

Abstract
Although non-steroidal anti-inflammatory drugs (NSAIDs), including sulindac, have been used extensively as chemopreventive agents for colorectal cancer (CRC), results are not consistent. NSAIDs, most reportedly sulindac, often do not cause a complete regression of adenomas and some patients develop resistance to NSAID treatment. In this study we evaluated the effect of sulindac on colon tumorigenesis in the ApcMin/+ mouse model. Sulindac (180 p.p.m.) given in drinking water for 9 weeks to ApcMin/+ mice significantly reduced the size of colon tumors, but actually caused an increase in colon tumor multiplicity relative to untreated controls (average of 5.5 vs. 1.6 tumors/mouse, respectively; P<0.0001). This indicated that the drug could inhibit colon tumor progression but not initiation. As expected, in the small intestine sulindac significantly reduced tumor size and multiplicity relative to untreated controls (average of 2.3 vs. 42.0 tumors/mouse, respectively; P<0.0001). Generation of a panel of prostanoids was comparably suppressed in the small intestine and colon by sulindac treatment. Sulindac is also known to exert its growth inhibitory effects through regulation of many non-COX targets, including p21, β-catenin, E-cadherin, mitochondrial apoptotic proteins and PPARγ. We found that sulindac treatment protected against E-cadherin loss in colon tumors, with associated inhibition of nuclear β-catenin accumulation. Importantly, p21WAF1/cip1 and PPARγ expression were absent in colon tumors from sulindac-treated mice, suggesting that loss of these proteins is necessary for drug resistance. Together, these observations may be translatable to designing novel clinical therapies utilizing combinations of agents that target multiple molecular pathways to overcome sulindac resistance.
Keywords: colon cancer, chemoprevention, sulindac, ApcMin/+, p21WAF1/cip1
Introduction
Sulindac and other NSAIDs have been shown to be effective chemopreventive agents for CRC (1, 2). In human familial adenomatous polyposis (FAP) patients, sulindac treatment causes an inhibition of aberrant crypt foci (ACF) formation as well as a reduction in the number and size of adenomas (3–7). Unfortunately, long-term sulindac treatment has several drawbacks. Side effects include gastrointestinal bleeding and ulceration, which limit its clinical use (8). In addition, numerous studies have found that sulindac often does not cause a complete regression of adenomas in FAP patients, and in some cases CRC develops in patients with FAP after receiving sulindac treatment (4, 9–13). Also, in the ApcMin/+ mouse, the murine model of FAP, sulindac was found to inhibit small intestinal tumors, but actually increased colon tumor incidence, multiplicity and volume (14).
The mechanism by which sulindac can inhibit intestinal tumorigenesis is strongly dependent on its ability to inhibit COX-1 and 2 enzymes, and therefore production of proliferative and inflammatory prostaglandins (PGs), including most notably PGE2 (15, 16). However, there is evidence that sulindac can also act via COX-1/2-independent mechanisms (17, 18). For example, the deregulation of Wnt/β-catenin signaling and subsequent accumulation of nuclear β-catenin is very common in colon adenomas and is mainly due to mutations in the Apc and β-catenin genes (19). It has been demonstrated, however, that sulindac treatment can induce the degradation of β-catenin protein in colon cancer cells, thus inhibiting its nuclear translocation (20). Furthermore, in normal differentiated cells, β-catenin is maintained as part of a protein complex at the plasma membrane, binding E-cadherin to the actin cytoskeleton (21). E-cadherin regulates cell adhesion in epithelial cells and is attached to the actin cytoskeleton through interactions with α- and β-catenins (22). It is believed that down-regulation of E-cadherin causes the initiation of an abnormal epithelial-mesenchymal transition (EMT) that occurs in invasive cancer cells (23). Interestingly, sulindac has been shown to increase production of E-cadherin protein in cancer cells (24). This may in fact be a result of the increased pool of β-catenin that is available to bind to E-cadherin (22).
Sulindac has also been shown to target other signaling pathways. For example, microarray experiments have demonstrated that the cdk inhibitor p21WAF1/cip1 is significantly up-regulated in response to sulindac treatment in colon cancer cell lines and rectal biopsies, resulting in an inhibition of cell proliferation (25). Subsequent studies using a variety of different mouse models, including ApcMin/+ or Apc1638+/−, p21 +/+, +/− or −/−, have shown that p21 may be critical for the tumor suppressive properties of sulindac, and that disruption of even one p21 allele may be sufficient to abrogate intestinal tumor inhibition by sulindac (26, 27). Other cellular targets may also mediate the effects of sulindac. For example, sulindac can bind to and activate peroxisome proliferator-activated receptors (PPARs γ or δ), which have been shown to act as either a tumor suppressor or oncogene in colon cancer, respectively (28, 29). Also, in prostate cells, it has been shown that PPARγ is required for both growth inhibition and p21 up-regulation by sulindac sulfide (30).
In the following study, we have evaluated the effects of sulindac on colon cancer using ApcMin/+ mice. As anticipated, sulindac treatment resulted in a profound suppression in the growth of tumors in the small intestine. However, in the colon the effects of sulindac treatment were less straightforward. While nine weeks of sulindac exposure inhibited growth of the tumors, tumor multiplicity was actually significantly increased. Importantly, PGE2 production was suppressed by sulindac to a comparable extent in both organs, indicating that regulation of non-COX targets of sulindac during tumorigenesis might be responsible for the observed increase in colon tumor multiplicity. The survival of tumors that have lost p21 and PPARγ expression following sulindac treatment may underlie the relative lack of efficacy of the drug in the colons of ApcMin/+ mice, and may provide new insights into the incomplete suppression of adenomas in a subset of human patients.
Materials and Methods
Sulindac dosing
Beginning at 5 weeks of age, ApcMin/+ mice were administered sulindac (Sigma-Aldrich, St. Louis, MO) at 180 p.p.m. in drinking water buffered with 40 mM sodium phosphate ad libitum. Control mice were given water without drug. All mice were maintained in a temperature-controlled, light-cycled room and allowed free access to drinking water (with or without sulindac) and standard diet (LM-485, Harlan Laboratories, Indianapolis, IN). Animals and food were weighed once weekly and mice were checked weekly for signs of weight loss or lethargy indicating intestinal obstruction or anemia associated with tumors. Animal experiments were conducted with approval from the Center for Laboratory Animal Care Committee, University of Connecticut Health Center.
Tumor Incidence and Multiplicity
Both control and sulindac treated ApcMin/+ mice were sacrificed at 14 weeks of age (9 weeks of sulindac treatment) for polyp scoring, histologic analyses and RNA/protein/lipid extraction from tissue. The entire small intestine and colon were harvested and flushed with ice-coldPBS and excised longitudinally. Specimens were fixed flat in 10% neutral buffered formalin solution for 4 hours and stored in 70% ethanol. Tissues were stained with 0.2% methylene blue and the number and size of tumors were scored under a dissecting microscope. Small intestines and colons were then Swiss-rolled, paraffin-embedded and sections were mounted onto glass slides and stained with H&E for histologic analysis. Tissues (normal and tumor) from a portion of control and sulindac treated mice were snap-frozen in liquid nitrogen at the time of sacrifice for RNA/protein extraction and eicosanoid analysis.
Cell culture
The wild-type HCT116 cells and a p21−/− variant of HCT116 cells were generously provided by Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD) and maintained in McCoy’s 5A medium supplemented with 10% (v/v) fetal bovine serum and 1% penicillin/streptamycin. Cells were treated with sulindac sulfide (Sigma-Aldrich) at varying concentrations (as indicated) for 24 hours.
Apoptosis assays
Apoptosis induced by sulindac sulfide in cell culture was assessed by (i) quantification of cytoplasmic histone-associated DNA fragmentation and (ii) flow cytometric analysis of cells with sub-G0/G1 DNA. The effect of sulindac on cytoplasmic histone-associated DNA fragmentation was determined using the Cell Death Detection ELISAPLUS kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions. The sub-G0/G1 fraction in control and sulindac treated cells was analyzed by flow cytometry. Briefly, cells were harvested with trypsin-EDTA, washed with 1x PBS, fixed in 70% ethanol and stored at −20°C overnight. Cells were then resuspended in a staining solution containing 500 μg/mL RNase A and 500 μg/mL propidium iodide and incubated at 37°C for 1 hour. Analysis of DNA content was performed using a BD FACSCalibur system and FlowJo software (Becton-Dickinson, Franklin Lakes, NJ).
Measurement of prostaglandins by gas chromatography-mass spectrometry (GC/MS)
PGD2, PGE2, PGF2α, 6-keto-PGF1α and thromboxane B2 (TxB2) were quantified in tissue samples by a modification of the method of Luderer JR et al., and Nichols FC, et al. (31, 32). 50–300 mg of tissue from the distal small intestine or colon was snap-frozen in liquid N2. Each homogenized tissue sample was added to 2 ml 100% methanol, 10 μM indomethacin and 25 ng of PGD2, PGE2, PGF2α, 6-keto-PGF1α and TxB2 D4 standard. After centrifugation, the supernatant was acidified with 1x PBS (pH 3) and prostaglandins were extracted twice with 2 ml of chloroform. Samples were dried down under N2 gas. Prostaglandins were derivatized using the method of Waddell, Blair and Welby (33). Prostaglandins were first treated with 2% methoxylamine hydrochloride in pyridine (30 μl). After incubating overnight at room temperature, the samples were dried under N2, dissolved in acetonitrile (30 μl) and treated with pentafluorobenzyl bromide (PFBBr; 35% v/v in acetonitrile; 10 μl) and diisopropylethylamine (10 μl). The samples were vortexed, incubated for 20 minutes at 40°C and evaporated under N2. The residue was treated with bistrimethylsilyl-trifluoroacetamide (BSTFA; 50 μl) and incubated at room temperature for 4–5 days. Gas chromatography/mass spectrometry (GC/MS) was carried out on a HP 5890 gas chromatograph interfaced with a 5988A mass spectrometer (Hewlett-Packard, Palo Alto, CA). Prostaglandin samples were applied to a SPB-1 column (12m × 0.2 mm, 0.33 μm film thickness; Supelco, Inc., Bellefonte, PA) held at 100°C. Prostaglandin samples were analyzed using a temperature program of 2°C/min from 100°C to 240°C. The injector block was held at 260°C and the transfer tube was maintained at 280°C. Prostaglandin derivatives were detected using electron capture-negative chemical ionization (34). Prostaglandin levels were quantified using selected ion monitoring of the characteristic base peak ions of the deuterated and authentic prostaglandins.
Quantitative real-time PCR
Polyps and normal-appearing mucosa were harvested from the colons of 14 week-old ApcMin/+ mice. Tissues were homogenized and total RNA was extracted with TRizol reagent (Invitrogen Corp., San Diego, CA). cDNA was synthesized using SuperScript III according to the manufacturer’s protocol (Invitrogen). mRNA expression levels were examined with TaqMan gene expression assays (Applied Biosystems Inc., Foster City, CA) using primer-probe combinations for Bax (Mm00432050_m1), Bcl-2 (Mm00477631_m1) or CDKN1a (p21; Mm00432448_m1). PCR amplification on a 7500 real-time PCR instrument (Applied Biosystems) was carried out by denaturing cDNA at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 60°C for 1 min. mRNA expression levels were normalized to hypoxanthine phosphoriboxyltransferase 1(HPRT1).
Immunoblotting
For protein extraction, tissue was incubated in SDS buffer (0.125 M Tris pH 6.8, 2% SDS, 10% glycerol) at 70°C for 10 minutes followed by sonication and centrifugation at 14000 rpm for 15 minutes at 4°C. The supernatant was removed and quantified for total protein. Cells were lysed in a buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1mM EDTA, 1 mM EGTA and 1% Triton X-100 supplemented with protease and phosphatase inhibitors (50 mM NaF, 10 mM Na β-glycerol PO4, 5 mM Na pyroPO4, 1 mM sodium vanadate, 1 μg/ml leupeptin and 1 mM PMSF). Following centrifugation at 14000 rpm for 15 minutes at 4°C the supernatant was removed and quantified for total protein. 30 μg of protein was incubated at 95°C for 5 minutes with 4x sample loading buffer containing 8% β-mercaptoethanol and loaded onto a 10–15% polyacrylamide gel. Proteins were transferred to an Immobilon-P membrane (Millipore Corp., Billerica, MA) and blocked in 5% non-fat dry milk in TBS-T (1x TBS, 0.1% Tween-20) for one hour. Blots were incubated at 4°C overnight with primary antibody (i.e. anti-p21 (1:4000; BD Biosciences), anti-PPARγ (1:1000; Cell Signaling, Danvers, MA), anti-PARP (1:1000, Cell Signaling), or anti-β actin (1:6000; Santa Cruz Biotechnology, Santa Cruz, CA). Blots were washed multiple times and incubated with goat α-mouse IgG HRP (1:10,000; Upstate Biotechnology, Billerica, MA) or donkey α-rabbit IgG HRP (1:4000; Santa Cruz Biotechnology) for 45 minutes at room temperature. HRP was visualized with enhanced luminal reagent (Millipore).
Immunohistochemistry
Staining was performed as described in our previous study (35). Briefly, small intestine and colon tissues were Swiss-rolled, paraffin-embedded and sectioned at 7-μm thickness. Tissue sections were de-paraffinized and incubated with 3% hydrogenperoxide for 20 min at room temperature. Sections were subjected to antigen retrieval and blocked with 10% normal goat serum in PBS. Sections were then incubated overnight at 4°C with anti-p21 (1:100, Neomarkers, Waltham, MA), anti-proliferating cell nuclear antigen (PCNA, 1:150, Novacastra Laboratories, Ipswich, MA), anti-cleaved caspase-3 (1:200, Cell Signaling) anti-E-cadherin (1:1000, Abcam, Cambridge, MA), anti-β-catenin (1:2000, Sigma-Aldrich) or anti-p53 (1:1000, Sigma-Aldrich) antibodies. Sections were washed and incubated with biotinylated anti-mouse secondary antibody for 30 min at room temperature. Sections were washed and then incubated with avidin-biotin complex reagent (Vector Laboratories, Burlingame, CA) for 30 min at room temperature, followed by signal detection with 3,3′-diaminobenzidine (DAB) solution (VectorLaboratories). Tissues were counterstained with hematoxylin.
Quantification of immunostaining
The area and density of E-cadherin and caspase-3 immunostaining was determined using Image-Pro Plus 7.0 software (Media Cybernetics, Bethesda, MD), using the area and density (sum) measurements.
Statistical analyses
For the comparison of size and multiplicityof polyps as well as analysis for PG levels, Bax/Bcl-2 expression and immunostaining pixel density, statistical analyses were performed using the Student’s t-test. P values were consideredstatistically significant at P < 0.05.
Results
Sulindac treatment significantly decreases colon adenoma size but increases colon tumor multiplicity in ApcMin/+ mice
To determine the effect of sulindac treatment on colon tumorigenesis in ApcMin/+ mice, we compared the multiplicity and size of colon polyps in sulindac-treated and control mice. Consistent with a previous study (14), analysis of methylene blue-stained colons revealed a 71% increase in colon tumor multiplicity following sulindac treatment (1.6 ± 0.4 vs. 5.5 ± 0.6 in control and sulindac treated, respectively; P < 0.0001; Figure 1A). However, morphometric analysis revealed a significant reduction in the size of the adenomas. There were no colon tumors over 5 mm in diameter present in the sulindac-treated mice (0 ± 0 vs. 0.9 ± 0.3 in the control group; P < 0.05). The number of small (1–2 mm) polyps increased by 97% (3.3 ± 0.3 vs. 0.1 ± 0.1, respectively; P < 0.0001) and the number of medium (2–5 mm) polyps increased by 73% (2.2 ± 0.3 vs. 0.6 ± 0.3, respectively; P < 0.01) in sulindac treated compared to control mice (Figure 1A). All tumors in both groups were in the distal region of the colon. These results suggest that although sulindac cannot prevent tumor initiation in the colons of ApcMin/+ mice, tumor progression is markedly suppressed.
Figure 1. Sulindac suppresses small intestinal tumor multiplicity and size but increases colon tumor multiplicity in ApcMin/+ mice.
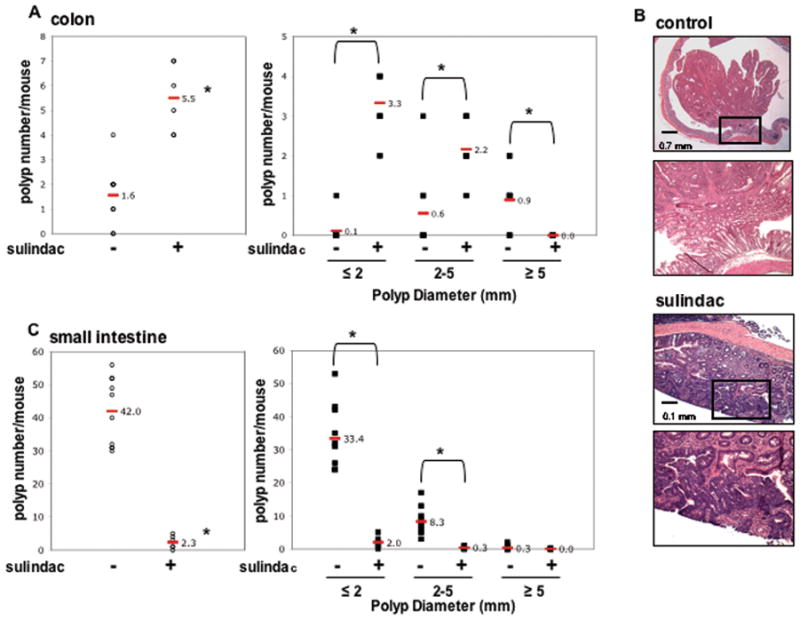
A & C, Total number and size of polyps per mouse in the colon (A) and small intestine (C) from ApcMin/+ control (n = 10) and sulindac treated (n = 10) mice. Polyps are classified by size as indicated. Each data point represents an individual mouse and the red bar and number indicate the mean value for each group. *, P < 0.05, compared with ApcMin/+ control mice (Student’s t-test). B, Representative H&E staining of colon polyps from 14-week-old ApcMin/+ control and sulindac treated mice. The bottom image in both groups is a magnification of the boxed region in the upper panel.
As shown in Figure 1B, further histologic examination of the colons revealed the presence of large, polypoid, non-invasive tumors in the untreated controls. However, polyps from sulindac-treated mice were generally smaller in size and largely sessile in appearance. Histologically, the polyps shared similar features, and in both groups there was no evidence of tumor invasion into the submucosa (Figure 1B).
As anticipated, sulindac treatment significantly reduced the total number of small intestinal polyps by 95% relative to untreated controls (2.3 ± 0.8 vs. 42.0 ± 3.3, respectively; P < 0.0001; Figure 1C). Sulindac treatment caused a similar reduction in the number of small (94%; 2.0 ± 0.7 vs. 33.4 ± 3.1, respectively; P < 0.0001), medium (96%; 0.3 ± 0.2 vs. 8.3 ± 1.3, respectively; P < 0.001) and large (100%; 0 ± 0 vs. 0.3 ± 0.2, respectively; N.S.) small intestinal adenomas (Figure 1C). This indicates that sulindac is able to inhibit both initiation and progression of tumors in the small intestine of ApcMin/+ mice.
Sulindac reduces prostanoid levels in the small intestine and colon of ApcMin/+ mice
To determine whether sulindac was effectively targeting COX-1/2 activity in the small intestine and colon, we measured the tissue levels of a panel of prostanoids by GC/MS, including PGE2, PGD2, PGF2α, thromboxane B2 and 6-keto-PGF1α. As shown in Figures 2A and B, PGE2 was the major prostaglandin produced in both organs. Sulindac treatment however, reduced the levels of PGE2 to a comparable extent in the small intestine (27%) and colon (38%; Figures 2A and B) relative to control tissue. Sulindac treatment also decreased the levels of PGD2 (43% in the small intestine and 64% in the colon), 6-keto-PGF1α (stable breakdown product of PGI2) (72% in the small intestine and 72% in the colon), and PGF2α (56% in the small intestine and 69% in the colon) to a comparable extent in both organs (Figures 2A and B). The equivalent suppression of prostanoids in the small intestine and colon, despite very different effects on tumorigenesis, suggests that the increased number of small adenomas in the colon may be a result of regulation of other events.
Figure 2. Sulindac reduces prostaglandin generation to similar extents in the small intestine and colon in ApcMin/+ mice.
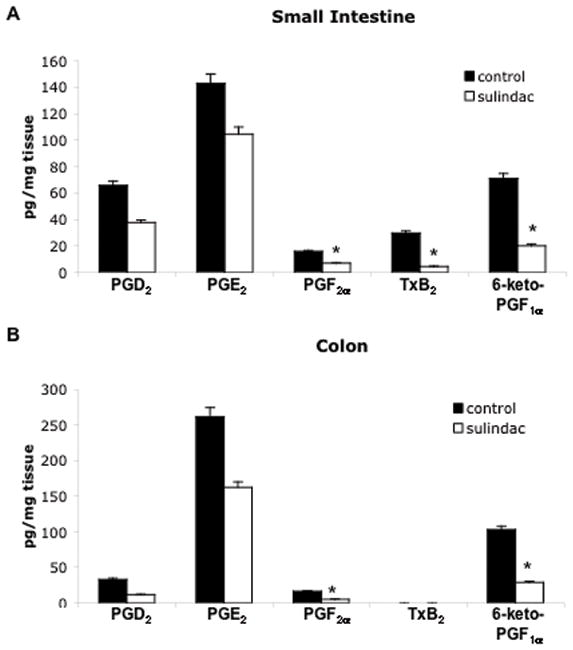
PGD2, PGE2, PGF2α, thromboxane B2 (TxB2), and 6-keto-PGF1αlevels were determined in the small intestine (A) and colon (B) of sulindac treated and untreated 14-week-old ApcMin/+ mice by gas chromatography-mass spectrometry as described in Materials and Methods. Concentrations are expressed as picograms of PG per milligram of tissue. Columns, mean of 6 samples (control) or 8 samples (sulindac treated); bars, SE. *, P < 0.05, compared with ApcMin/+ control mice (Student’s t-test).
Effect of sulindac treatment on intestinal cell turnover
To determine whether sulindac treatment affected cell turnover in the colon, immunohistochemical analyses of PCNA and caspase-3 were performed on sulindac-treated and untreated control tissues. As shown in representative images in Figure 3A(a-d), sulindac treatment did not affect the extent of PCNA staining in either colon polyps nor in normal colon crypts relative to untreated controls (56.8% vs. 65.6%, respectively; P>0.05). Also shown in Figure 3A(e & f), is caspase-3 immunostaining in colon polyps from both control and sulindac treated mice. Sulindac treatment, however, had no effect on the levels of caspase-3 staining (P>0.05).
Figure 3. The effects of sulindac on colon cell turnover in ApcMin/+ mice.
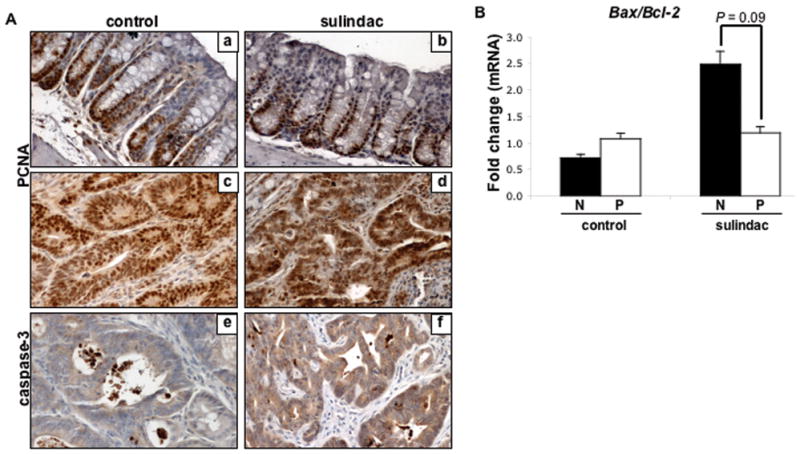
A, Representative examples of immunohistochemical analysis of proliferating cell nuclear antigen (PCNA) (a–d) and cleaved caspase-3 (e & f) in 14-week-old ApcMin/+ control and sulindac treated normal tissue (a & b) and colon polyps (c–f) performed as described in Materials and Methods. Quantification of the proliferative zone was not found to be statistically significant between groups in normal tissue (P=0.21, Student’s t-test). Quantification of caspase-3 immunostaining area and density (sum) using Image-Pro software was not found to be statistically significant between sulindac treated and untreated polyp tissue (P=0.14, Student’s t-test). B, cDNA was isolated from the colon of 14-week-old ApcMin/+ mice and subjected to quantitative real-time PCR as described in Materials and Methods. Bax and Bcl-2 mRNA expression levels were assayed in control and sulindac treated normal colonic mucosa (N) and polyps (P). Expression levels were normalized to HPRT1. Columns, mean of Bax/Bcl-2 expression of 3 samples per group; bars, SE. P = 0.09 between groups (Student’s t-test).
Sulindac-induced apoptosis has been shown to be dependent on mitochondrial apoptotic proteins (36). As shown in Figure 3B, sulindac treatment moderately decreased the Bax (pro-apoptotic protein)/Bcl-2 (anti-apoptotic protein) mRNA expression ratio in colon polyps compared to normal mucosa by 52% (1.19 vs. 2.48, respectively; P = 0.1). In comparison, in untreated control tissues, the Bax/Bcl-2 expression ratio was slightly increased (33%) in colon polyps relative to normal mucosa (1.08 vs. 0.72, respectively; P > 0.05), indicating a modest decrease in apoptosis within polyps from sulindac-treated mice relative to normal mucosa
The expression of E-cadherin and β-catenin is altered in sulindac treated ApcMin/+ colon polyps
Suppression of PGE2 production has been associated with reduced accumulation of nuclear β-catenin in colon cancer cells as well as in polyps (35, 37). Thus, to gain additional insight into the increased colon tumor multiplicity found in the colon after sulindac exposure, the cellular localization of β-catenin and E-cadherin was evaluated. As shown in Figure 4A (panels a & c) & B, immunohistochemical analysis of E-cadherin showed a dramatic reduction in membrane associated E-cadherin staining in untreated ApcMin/+ polyps compared to normal adjacent mucosa (4.0 × 106 pixels/frame vs. 6.3 × 106 pixels/frame, respectively; P<0.01). However, sulindac treatment resulted in a profound protection against this tumor-associated loss of E-cadherin (Figure 4A, panels b & d). In fact, as shown in Figure 4B, the loss of membrane staining of E-cadherin in polyp tissue was completely inhibited by sulindac treatment.
Figure 4. Sulindac affects E-cadherin and β-catenin expression in ApcMin/+ colon polyps.
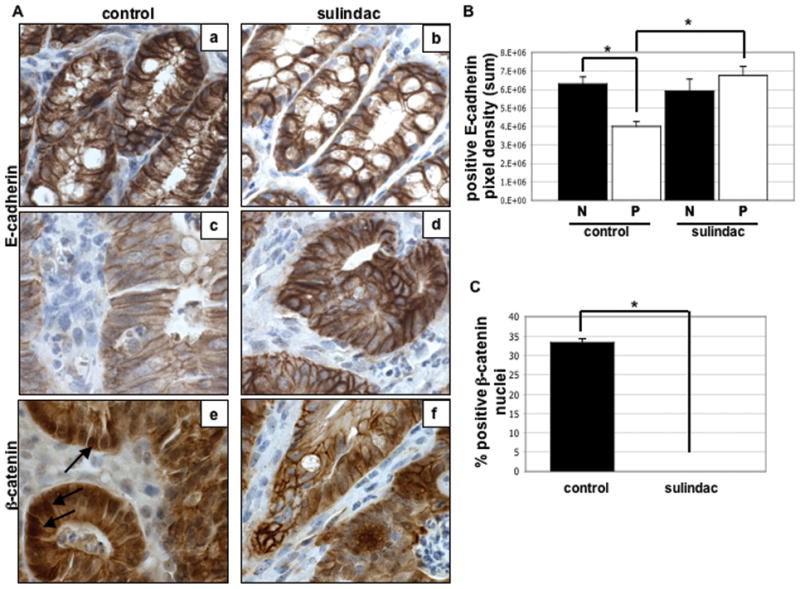
A, Representative examples of immunohistochemical analysis of E-cadherin (a–d) and β-catenin (e & f) in 14-week-old ApcMin/+ control and sulindac treated normal tissue (a & b) and colon polyps (c– f) performed as described in Materials and Methods. Arrows, nuclear β-catenin staining. B, Quantification of the intensity of E-cadherin immunostaining in normal (N) and polyp (P) colon tissue using Image-Pro software as described in Materials and Methods. Columns, mean of E-cadherin staining pixel density (sum) of 5 samples per group; bars, SE. *, P < 0.01 between groups (Student’s t-test). C, Quantification of nuclear β-catenin staining in sulindac treated and untreated colon polyps. Columns, mean % positive nuclei of 12 samples per group; bars, SE. *, P<0.0001 between groups (Student’s t-test).
Since E-cadherin and β-catenin exist in a protein complex at the plasma membrane (22), we next wanted to determine whether altered E-cadherin levels might impact β-catenin localization. Although the accumulation of nuclear β-catenin is clearly associated with increased cell proliferation (19), there is evidence that elevated nuclear levels might actually become pro-apopototic (38). As expected (Figure 4A (panels e & f)), intense nuclear staining of β-catenin was found consistently within polyp tissue of untreated controls. However, sulindac treatment resulted in the complete abolition of nuclear β-catenin within the colon polyps (P<0.0001, Figure 4C). In addition, there was no evidence of nuclear β-catenin staining in the normal colon mucosa from either group (data not shown). In the small intestine, sulindac treatment had no effect on the expression of nuclear β-catenin. Strong nuclear staining was observed in both sulindac treated and control polyp tissue while normal small intestinal mucosa did not exhibit nuclear β-catenin expression (data not shown). Taken together, these data suggest that the ability of sulindac to inhibit loss of E-cadherin in colon polyps may contribute to the significant reduction in nuclear β-catenin.
Sulindac treated ApcMin/+ colon polyps do not express p21WAF1/cip1 or PPARγ protein
The increase in colon polyp multiplicity occurred despite continuous exposure to sulindac, suggesting that a subset of lesions have developed sulindac resistance. It is possible that the failure to respond to sulindac may be a result of the acquisition of an altered molecular signature within the polyps. p21 has been shown to be up-regulated by sulindac treatment, and its expression has been demonstrated to be required for tumor suppression in response to sulindac (25, 26). As shown in Figure 5(A, B), we performed immunoblot and immunohistochemical (IHC) analysis of p21 in control and sulindac-treated colons. In the absence of sulindac, p21 expression was up-regulated in colon polyps relative to normal mucosa (4/4 cases). Following sulindac treatment, however, p21 levels were not detectable in the polyps (Fig. 5A & B). However, p21 mRNA analysis did not reveal expression differences between the control and sulindac treated groups (data not shown). p21 expression in the small intestine, assessed by IHC, was found to be highly variable in tumors from both groups (data not shown). In addition, we did not detect any differences in p53 expression between the control and sulindac treated groups (data not shown).
Figure 5. ApcMin/+ colon polyps that are not suppressed by sulindac do not express p21WAF1/cip1 or PPARγ.
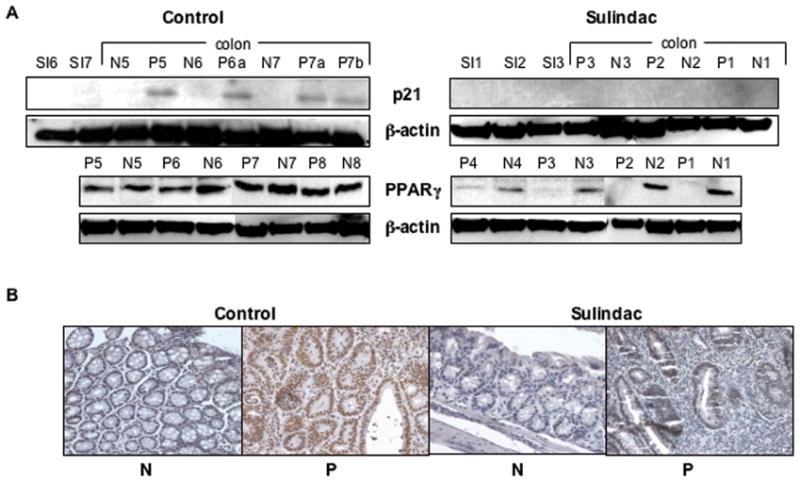
A, Immunoblot analysis of p21 and PPARγ in total cell lysates from normal appearing colonic mucosa (N), polyps (P) and small intestinal (SI) tissue in 14 week old control and sulindac treated ApcMin/+ mice performed as described in Materials and Methods. The blots were reprobed using β-actin as a loading control. B, Representative examples of immunohistochemical analysis of p21 in 14-week-old ApcMin/+ sulindac treated and control normal colonic mucosa (N) and polyps (P) performed as described in Materials and Methods.
It has recently been reported that up-regulation of p21, with associated growth suppression, following sulindac treatment may be mediated in part through activation of the nuclear receptor protein PPARγ (30). As shown in Figure 5A, PPARγ expression was maintained in polyp tissue in the absence of sulindac treatment. However, exposure to sulindac resulted in the growth of polyps that failed to express PPARγ (Fig. 5A), suggesting that sulindac-resistant polyps may also be able to survive because they have lost PPARγ. These data suggest that during tumorigenesis a subset of cells will lose expression of p21 and PPARγ, possibly facilitating their clonal expansion and contributing to their resistance to sulindac.
p21 null colon cancer cells are resistant to sulindac induced apoptosis and cell cycle arrest
Based on our observation that colon polyps that lose p21 can survive in the presence of sulindac, we wanted to further investigate the mechanism for this resistance in vitro using HCT116 wild-type and p21−/− cells. As shown in Figure 6A, sulindac sulfide (SS) increased PARP cleavage in HCT116 wild-type cells in a dose-related manner, consistent with previous studies (39). This response was abrogated in the absence of p21. Apoptosis as assessed by enrichment of histone/DNA cytoplasmic complexes was also significantly inhibited in p21−/− cells compared to wild-type cells in response to 90 and 120 μM SS (Figure 6B). In addition, the % sub-G0/G1 phase cells (apoptotic) were reduced in p21−/− cells compared to wild-type cells in response to 90 and 120 μM SS (Figure 6C). These data indicate the cells that lose p21 do not respond to sulindac sulfide due to inhibition of apoptosis and cell cycle arrest.
Figure 6. HCT116 p21−/− cells are resistant to sulindac sulfide induced apoptosis.
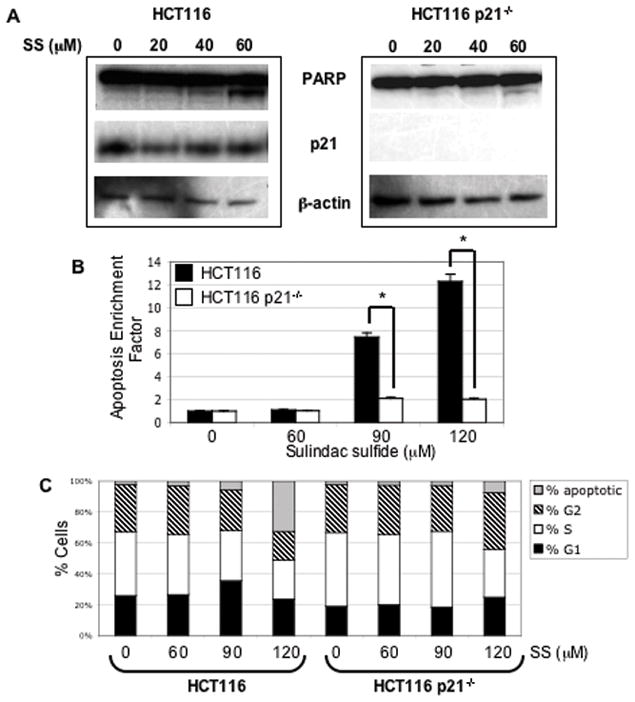
A, Western blot analysis of PARP and p21 in total cell lysates from HCT116 WT and HCT116 p21−/− cells treated with varying concentrations (as indicated) of sulindac sulfide (SS) for 24 hours. The blots were re-probed using β-actin as a loading control. B, Analysis of apoptosis as assessed by quantification of cytoplasmic histone-associated DNA fragmentation. HCT116 WT (black bars) and HCT116 p21−/− cells (white bars) were treated with varying concentrations (as indicated) of sulindac sulfide for 24 hours and relative levels of apoptosis were assayed by ELISA as described in Materials and Methods. Columns, mean of triplicate samples; bars, SE. *, P < 0.05, between groups (Student’s t-test). C, Cell cycle analysis of HCT116 WT and HCT116 p21−/− cells treated with varying concentrations (as indicated) of sulindac sulfide (SS) for 24 hours. Cells were stained with propidium iodide and analyzed by flow cytometry as described in Materials and Methods. Stacked columns, mean of triplicate samples.
Discussion
Human clinical trails have shown that NSAIDs, including sulindac, which inhibit COX-1 and 2, are effective in reducing the recurrence of colorectal adenomas (4, 5). Recent reports, however, suggest that sulindac therapy may be associated with the development of resistance among a subset of patients. This outcome includes the failure of sulindac to promote the regression of adenomas and/or development of a ‘break-through’ carcinoma in patients while on sulindac treatment (4, 9–13). Thus, it becomes of paramount importance to begin to elucidate the mechanisms of sulindac resistance in order to predict those individuals who will benefit most from NSAID treatment. In addition, gaining a better understanding of underlying molecular mechanisms of this effect may facilitate the development of new chemoprevention strategies. For these reasons, we have tested the efficacy of sulindac in inhibiting tumorigenesis in a widely used mouse model of intestinal cancer, ApcMin/+, with a focus on colonic rather than small intestinal tumors. We report that sulindac is effective at reducing the size of colon adenomas, indicating inhibition of progression. However, the number of colon adenomas is significantly increased by sulindac exposure, consistent with an earlier study (14). Together, these results suggest that sulindac fails to block early events associated with tumor initiation.
To begin to evaluate potential mechanisms by which sulindac can inhibit colon tumor progression, we measured the concentrations of a panel of prostanoids in the small intestine and colon. Sulindac is a potent inhibitor of both COX-1 and COX-2, and has been shown to effectively suppress PGE2 production within the intestine (15, 16). As expected, PGE2 levels were markedly reduced in the small intestine by sulindac, consistent with its tumor suppressive properties (Figure 1C). However, in the colon, where tumor numbers were actually increased by sulindac treatment, PGE2 production was inhibited to a comparable extent as in the small intestine, suggesting that resistance to drug treatment in this organ is not directly related to the degree of PGE2 suppression. Thus, we began to consider the possibility that regulation of other events might account for the drug-associated increase in colon tumor multiplicity and decrease in size.
Sulindac is known to affect a number of non-COX cellular pathways that may directly impact tumor cell growth (40). For example, it has been shown to disrupt Wnt signaling by inhibiting the nuclear translocation of β-catenin, thus impairing the transcription of TCF/LEF target genes (20). We found that sulindac abolished nuclear β-catenin within the colon polyps while increasing its membrane localization. To better define a mechanism for this effect, we examined the levels of E-cadherin expression in the presence and absence of sulindac since E-cadherin can anchor β-catenin at cell-cell junctions (22). Repression of E-cadherin, often occurring during tumor cell invasion, results in nuclear accumulation of β-catenin and activation of target genes (41, 42). In the present study, we found a dramatic reduction in E-cadherin levels in untreated ApcMin/+ colon polyps, suggesting a high potential for growth and invasion. Interestingly, sulindac completely protected against this loss of E-cadherin. Thus, it seems likely that the sulindac-induced maintenance of E-cadherin in colon polyps is causing an increase in membrane bound β-catenin, inhibiting its nuclear accumulation and suppressing growth of the tumors.
Recently, a “just right signaling model” for CRC has been proposed by several research groups (38, 43). This model suggests that during colon carcinogenesis, APC must acquire specific mutations that will cause sufficient accumulation of nuclear β-catenin to promote transcription of proliferative target genes. However, excessive nuclear β-catenin accumulation has also been shown to promote apoptosis, and is therefore unlikely to provide a selective advantage during tumorigenesis (44). It is thought that these elevated levels of nuclear β-catenin will result in a broader change in gene expression, which increases the likelihood of conflicting downstream signals, thus inducing an apoptotic response (43). This model holds true in FAP and sporadic tumors as well as in APC mutant mouse models, although there is tissue- and species-specificity (45–47). Therefore, it is possible that the observed sulindac-mediated suppression of nuclear β-catenin and increased membrane localization in ApcMin/+ colon polyps might have duel effects of inhibiting growth while simultaneously creating a resistance to apoptosis, thus increasing tumor multiplicity (Figures 1A & 4A). In contrast, nuclear β-catenin expression in the small intestinal tumors was not affected by sulindac treatment (data not shown). In the small intestinal tissue, the level of β-catenin/TCF signaling may be enough to maintain a balance of proliferation and apoptosis. Recently, there has been interest in developing chemoprevention strategies to target Wnt/β-catenin-mediated signaling (48). Our study points to a potential caveat to developing therapies that completely inhibit the nuclear translocation or enhance the proteosomal degradation of β-catenin as a means for tumor prevention. The possibility exists that complete suppression of nuclear β-catenin may ultimately weaken the apoptotic response to sulindac, paradoxically facilitating tumor initiation. The increased formation of colon tumors in sulindac-treated ApcMin/+ mice may arise from excessive β-catenin inhibition.
Sulindac is known to induce transcription of p21WAF1/cip1, a key inhibitor of the cell cycle, which has been shown to be required for drug-dependent suppression of tumor growth (25, 26). In accordance with these earlier findings, we also observed that HCT116 p21−/− cells have reduced levels of apoptosis and cell cycle arrest in comparison to their wild-type counterparts in response to treatment with sulindac sulfide. In addition, the nuclear receptor PPARγ can be activated by direct sulindac binding and has been implicated in sulindac-induced growth arrest and transcriptional activation of p21 (30). In fact, PPARγ activation by other ligands (e.g. pioglitazone, bezafibrate) has also been shown to cause a significant reduction in adenoma multiplicity in ApcMin/+ mice (49). Our study found that the majority of remaining colon polyps sustained a loss of p21 and PPARγ expression, suggesting the evolution of an altered molecular signature within non-responding tumors that have been chronically exposed to sulindac. While the underlying mechanisms that may account for this loss of protein expression remain to be determined, these data do suggest that increasing p21 and/or PPARγ levels in tumor cells may provide a pathway for these cells to overcome resistance to sulindac. One limiting factor to these observations was the inability to compare small size vs. large size tumors in the sulindac treated group because there were no large size tumors (> 5 mm) present. These data would have been more complete if it had been possible to perform a size-matched comparison of colon tumors between the two groups.
Taken together, our data suggest that colon tumors that evade sulindac suppression possess an altered molecular signature that potentially confers drug resistance. In particular, the loss of p21 and PPARγ expression may provide a fundamental mechanism for incomplete sulindac chemoprevention. In fact, such a mechanism may extend to other chemopreventive agents that induce cell cycle arrest. For example, celecoxib, like sulindac, has been shown to induce transcription of p21 (50). Therefore, loss of p21 and/or PPARγ in tumors may impart chemo-resistance to a large number of therapeutic agents. These findings may provide new insights into designing novel clinical therapies targeting multiple molecular pathways to overcome sulindac resistance.
Footnotes
This work was supported by National Institute of Health Grant 5R01CA114635 (to D.W.R.)
References
- 1.Smalley WE, DuBois RN. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv Pharmacol. 1997;39:1–20. doi: 10.1016/s1054-3589(08)60067-8. [DOI] [PubMed] [Google Scholar]
- 2.Williams CS, Smalley W, DuBois RN. Aspirin use and potential mechanisms for colorectal cancer prevention. J Clin Invest. 1997;100(6):1325–9. doi: 10.1172/JCI119651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cruz-Correa M, Hylind LM, Romans KE, Booker SV, Giardiello FM. Long-term treatment with sulindac in familial adenomatous polyposis: a prospective cohort study. Gastroenterology. 2002;122(3):641–5. doi: 10.1053/gast.2002.31890. [DOI] [PubMed] [Google Scholar]
- 4.Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328(18):1313–6. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 5.Rigau J, Pique JM, Rubio E, Planas R, Tarrech JM, Bordas JM. Effects of long-term sulindac therapy on colonic polyposis. Ann Intern Med. 1991;115(12):952–4. doi: 10.7326/0003-4819-115-12-952. [DOI] [PubMed] [Google Scholar]
- 6.Takayama T, Katsuki S, Takahashi Y, et al. Aberrant crypt foci of the colon as precursors of adenoma and cancer. N Engl J Med. 1998;339(18):1277–84. doi: 10.1056/NEJM199810293391803. [DOI] [PubMed] [Google Scholar]
- 7.Waddell WR, Loughry RW. Sulindac for polyposis of the colon. J Surg Oncol. 1983;24(1):83–7. doi: 10.1002/jso.2930240119. [DOI] [PubMed] [Google Scholar]
- 8.Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340(24):1888–99. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- 9.Giardiello FM, Offerhaus JA, Tersmette AC, et al. Sulindac induced regression of colorectal adenomas in familial adenomatous polyposis: evaluation of predictive factors. Gut. 1996;38(4):578–81. doi: 10.1136/gut.38.4.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giardiello FM, Yang VW, Hylind LM, et al. Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med. 2002;346(14):1054–9. doi: 10.1056/NEJMoa012015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niv Y, Fraser GM. Adenocarcinoma in the rectal segment in familial polyposis coli is not prevented by sulindac therapy. Gastroenterology. 1994;107(3):854–7. doi: 10.1016/0016-5085(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 12.Thorson AG, Lynch HT, Smyrk TC. Rectal cancer in FAP patient after sulindac. Lancet. 1994;343(8890):180. doi: 10.1016/s0140-6736(94)90974-1. [DOI] [PubMed] [Google Scholar]
- 13.Tonelli F, Valanzano R, Messerini L, Ficari F. Long-term treatment with sulindac in familial adenomatous polyposis: is there an actual efficacy in prevention of rectal cancer? J Surg Oncol. 2000;74(1):15–20. doi: 10.1002/1096-9098(200005)74:1<15::aid-jso4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 14.Yang K, Fan K, Kurihara N, et al. Regional response leading to tumorigenesis after sulindac in small and large intestine of mice with Apc mutations. Carcinogenesis. 2003;24(3):605–11. doi: 10.1093/carcin/24.3.605. [DOI] [PubMed] [Google Scholar]
- 15.Shiff SJ, Rigas B. Nonsteroidal anti-inflammatory drugs and colorectal cancer: evolving concepts of their chemopreventive actions. Gastroenterology. 1997;113(6):1992–8. doi: 10.1016/s0016-5085(97)99999-6. [DOI] [PubMed] [Google Scholar]
- 16.Subbaramaiah K, Zakim D, Weksler BB, Dannenberg AJ. Inhibition of cyclooxygenase: a novel approach to cancer prevention. Proc Soc Exp Biol Med. 1997;216(2):201–10. doi: 10.3181/00379727-216-44170. [DOI] [PubMed] [Google Scholar]
- 17.Babbar N, Ignatenko NA, Casero RA, Jr, Gerner EW. Cyclooxygenase-independent induction of apoptosis by sulindac sulfone is mediated by polyamines in colon cancer. J Biol Chem. 2003;278(48):47762–75. doi: 10.1074/jbc.M307265200. [DOI] [PubMed] [Google Scholar]
- 18.Soh JW, Weinstein IB. Role of COX-independent targets of NSAIDs and related compounds in cancer prevention and treatment. Prog Exp Tumor Res. 2003;37:261–85. doi: 10.1159/000071377. [DOI] [PubMed] [Google Scholar]
- 19.Behrens J. The role of the Wnt signalling pathway in colorectal tumorigenesis. Biochem Soc Trans. 2005;33(Pt 4):672–5. doi: 10.1042/BST0330672. [DOI] [PubMed] [Google Scholar]
- 20.Rice PL, Kelloff J, Sullivan H, et al. Sulindac metabolites induce caspase- and proteasome-dependent degradation of beta-catenin protein in human colon cancer cells. Mol Cancer Ther. 2003;2(9):885–92. [PubMed] [Google Scholar]
- 21.Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci. 1999;112 (Pt 8):1237–45. doi: 10.1242/jcs.112.8.1237. [DOI] [PubMed] [Google Scholar]
- 22.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303(5663):1483–7. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ. Wnt-dependent regulation of the E-cadherin repressor snail. J Biol Chem. 2005;280(12):11740–8. doi: 10.1074/jbc.M413878200. [DOI] [PubMed] [Google Scholar]
- 24.Jiang MC, Liao CF, Lee PH. Aspirin inhibits matrix metalloproteinase-2 activity, increases E-cadherin production, and inhibits in vitro invasion of tumor cells. Biochem Biophys Res Commun. 2001;282(3):671–7. doi: 10.1006/bbrc.2001.4637. [DOI] [PubMed] [Google Scholar]
- 25.Yang W, Velcich A, Mariadason J, et al. p21(WAF1/cip1) is an important determinant of intestinal cell response to sulindac in vitro and in vivo. Cancer Res. 2001;61(16):6297–302. [PubMed] [Google Scholar]
- 26.Yang W, Bancroft L, Augenlicht LH. Methylation in the p21WAF1/cip1 promoter of Apc+/−, p21+/− mice and lack of response to sulindac. Oncogene. 2005;24(12):2104–9. doi: 10.1038/sj.onc.1208444. [DOI] [PubMed] [Google Scholar]
- 27.Yang WC, Mathew J, Velcich A, et al. Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a Western-style high-risk diet by altering cell maturation in the intestinal mucosal. Cancer Res. 2001;61(2):565–9. [PubMed] [Google Scholar]
- 28.Park BH, Vogelstein B, Kinzler KW. Genetic disruption of PPARdelta decreases the tumorigenicity of human colon cancer cells. Proc Natl Acad Sci U S A. 2001;98(5):2598–603. doi: 10.1073/pnas.051630998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarraf P, Mueller E, Jones D, et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat Med. 1998;4(9):1046–52. doi: 10.1038/2030. [DOI] [PubMed] [Google Scholar]
- 30.Jarvis MC, Gray TJ, Palmer CN. Both PPARgamma and PPARdelta influence sulindac sulfide-mediated p21WAF1/CIP1 upregulation in a human prostate epithelial cell line. Oncogene. 2005;24(55):8211–5. doi: 10.1038/sj.onc.1208983. [DOI] [PubMed] [Google Scholar]
- 31.Luderer JR, Riley DL, Demers LM. Rapid extraction of arachidonic acid metabolites utilizing octadecyl reversed-phase columns. J Chromatogr. 1983;273(2):402–9. doi: 10.1016/s0378-4347(00)80961-5. [DOI] [PubMed] [Google Scholar]
- 32.Nichols FC, Riep B, Mun J, et al. Structures and biological activities of novel phosphatidylethanolamine lipids of Porphyromonas gingivalis. J Lipid Res. 2006;47(4):844–53. doi: 10.1194/jlr.M500542-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Waddell KA, Barrow SE, Robinson C, Orchard MA, Dollery CT, Blair IA. Quantitative analysis of prostanoids in biological fluids by combined capillary column gas chromatography negative ion chemical ionization mass spectrometry. Biomed Mass Spectrom. 1984;11(2):68–74. doi: 10.1002/bms.1200110205. [DOI] [PubMed] [Google Scholar]
- 34.Nichols FC, Riep B, Mun J, et al. Structures and biological activity of phosphorylated dihydroceramides of Porphyromonas gingivalis. J Lipid Res. 2004;45(12):2317–30. doi: 10.1194/jlr.M400278-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Nakanishi M, Montrose DC, Clark P, et al. Genetic deletion of mPGES-1 suppresses intestinal tumorigenesis. Cancer Res. 2008;68(9):3251–9. doi: 10.1158/0008-5472.CAN-07-6100. [DOI] [PubMed] [Google Scholar]
- 36.Sinicrope FA, Penington RC. Sulindac sulfide-induced apoptosis is enhanced by a small-molecule Bcl-2 inhibitor and by TRAIL in human colon cancer cells overexpressing Bcl-2. Mol Cancer Ther. 2005;4(10):1475–83. doi: 10.1158/1535-7163.MCT-05-0137. [DOI] [PubMed] [Google Scholar]
- 37.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310(5753):1504–10. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 38.Bordonaro M, Lazarova DL, Sartorelli AC. Hyperinduction of Wnt activity: a new paradigm for the treatment of colorectal cancer? Oncol Res. 2008;17(1):1–9. doi: 10.3727/096504008784046108. [DOI] [PubMed] [Google Scholar]
- 39.Flis S, Soltysiak-Pawluczuk D, Jedrych A, Jastrzebski Z, Remiszewska M, Splawinski J. Antiangiogenic effect of sulindac sulfide could be secondary to induction of apoptosis and cell cycle arrest. Anticancer Res. 2006;26(4B):3033–41. [PubMed] [Google Scholar]
- 40.Piazza GA, Rahm AK, Finn TS, et al. Apoptosis primarily accounts for the growth-inhibitory properties of sulindac metabolites and involves a mechanism that is independent of cyclooxygenase inhibition, cell cycle arrest, and p53 induction. Cancer Res. 1997;57(12):2452–9. [PubMed] [Google Scholar]
- 41.Conacci-Sorrell M, Simcha I, Ben-Yedidia T, Blechman J, Savagner P, Ben-Ze’ev A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J Cell Biol. 2003;163(4):847–57. doi: 10.1083/jcb.200308162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jamora C, DasGupta R, Kocieniewski P, Fuchs E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature. 2003;422(6929):317–22. doi: 10.1038/nature01458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Albuquerque C, Breukel C, van der Luijt R, et al. The ‘just-right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Hum Mol Genet. 2002;11(13):1549–60. doi: 10.1093/hmg/11.13.1549. [DOI] [PubMed] [Google Scholar]
- 44.Kim K, Pang KM, Evans M, Hay ED. Overexpression of beta-catenin induces apoptosis independent of its transactivation function with LEF-1 or the involvement of major G1 cell cycle regulators. Mol Biol Cell. 2000;11(10):3509–23. doi: 10.1091/mbc.11.10.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shoemaker AR, Luongo C, Moser AR, Marton LJ, Dove WF. Somatic mutational mechanisms involved in intestinal tumor formation in Min mice. Cancer Res. 1997;57(10):1999–2006. [PubMed] [Google Scholar]
- 46.Smits R, Hofland N, Edelmann W, et al. Somatic Apc mutations are selected upon their capacity to inactivate the beta-catenin downregulating activity. Genes Chromosomes Cancer. 2000;29(3):229–39. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1033>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 47.Smits R, Kartheuser A, Jagmohan-Changur S, et al. Loss of Apc and the entire chromosome 18 but absence of mutations at the Ras and Tp53 genes in intestinal tumors from Apc1638N, a mouse model for Apc-driven carcinogenesis. Carcinogenesis. 1997;18(2):321–7. doi: 10.1093/carcin/18.2.321. [DOI] [PubMed] [Google Scholar]
- 48.Clapper ML, Coudry J, Chang WC. beta-catenin-mediated signaling: a molecular target for early chemopreventive intervention. Mutat Res. 2004;555(1–2):97–105. doi: 10.1016/j.mrfmmm.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 49.Niho N, Takahashi M, Kitamura T, et al. Concomitant suppression of hyperlipidemia and intestinal polyp formation in Apc-deficient mice by peroxisome proliferator-activated receptor ligands. Cancer Res. 2003;63(18):6090–5. [PubMed] [Google Scholar]
- 50.Bock JM, Menon SG, Sinclair LL, et al. Celecoxib toxicity is cell cycle phase specific. Cancer Res. 2007;67(8):3801–8. doi: 10.1158/0008-5472.CAN-06-3780. [DOI] [PubMed] [Google Scholar]