

Abstract
No clinically effective chemoprevention for lung cancer has been found. Angiogenesis is an early feature of both adenocarcinoma and squamous cell lung cancer. We investigated the effects of VEGFR-2 inhibition on lung carcinogenesis in a murine model of adenocarcinoma.
The VEGFR-2 tyrosine kinase inhibitor, vandetanib, was administered to FVB/N mice in chow for 7 days at varying doses in order to demonstrate pharmacologic activity by inhibition of VEGF mediated VEFGR-2 and ERK phosphorylation. Plasma levels corroborated adequate dosage. For chemoprevention experiments, mice were injected i.p. with 1 mg/gm urethane, a carcinogen found in tobacco smoke. Chow containing vandetanib, 75 mg/kg/d, or control chow was given to mice, starting 7 days after urethane administration. Sixteen weeks after urethane injection, mice were sacrificed, tumors enumerated and measured. Vandetanib resulted in reductions in tumor multiplicity (6.5 +/− 0.86 vs 1.0 +/− 0.30, p = 0.001) and average tumor volume (0.85 +/− 0.10 mm3 vs. 0.15 +/− 0.09 mm3, p = 0.001), but not incidence (71% vs. 100%, p = ns), compared to control. As vandetanib has other activities besides VEGFR-2 tyrosine kinase inhibition, we administered the anti-VEGFR-2 monoclonal antibody, DC101, for weeks 11–15 of a urethane carcinogenesis protocol with an arrest in tumor volume increase, but no change in multiplicity or incidence. Further investigation of the chemopreventive effect of vandetanib and other VEGF signaling inhibitors is needed.
Introduction
Lung cancer is the leading cause of cancer death in the world(1). Tobacco smoking is the major cause of lung cancer and smoking cessation is an effective means to decrease lung cancer risk(2). However, significant risk of lung cancer persists after smoking cessation, such that in the United States, lung cancer is now diagnosed in approximately equal numbers of current and ex-smokers(3). Chemoprevention of lung cancer has the potential to significantly reduce morbidity and mortality. Unfortunately, no effective chemoprevention for lung cancer in humans has been found.
Angiogenesis has long been recognized as necessary for tumor growth(4). After reaching a diameter of 1–2 mm, tumors are dependent on recruitment of new vessels and remain in a dormant state until the “angiogenic switch” occurs and new vessels are recruited. The molecular mechanisms of the angiogenic switch have been partially defined and include activating ras mutations as well as inactivation of p53, PTEN and Smad4(5). The hypoxia inducible factors, HIF-1α and HIF-2α, induce expression of a variety of angiogenic factors, including VEGF, FGF, (ELR+) CXC chemokines (IL-8, CXCL12 and others), PDGF, endothelins, angiopoetins, and others(6). Conventionally thought of as critical when a tumor reaches 1–2 mm in diameter, angiogenesis is not commonly considered a feature of premalignancy. However, in the central airways a premalignant lesion in which capillaries invade the overlying dysplastic endobronchial epithelium has been described and termed angiogenic squamous dysplasia (Figure 1)(7). This lesion occurs primarily in current or ex-smokers with endobronchial dysplasia and contains elevated levels of mRNAs for both VEGF-A and VEGFR-2(8). The elevated levels of VEGF-A occur at multiple sites in individuals with angiogenic squamous dysplasia, suggesting a field effect. Angiogenesis also occurs in the evolution of at least some peripheral adenocarcinomas of the lung, which are thought to progress from atypical alveolar hyperplasia to bronchioloalveolar carcinoma to papillary adenocarcinoma and solid adenocarcinoma (Figure 2). In papillary adenocarcinoma, malignant epithelial cells grow on an underlying capillary scaffold. Mouse lung adenomas are histologically similar to the papillary stage of human adenocarcinoma, with more advanced lesions displaying solid features (Figure 3)
Figure 1.
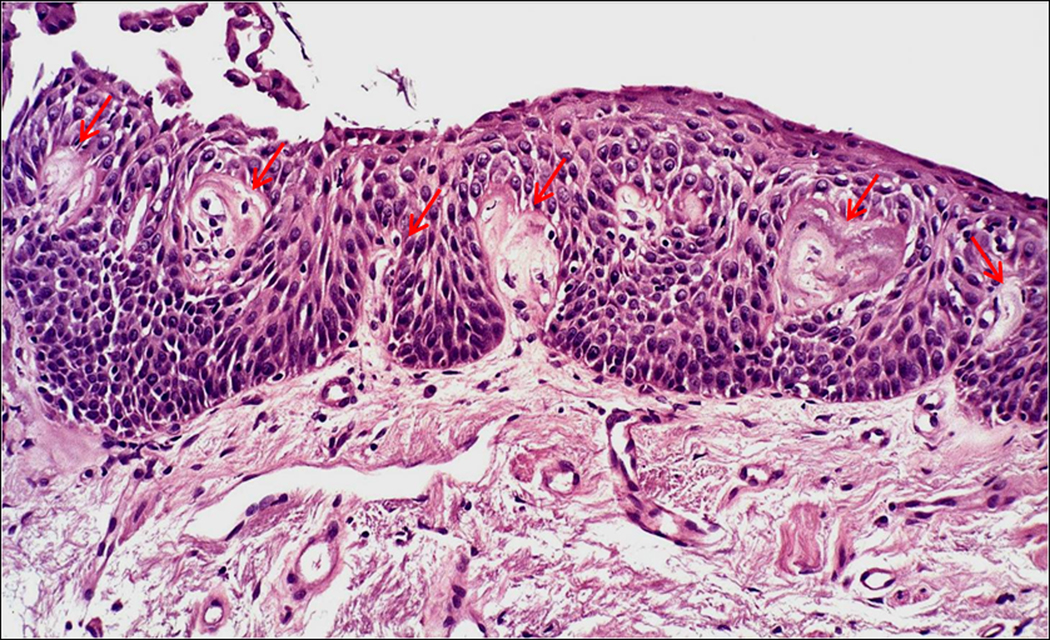
Angiogenic squamous dysplasia in a human endobronchial biopsy. Note the capillary loops closely associated with the dysplastic squamous epithelium, designated by arrows.
Figure 2.
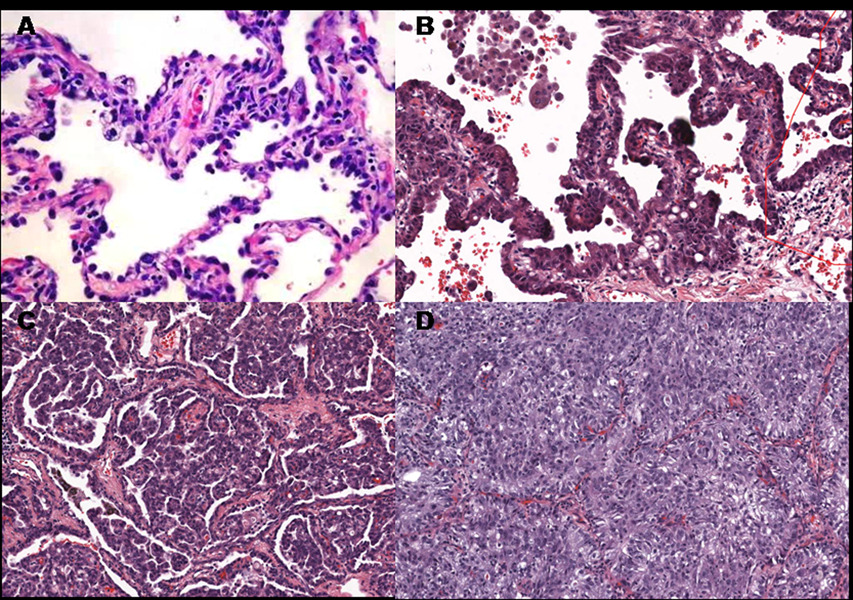
Stages of human lung adenocarcinoma progression: A.) atypical alveolar hyperplasia; B.) bronchioloalveolar carcinoma; C.) papillary adenocarcinoma and D.) solid adenocarcinoma. The last 3 images were taken from different areas of the same tumor of a single patient. Note that the neoplastic cells in bronchioloalveolar and papillary carcinomas are arrayed on the surface of cores of mesenchymal cells containing central capillaries. It is apparent that in papillary adenocarcinoma, these structures have proliferated and fill alveolar spaces.
Figure 3.
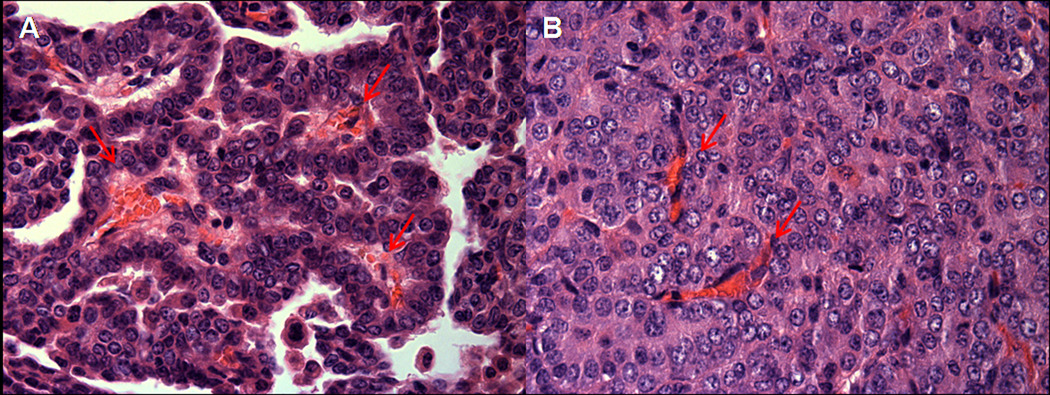
A. Early mouse lung adenoma with papillary structures showing prominent central vascular core, designated by arrows. B. Advanced mouse lung adenoma with solid tumor growth pattern and disorganized vascular network, designated by arrows.
Several natural substances under investigation for cancer chemoprevention, including silibinin, resveratrol and green tea extract, have antiangiogenic properties(9–11). However, few published studies have examined the chemopreventive properties of targeted antiangiogenic agents. We hypothesized that inhibition of angiogenesis might be an effective chemoprevention strategy for lung cancer in a murine model that has features of bronchioloalveolar carcinoma and adenocarcinoma. Chemical and genetic murine models of bronchioloalveolar carcinoma and adenocarcinoma have been investigated for many years and have many histologic, mutational and gene expression features in common with human adenocarcinoma(12, 13). Murine models of squamous cell carcinoma are less well developed and have not yet been widely utilized for preclinical testing of chemopreventive agents(14, 15).
Materials and Methods
Human Tissues
Human lung tumor tissues were harvested under protocols approved by the Colorado Multiple Institutional Review Board.
Mouse maintenance
Female FVB/N mice were purchased (Harlan, Indianapolis) at ages 6–8 weeks and maintained in conventional caging in a controlled environment (12 h light-dark cycle, food and water ad libitum) in the Denver Veterans Affairs Medical Center Animal Care Facility. All animal procedures were approved by the Denver Veteran Affairs Medical Center Institutional Animal Care and Use Committee.
Demonstration of Pharmacologic Activity of Vandetanib
Mice were fed powdered AIN-76A chow to which varying concentrations of vandetanib (a kind gift of Astra Zeneca, Macclesfield, UK) was added (Bio-Serv, Frenchtown, NJ). Dosage was determined by observation of the chow consumption by mice and calculation to achieve final doses of vandetanib of 50, 100 and 150 mg/kg (BW)/d. After 7 days on this diet, mice were injected i.p. with 20 ng VEGF (Sigma, St. Louis, MO) dissolved in PBS, then sacrificed 20 minutes later by i.p. injection of a mixture of pentobarbital (10mg) and heparin (100unit)/mouse in a 200ul volume of saline. The aorta and inferior vena cava were then identified and transected, blood aspirated from the abdominal cavity and lungs removed, dissected from the major bronchi, and snap-frozen in liquid nitrogen. Lung homogenates were prepared in buffer containing 50 mM Tris HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 10% glycerol, 1% Nonidet P-40, 1 mM dithiothreitol, 10 mM β-glycerophosphate, 10 mM NaF, 1 mM sodium orthovanadate, 10 µg/ml leupeptin, 10 µg/ml aprotinin, 10 µg/ml pepstatin A, and 1 mM phenylmethylsulfonyl fluoride. The homogenates were centrifuged for 10 min at 10,000 rpm and the supernatant collected. Protein concentration was determined using the BCA protein assay. Proteins were separated on SDS-PAGE, transferred to nitrocellulose membranes (GE HealthCare, Buckinghamshire, UK). Membranes were blocked in PBS containing 0.1% Tween 20 and 1% BSA for 1 h. Membranes were incubated with primary antibodies overnight at 4 °C, and with secondary antibodies for 1 h at room temperature. Primary antibodies to VEGFR-2, p-VEGFR-2, Erk and p-Erk were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Vandetanib levels were measured in plasma taken at the time of sacrifice by the University of Colorado Cancer Center Pharmacology Core(16).
Urethane carcinogenesis and chemoprevention
Mice were injected i.p. with 1 mg/gm urethane dissolved in saline. Serial body weights were followed during the experiment. One week after urethane injection, mice were placed on vandetanib or control AIN-76A chow, 75 mg/kg (BW)/d and maintained until the end of experiment, 16 weeks after urethane injection. Mice were sacrificed by i.p. injection of a mixture of pentobarbital (10mg) and heparin (100unit)/mouse in a 200ul volume of saline. The abdominal cavity was opened and the aorta and inferior vena cava identified and transected. Blood was harvested by aspiration from the abdominal cavity, then lungs removed. Tumors were identified under a dissecting microscope, dissected free from the lung tissue and measured using a digital calipers. The volume of tumors was calculated based on the formula v = 4/3πr3. Tumors were either snap-frozen in liquid nitrogen or fixed in 10% buffered formalin and paraffin embedded. In a subsequent experiment to determine the effects of intermittent vandetanib treatment, mice were treated either for the first or last 10 weeks of a 20 week carcinogenesis experiment. Mice on control chow were sacrificed at 10 or 20 week time points.
Chemoprevention using anti-VEGFR-2 monoclonal antibody
The IgG1 rat anti-mouse VEGFR-2 monoclonal antibody DC101 was obtained from BioXcell (West Lebanon, NH). Eleven weeks after i.p. urethane (1 mg/gm) injection, mice were administered 700 ug/mouse DC101 or monoclonal rat IgG1 in 200ul PBS, or PBS alone, i.p. three times weekly for 4 weeks, then sacrificed, tumors enumerated and measured.
Immunohistochemistry
Tumors and lungs were fixed in 10% buffered formalin overnight, embedded in paraffin and cut in serial 4 µm sections which were mounted on glass slides. All sections were pretreated with 10 mM Tris EDTA, pH 9.0 for 20’ at 95° C. Sections were incubated with primary antibodies overnight at 4° C in a humidified chamber on a shaker. The sections were subsequently washed 3x in TBS and incubated with secondary antibodies, either biotinylated anti rat IgG or anti rabbit IgG (both at 1:200 dilution, Vector Laboratories, Burlingame, CA) for one hour. Tissues were then washed 3x in TBS and incubated with HRP conjugated Streptavidin (Vector Laboratories) for 30’. Tissues were washed 3x in TBS and developed in liquid DAB (Biogenix, San Ramon, CA) for 5–10’ until a brown color appeared. Tissues were then washed 3x in water, counterstained with modified Harris’ hematoxylin (Fisher Diagnostics, Middletown, VA) for 30’, then washed 3x in water, dipped in acid ammonia, washed 3x in water, dehydrated in serial ethanol baths, cleared in CitriSolv (Fisher Diagnostics) and mounted on slides. Primary antibodies included: 1:50 rat anti mouse Ki67 (Dako A/S, Glostrup, Denmark); 1:100 rat anti mouse CD34 (Santa Cruz Biotechnology); 1:100 rabbit anti mouse pEGFR (Y1086); 1:100 rabbit anti mouse pVEGFR2 (Cell Signalling, Beverly, MA). Controls included no primary antibody or same species IgG.
Statistical tests
Data were analyzed using two-sided, non-parametric t-tests.
Results
Dose determination
Mice tolerated vandetanib well and gained weight as did those fed control chow. Injection of 20 ng VEGF i.p. resulted in increased phosphorylation of VEGFR-2 and Erk in lungs of controls. Following 7 days of treatment, vandetanib at a dose of 100 or 150 mg/kg/d was effective in blocking VEGF induced VEGFR-2 phosphorylation. Vandetanib at 50, 100 and 150 mg/kg/d blocked Erk phosphorylation in response to VEGF injection. After 7 days of treatment, plasma levels of vandetanib at doses of 50, 100 and 150 mg/kg/d were 2.1, 4.5 and 5.7 µM respectively. For the remainder of the study, vandetanib was administered in solid chow at a dose of 75 mg/kg/d.
Chemoprevention by vandetanib
In a 16 week carcinogenesis protocol, vandetanib chow (n = 7) had a major chemopreventive effect compared to control chow (n = 9). Mean tumor multiplicity and volume were reduced from 6.5 to 1.0 tumors/mouse (p = 0.0001) and 0.85 +/− 0.10 mm3 vs. 0.15 +/− 0.09 mm3, (p = 0.001) respectively (Figure 4). Tumor incidence was 100% with control chow and 71% with vandetanib (p = ns).
Figure 4.

Tumor multiplicity and volume of mice treated with vandetanib (n = 7) or control (n = 9) chow from weeks 1–16 of a 16 week urethane carcinogenesis protocol.
Vandetanib is effective administered both early and late
To compare early and late chemoprevention with vandetanib, we exposed mice to vandetanib containing chow for either the first (n = 8) or last (n = 9) 10 weeks of a 20 week urethane carcinogenesis protocol. Mice fed control chow were sacrificed either at 10 (n = 8) or 20 (n = 10) weeks after urethane administration. Vandetanib given for either the first or last half of the carcinogenesis protocol was effective in reducing tumor multiplicity and volume in comparison to mice after 20 weeks of urethane carcinogenesis protocol (Figure 5). Of interest, vandetanib given for either the first or last 10 weeks of the 20 week carcinogenesis protocol reduced tumor multiplicities below that of mice fed control chow and sacrificed at 10 weeks after urethane, suggesting a therapeutic effect that persisted after vandetanib treatment was stopped.
Figure 5.

Comparison of early and late chemoprevention with vandetanib. Mice were injected with urethane (week 0) and treated from weeks 1–10 (n = 8) or 10–20 (n = 9) with vandetanib containing chow, with control chow during the untreated period. Controls include mice sacrificed at week 10 (n = 8) or 20 (n = 10). Both early and late treatment are effective in reducing tumor multiplicity and volume compared to 20 week control mice. A reduction in tumor mulitiplicity below that of mice sacrificed at week 10 is seen for both 1–10 week (p = 0.009) and 10–20 week (p = 0.008) treatment periods.
Antibody inhibition of VEGFR-2 for chemoprevention
Vandetanib is not a specific inhibitior of VEGFR-2 tyrosine kinase inhibition. It also has significant inhibitory effects against other receptor tyrosine kinases including EGFR, VEGFR-3 and RET. The DC101 rat monoclonal antibody is a blocking antibody specific to the murine VEGFR-2(17). Administration of DC101 is limited by the development of mouse anti-rat antibodies, so we administered DC101 between weeks 11–15 after urethane injection, a time period when the lung tumors begin to grow rapidly. Mice tolerated DC101 or an isotype control IgG1 rat monoclonal well, with no differences in body weights during antibody or PBS administration. DC101 was effective in reducing tumor volume, but not multiplicity or incidence compared to PBS injection, in this modified protocol (Figure 6). DC 101 did prevent any tumor volume increase compared to controls sacrificed at 11 weeks.
Figure 6.

Tumor volume and multiplicity for mice treated with DC101 (n = 10), compared with IgG1 (n = 10) or PBS (n = 4) from weeks 11–15 of a 15 week urethane carcinogenesis protocol. A significant reduction in tumor volume is seen, but there is no reduction in tumor number. Note that tumor volume of control mice (n = 4) sacrificed at 11 weeks is similar to that of the group treated with DC101 from weeks 11–15, suggesting that this treatment arrests tumor growth.
Effects of treatment on tumors developing during chemoprevention
We chose to examine characteristics of tumors that occurred in spite of chemoprevention by vandetanib (weeks 1–16) or DC101 (weeks 11–15) using immunohistochemistry, due to the significant limitation of tissue availability for immunoblotting resulting from the reduction in tumor number and volume by vandetanib. Histologically, tumors occurring in control, vandetanib and DC101 treated mice appeared similar. Capillaries within the tumor were reduced by both vandetanib and DC101 (Figure 7). Immunostaining for phosphorylated EGFR and VEGFR was reduced below control by vandetanib, but not DC101 (Figure 7). Expression of both total EGFR and VEGFR did not appear to be markedly altered by treatment with either vandetanib or DC101 (data not shown). Ki-67 index was reduced by approximately 50% in both the vandetanib (p = ns) and DC101 (p = 0.003) treated mouse tumors compared to control; in the case of vandetanib treated tumors, the number of tumors evaluable was limited by lack of tissue for Ki-67 analysis.
Figure 7.
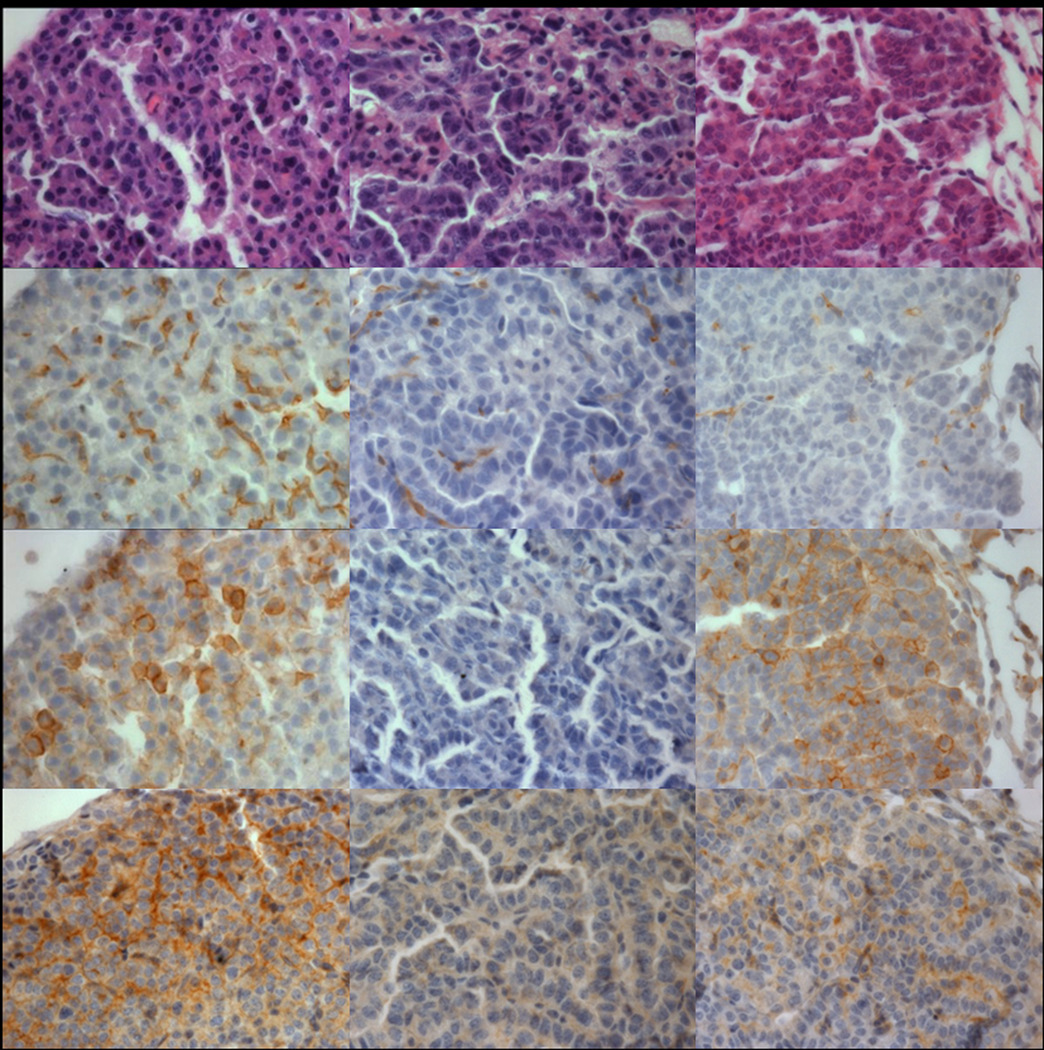
Comparison of tumors from mice treated with control or vandetanib chow (weeks 1–15) or DC101 injection (weeks 11–15). There are no significant changes in tumor histology. Tumor microvessels demonstrated by CD34 immunostaining are reduced in both vadetanib and DC101 treated mice, more completely in the latter. pEGFR immunostaining is abolished in vandetanib treated tumors, but not control or DC101 treated. pVEGFR2 immunostaining is reduced in both vandetanib and DC101 treated tumors, more completely in the former.
Discussion
We believe that this is the first report to determine the effect of a targeted angiogenesis inhibitor for lung cancer chemoprevention. Vandetanib has previously been demonstrated to be effective in reducing intestinal adenomas in the Apc null mouse model(18). Vandetanib, given either continuously or intermittently, was highly effective in reducing lung tumor multiplicity and volume, but did not have a statistically significant effect on tumor incidence. This reduction in multiplicity and volume is significantly greater than our group has previously observed with SPC promoter driven prostacyclin synthase overexpression, iloprost chow, or gefitinib injection(19–21). The efficacy of vandetanib given either early or late in a 20 week urethane carcinogenesis model demonstrates that it does not have to be given continuously for chemopreventive efficacy. It is possible that further limited dosing strategies may limit toxicities, which include rash, diarrhea and QT prolongation, in human studies while maintaining chemopreventive efficacy.
Vandetanib is not completely specific for the VEGFR-2 and inhibits other receptor tyrosine kinases, including the EGFR TK, VEGFR-3 TK, RET and PDGF TK. We used a second agent, the anti-mouse VEGFR-2 monoclonal DC101 and demonstrated significant effects on tumor volume when given for weeks 11–15 of a 15 week urethane carcinogenesis protocol, but no effect on tumor multiplicity, compared to control IgG1 or PBS injection from week 11–15. Moreover, DC101 administration prevented tumor volume increase compared to control mice sacrificed at 11 weeks. It is possible that earlier treatment with DC101, at a time when tumors were of a small enough volume as to be undetectable, might have reduced multiplicity as well. The development of mouse anti-rat antibodies limit the administration of DC101 to 4 weeks or less, making it difficult to directly compare the antibody and vandetanib experiments. Our previous results with the EGFR TKI gefitinib in a similar urethane model demonstrated no chemopreventive activity in wildtype FVB/N mice and only a possible minor additive effect in prostacyclin synthase overexpressor mice (Lung Cancer, in press). Therefore, we do not believe that the chemopreventive effects of vandetanib are solely due to EGFR inhibition, although this could be a contributing factor. Furthermore, of the 4 other groups who have previously published chemoprevention experiments with EGFR TKIs, only one has reported a large effect in a ras driven model, such as that produced by urethane, a carcinogen found in tobacco smoke(22–25). Further investigation into the mechanisms by which vandetanib produces chemoprevention will require more specific, molecular or genetic, strategies, which are beyond the scope of this report.
Activating EGFR mutations have been described as occurring frequently in histologically normal respiratory epithelium adjacent to human lung adenocarcinomas containing the same mutation(26). This supports EGFR mutation as an early premalignant event in some adenocarcinomas. A transgenic mouse model based on SPC promoter driven mutant EGFR was amenable to highly effective chemoprevention by gefitinib(25, 27). However, most groups studying the chemopreventive effect of gefitinib in K-ras driven models have found a small or no therapeutic effect(23, 24). In contrast, vandetanib is highly effective in the K-ras driven urethane model. The dual VEGFR-2/EGFR inhibitory effects of vandetanib might make it or a similar agent effective in chemoprevention of lung tumors driven by either mutant EGFR or K-ras.
Experiments characterizing the effects of vandetanib or DC101 on tumors were limited to tumors that developed during treatment. Comparability to tumors in control animals is difficult due to differences in volume and the possibility of acquired resistance to the agents. Both vandetanib and DC101 appeared to reduce capillary density and Ki-67 index. Vandetanib inhibited phosphorylation of both EGFR and VEGFR-2, while DC101 only affected the latter.
Chemoprevention in humans requires the identification of high risk populations and the availability of effective agents with minimal side effects. Populations at high (up to 2% incidence/year) risk for lung cancer can be readily identified using simple clinical features, including previous history of a tobacco induced cancer, tobacco exposure history, coexistent chronic obstructive lung disease and family history(28–30). Emerging genetic polymorphisms and susceptibility biomarkers may further improve the ability to identify high risk subjects. However, effective agents with minimal side effects have not been discovered. Targeted agents often have toxicities that decrease their tolerability for chemoprevention. However, creative study designs, such as evaluating the effect on either surrogate endpoints or second primary tumors when given in a neoadjuvant setting, may allow the clinical evaluation of targeted agents for chemoprevention.
Acknowledgments
Supported by Department of Veterans Affairs Merit Review and NCI P50 CA58187 (YEM); NHLBI RO1 HL078929 and PPG HL14985 (VK and ECD)
Footnotes
Conflict of Interest: YEM is a co-inventor of a patent for the use of prostacyclin analogs for the prevention of cancer. No other authors have potential conflicts of interest.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J. Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Anthonisen NR, Skeans MA, Wise RA, Manfreda J, Kanner RE, Connett JE. The effects of a smoking cessation intervention on 14.5-year mortality: a randomized clinical trial. Ann. Intern. Med. 2005;142:233–239. doi: 10.7326/0003-4819-142-4-200502150-00005. [DOI] [PubMed] [Google Scholar]
- 3.Tong L, Spitz MR, Fueger JJ, Amos CA. Lung carcinoma in former smokers. Cancer. 1996;78:1004–1010. doi: 10.1002/(SICI)1097-0142(19960901)78:5<1004::AID-CNCR10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339:58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 5.Abdollahi A, Schwager C, Kleeff J, et al. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12890–12895. doi: 10.1073/pnas.0705505104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otrock ZK, Hatoum HA, Awada AH, Ishak RS, Shamseddine AI. Hypoxia-inducible factor in cancer angiogenesis: structure, regulation and clinical perspectives. Crit Rev. Oncol. Hematol. 2009;70:93–102. doi: 10.1016/j.critrevonc.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Keith RL, Miller YE, Gemmill RM, et al. Angiogenic squamous dysplasia in bronchi of individuals at high risk for lung cancer. Clin. Cancer Res. 2000;6:1616–1625. [PubMed] [Google Scholar]
- 8.Merrick DT, Haney J, Petrunich S, et al. Overexpression of vascular endothelial growth factor and its receptors in bronchial dypslasia demonstrated by quantitative RT-PCR analysis. Lung Cancer. 2005;48:31–45. doi: 10.1016/j.lungcan.2004.07.049. [DOI] [PubMed] [Google Scholar]
- 9.Bhat TA, Singh RP. Tumor angiogenesis--a potential target in cancer chemoprevention. Food Chem. Toxicol. 2008;46:1334–1345. doi: 10.1016/j.fct.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 10.Jiang C, Jiang W, Ip C, Ganther H, Lu J. Selenium-induced inhibition of angiogenesis in mammary cancer at chemopreventive levels of intake. Mol. Carcinog. 1999;26:213–225. doi: 10.1002/(sici)1098-2744(199912)26:4<213::aid-mc1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 11.Noonan DM, Benelli R, Albini A. Angiogenesis and cancer prevention: a vision. Recent Results Cancer Res. 2007;174:219–224. doi: 10.1007/978-3-540-37696-5_19. [DOI] [PubMed] [Google Scholar]
- 12.Malkinson AM. Primary lung tumors in mice: an experimentally manipulable model of human adenocarcinoma. Cancer Res. 1992;52:2670s–2676s. [PubMed] [Google Scholar]
- 13.Stearman RS, Dwyer-Nield L, Zerbe L, et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am. J Pathol. 2005;167:1763–1775. doi: 10.1016/S0002-9440(10)61257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Zhang Z, Yan Y, et al. A chemically induced model for squamous cell carcinoma of the lung in mice: histopathology and strain susceptibility. Cancer Res. 2004;64:1647–1654. doi: 10.1158/0008-5472.can-03-3273. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Zhang Z, Garbow JR, et al. Chemoprevention of lung squamous cell carcinoma in mice by a mixture of Chinese herbs. Cancer Prev. Res. 2009;2:634–640. doi: 10.1158/1940-6207.CAPR-09-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gustafson DL, Frederick B, Merz AL, Raben D. Dose scheduling of the dual VEGFR and EGFR tyrosine kinase inhibitor vandetanib (ZD6474, Zactima) in combination with radiotherapy in EGFR-positive and EGFR-null human head and neck tumor xenografts. Cancer Chemother. Pharmacol. 2008;61:179–188. doi: 10.1007/s00280-007-0460-5. [DOI] [PubMed] [Google Scholar]
- 17.Witte L, Hicklin DJ, Zhu Z, et al. Monoclonal antibodies targeting the VEGF receptor-2 (Flk1/KDR) as an anti-angiogenic therapeutic strategy. Cancer Metastasis Rev. 1998;17:155–161. doi: 10.1023/a:1006094117427. [DOI] [PubMed] [Google Scholar]
- 18.Alferez D, Wilkinson RW, Watkins J, et al. Dual inhibition of VEGFR and EGFR signaling reduces the incidence and size of intestinal adenomas in Apc(Min/+) mice. Mol. Cancer Ther. 2008;7:590–598. doi: 10.1158/1535-7163.MCT-07-0433. [DOI] [PubMed] [Google Scholar]
- 19.Keith RL, Miller YE, Hoshikawa Y, et al. Manipulation of pulmonary prostacyclin synthase expression prevents murine lung cancer. Cancer Res. 2002;62:734–740. [PubMed] [Google Scholar]
- 20.Keith RL, Miller YE, Hudish TM, et al. Pulmonary prostacyclin synthase overexpression chemoprevents tobacco smoke lung carcinogenesis in mice. Cancer Res. 2004;64:5897–5904. doi: 10.1158/0008-5472.CAN-04-1070. [DOI] [PubMed] [Google Scholar]
- 21.Nemenoff R, Meyer AM, Hudish TM, et al. Prostacyclin prevents murine lung cancer independent of the membrane receptor by activation of peroxisomal proliferator--activated receptor gamma. Cancer Prev. Res. 2008;1:349–356. doi: 10.1158/1940-6207.CAPR-08-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan Y, Lu Y, Wang M, et al. Effect of an epidermal growth factor receptor inhibitor in mouse models of lung cancer. Mol. Cancer Res. 2006;4:971–981. doi: 10.1158/1541-7786.MCR-06-0086. [DOI] [PubMed] [Google Scholar]
- 23.Fujimoto N, Wislez M, Zhang J, et al. High expression of ErbB family members and their ligands in lung adenocarcinomas that are sensitive to inhibition of epidermal growth factor receptor. Cancer Res. 2005;65:11478–11485. doi: 10.1158/0008-5472.CAN-05-1977. [DOI] [PubMed] [Google Scholar]
- 24.Kishino D, Kiura K, Takigawa N, et al. Effect of gefitinib on N-nitrosamine-4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induced lung tumorigenesis in A/J mice. Lung Cancer. 2009;64:284–289. doi: 10.1016/j.lungcan.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 25.Ohashi K, Takigawa N, Osawa M, et al. Chemopreventive effects of gefitinib on nonsmoking-related lung tumorigenesis in activating epidermal growth factor receptor transgenic mice. Cancer Res. 2009;69:7088–7095. doi: 10.1158/0008-5472.CAN-08-4205. [DOI] [PubMed] [Google Scholar]
- 26.Tang X, Varella-Garcia M, Xavier AC, et al. Epidermal growth factor receptor abnormalities in the pathogenesis and progression of lung adenocarcinomas. Cancer Prev. Res. 2008;1:192–200. doi: 10.1158/1940-6207.CAPR-08-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohashi K, Rai K, Fujiwara Y, et al. Induction of lung adenocarcinoma in transgenic mice expressing activated EGFR driven by the SP-C promoter. Cancer Sci. 2008;99:1747–1753. doi: 10.1111/j.1349-7006.2008.00875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Byers T, Wolf HJ, Franklin WA, et al. Sputum cytologic atypia predicts incident lung cancer: defining latency and histologic specificity. Cancer Epidemiol. Biomarkers Prev. 2008;17:158–162. doi: 10.1158/1055-9965.EPI-07-0436. [DOI] [PubMed] [Google Scholar]
- 29.Bach PB, Kattan MW, Thornquist MD, et al. Variations in lung cancer risk among smokers. J. Natl. Cancer Inst. 2003;95:470–478. doi: 10.1093/jnci/95.6.470. [DOI] [PubMed] [Google Scholar]
- 30.Jonsson S, Thorsteinsdottir U, Gudbjartsson DF, et al. Familial risk of lung carcinoma in the Icelandic population. JAMA. 2004;292:2977–2983. doi: 10.1001/jama.292.24.2977. [DOI] [PubMed] [Google Scholar]