedt S, Latt J, Larsson EM. Diffusion tensor MRI post mortem demonstrated cerebral white matter pathology. J Neurol. 2004;251:350–352. doi: 10.1007/s00415-004-0318-2. [DOI] [PubMed] [Google Scholar]
- 85.Snook L, Plewes C, Beaulieu C. Voxel based versus region of interest analysis in diffusion tensor imaging of neurodevelopment. Neuroimage. 2007;34:243–252. doi: 10.1016/j.neuroimage.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 86.Chao TC, Chou MC, Yang P, Chung HW, Wu MT. Effects of interpolation methods in spatial normalization of diffusion tensor imaging data on group comparison of fractional anisotropy. Magn Reson Imaging. 2009;27:681–690. doi: 10.1016/j.mri.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 87.Yarkoni T. Big correlations in little studies: Inflated fMRI correlations reflect low statistical power. Perspectives on Psychological Science. 2009 doi: 10.1111/j.1745-6924.2009.01127.x. Commentary on Vul et al. In press. [DOI] [PubMed] [Google Scholar]