

Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disorder that affects the aging population. A progressive loss of motor neurons in the spinal cord and brain leads to muscle paralysis and death. As in other common neurodegenerative diseases, aging-related mitochondrial dysfunction is increasingly being considered among the pathogenic factors. Mitochondria are critical for cell survival: they provide energy to the cell, buffer intracellular calcium, and regulate apoptotic cell death. Whether mitochondrial abnormalities are a trigger or a consequence of the neurodegenerative process and the mechanisms whereby mitochondrial dysfunction contributes to disease are not clear yet. Calcium homeostasis is a major function of mitochondria in neurons, and there is ample evidence that intracellular calcium is dysregulated in ALS. The impact of mitochondrial dysfunction on intracellular calcium homeostasis and its role in motor neuron demise are intriguing issues that warrants in depth discussion. Clearly, unraveling the causal relationship between mitochondrial dysfunction, calcium dysregulation, and neuronal death is critical for the understanding of ALS pathogenesis. In this review, we will outline the current knowledge of various aspects of mitochondrial dysfunction in ALS, with a special emphasis on the role of these abnormalities on intracellular calcium handling.
Keywords: Amyotrophic lateral sclerosis, ALS, motor neurons, mitochondria, astrocytes, calcium
1. Introduction
Mitochondria are essential organelles for every cell as the powerhouse to provide ATP for a multitude of cellular processes. They are also the hub of metabolic pathways, primary sources of reactive oxygen species, regulators of apoptosis, and buffers of intracellular calcium. Simply put, mitochondrial function is central to cellular life and death. Mitochondrial dysfunction is well documented in a wide variety of age-related neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS). In ALS, degenerating motor neurons (MNs) in the spinal cord show abnormal mitochondrial morphology, bioenergetics, calcium handling, organelle transport and dynamics, before or at the time of paralysis onset. Description of these mitochondrial abnormalities have been extensively documented and reviewed previously (Bacman et al., 2006; Hervias et al., 2006; Magrane and Manfredi, 2009; Shi et al., 2009). In this article, we will first briefly review some of the more recent advances on the mitochondrial involvement in ALS, and then we will focus on mitochondrial handling of calcium for maintenance of intracellular calcium homeostasis, and its relevance to the pathogenesis of ALS. Intracellular calcium dysregulation is recently receiving increasing attention in the field of aging-related neurodegenerative disorders, because it represents a potential link among converging pathogenic pathways, from signaling to bioenergetic failure and apoptosis. The widespread consequences of calcium dysregulation can extend, not only within the intracellular domain, but also to intercellular communication, at the tissue and organ levels.
2. Amyotrophic lateral sclerosis
ALS is characterized by selective and progressive degeneration of MNs in the spinal cord and brain, leading to weakness, atrophy, and paralysis of voluntary muscles. ALS is an adult onset disease, affecting men and women of all races in their 50s and 60s, with incidence increasing with age. The disease progression is very aggressive with fatal outcome usually within 5 years of onset. Currently, there are no effective cures and very few treatments are available for this devastating MN disease.
The majority of ALS cases are sporadic, with no known etiology, but in 10% of the cases, the disorder is inherited. Mutations in Cu,Zn,superoxide dismutase (SOD1) are the most common and constitute about 20% of familial ALS (fALS) cases (Rosen et al., 1993). More than one hundred SOD1 mutations result in fALS, whose clinical and pathological features are very similar to those of sporadic ALS. SOD1 is a ubiquitously expressed abundant enzyme that converts superoxide to hydrogen peroxide and thus protects cells against oxidative stress. In SOD1-fALS, a toxic gain-of-function of the mutant protein is pathogenic, rather than an enzymatic defect, because superoxide dismutase activity is not a disease determinant and the mutations cause an autosomal dominant disease. Leading hypotheses for the toxic mechanisms of mutant SOD1 include mitochondrial dysfunction, aberrant production of free radical species, protein aggregation, neurofilament dysregulation, impaired axonal transport, glutamate excitotoxicity, and proteasome dysfunction (Pasinelli and Brown, 2006; Rothstein, 2009). It is likely that multiple mechanisms are involved, rather than a single one.
While there is still no consensus as to how mutant SOD1 causes selective toxicity to MNs, one aspect that is apparent is that MN death is non-cell autonomous, and involves other cell types in the vicinity of MNs. In this respect, the involvement of astrocytes and microglia is well documented, and mutant SOD1 expression in glial cells has been proposed to accelerate disease progression (Boillee et al., 2006b; Yamanaka et al., 2008).
The development of multiple transgenic mouse lines expressing various mutant human SOD1 proteins that recapitulate MN degeneration in SOD1-fALS (Turner and Talbot, 2008) has produced ample evidence of mitochondrial dysfunction (Bacman et al., 2006; Hervias et al., 2006; Magrane and Manfredi, 2009; Shi et al., 2009). Recently, other genetic causes of fALS have been identified, such as mutations in the 43kDa TAR DNA-binding protein (TDP-43) (Kabashi et al., 2008; Sreedharan et al., 2008) and in fused in sarcoma (FUS) (Kwiatkowski et al., 2009; Vance et al., 2009), which have stimulated new intriguing hypotheses for a role of DNA/RNA binding proteins in the pathogenesis of ALS. However, studies on these proteins are still at their infancies, and whether mitochondrial functions are affected in ALS associated with these mutations is completely unexplored and is an important issue to be addressed in the future for the understanding of etiology of ALS. Since to date most of our understanding of mitochondrial dysfunction in ALS comes from studies of SOD1-fALS, in this article we concentrate on the toxicity of mutant SOD1 to mitochondria.
3. Mitochondrial abnormalities in ALS
As commonly observed in other aging-related neurodegenerative disorders, like Alzheimer and Parkinson diseases, also in ALS, mitochondria display various abnormalities (summarized in Table 1). Importantly, in the mutant SOD1 mouse model, mitochondrial abnormalities appear before symptoms of paralysis and MN degeneration manifest, suggesting that they are actively involved in disease pathogenesis. Morphological abnormalities in the form of swollen and vacuolated mitochondria of MNs were recognized early and have been described extensively in mutant SOD1 transgenic mice (Dal Canto and Gurney, 1994; Doi et al., 2008; Higgins et al., 2002; Higgins et al., 2003; Kong and Xu, 1998; Sasaki et al., 2005). Similar abnormalities have been observed in human sporadic ALS tissue (Hirano et al., 1984; Sasaki and Iwata, 1996, 2007), highlighting commonalities between sporadic and familial ALS pathogenesis. At the time of disease onset, mitochondrial respiration and ATP synthesis are defective in brain and spinal cord of G93A mutant SOD1 transgenic mice (Jung et al., 2002; Kirkinezos et al., 2005; Mattiazzi et al., 2002). In addition, mitochondria may be involved in the apoptotic execution of diseased MNs (Guegan et al., 2002; Oh et al., 2006; Pasinelli et al., 2000; Wootz et al., 2004), but nonapoptotic death has also been described in SOD1-fALS (Martin et al., 2007). While manipulation of proteins that modulate apoptosis have some beneficial impact on disease phenotype (Friedlander et al., 1997; Gould et al., 2006; Kieran et al., 2007; Vukosavic et al., 2000; Wootz et al., 2006), in some cases, inhibiting caspase-mediated apoptotic pathways had no effect (Kang et al., 2003). Defects in axonal transport of mitochondria have also been described in SOD1-fALS. These abnormalities are likely to play a role in impaired mitochondrial localization at crucial cellular sites, such as synapses and Ranvier nodes, thus contributing to the denervation process (De Vos et al., 2008; Magrane and Manfredi, 2009; Shi et al., 2009).
Table 1.
Reported mitochondrial abnormalities in ALS. The types of mitochondrial changes and the model system or patient's tissue, in which they were found, are indicated. The findings listed at the bottom of the table refer to mitochondrial changes induced by mutant SOD1 specifically targeted to mitochondria. NO, nitric oxide; ETC, electron transfer chain; lysRS, lysyl-tRNA sythetase; UCP3, uncoupling protein 3.
| Mitochondrial abnormalities | Tissue and disease model | References |
|---|---|---|
| Swollen and vacuolated mitochondria | MN of mutant SOD1 mice | Dal Canto and Gurney, 1994; Kong and Xu, 1998; Higgins et al., 2002; Higgins et al., 2003; Sasaki et al., 2005; Doi et al., 2008 |
| Aggregated and swollen mitochondria | Post-mortem sporadic ALS spinal cord | Sasaki and Iwata, 1996; Sasaki and Iwata, 2007 |
| Swollen and fragmented mitochondria | N2A and NSC34 cells | Menzies et al., 2002; Magrane et al., 2009 |
| Decreased oxygen consumption | Brain, spinal cord, G93A SOD1 mice | Jung et al., 2002; Kirkinezos et al., 2005; Mattiazzi et al., 2002 |
| Decreased ATP synthesis | Brain, spinal cord, G93A SOD1 mice | Mattiazzi et al., 2002 |
| Reduced ETC activity, respiration, mitochondrial redox state alteration | NSC34 cells | Ferri et al., 2006 |
| Reduced ETC activity, respiration, ROS production | Mutant G93A mice, primary astrocytes | Cassina et al., 2008 |
| Reduced ETC activity, complex IV | Brain, spinal cord, G93A SOD1 mice, G93A×CCS mice | Jung et al., 2002; Mattiazzi et al., 2002; Kirkinezos et al., 2005; Son et al., 2007 |
| Complex IV assembly defect | G93A×CCS mice | Son et al., 2008 |
| Decreased complex II, complex IV activities | NSC34 cells | Menzies et al., 2002 |
| Decreased complex IV activity involving NO | NSC34 cells | Arciello et al., 2009 |
| Apoptosis activation | Mutant SOD1 mice, N2A cells | Pasinelli et al., 2000; Guegan et al., 2002; Wootz et al., 2004; Oh et al., 2006 |
| Mitochondrial axonal transport defect | Mutant G93A mice, primary motor neurons | De Vos et al., 2007 |
| Mislocalization of mitochondria | Mutant G93A mice, motor axons | Sotelo-Silveira et al., 2009 |
| Mutant SOD1 aggregation in mitochondria | Mutant SOD1 mice, brain, spinal cord | Higgins et al., 2001; Jaarsma et al., 2001; Okado-Matsumoto and Fridovich, 2001; Mattiazzi et al., 2002; Higgins et al., 2002; Liu et al., 2004; Pasinelli et al., 2004; Vijayvergiya et al., 2005; Deng et al., 2006; Vande Velde et al., 2008 |
| Mutant SOD1 aggregation in mitochondria | NSC 34 cells | Ferri et al., 2006; Cozzolino et al., 2009 |
| Mitochondrial protein import of lysRS | COS cells | Kawamata et al., 2008 |
| Calcium uptake defect | G93A and G85R mice, brain, spinal cord | Damiano et al., 2006 |
| Prolonged calcium-induced membrane potential depolarization | Mutant SOD1 mice, motor nerve terminals | Nguyen et al., 2009 |
| Calcium uptake defect | G93A mice, brainstem slice | Jaiswal and Keller, 2009 |
| Impaired calcium stores in mitochondria | SH-SY5Y cells | Jaiswal et al., 2009 |
| Mitochondrial calcium handling defect | G93A mice, muscle | Zhou et al., 2009 |
| Decreased respiratory chain activity | ALS patients, skeletal muscle | Wiedemann et al., 1998; Vielhaber et al., 1999; Vielhaber et al., 2000 |
| UCP3 upregulation | ALS patients, G93A mice, muscle | Dupuis et al., 2003 |
| Oxidative damage, SOD1 enzyme upregulation | Mutant SOD1 mice, skeletal muscle | Mahoney et al., 2006 |
| Mitochondrial morphological abnormalities | Mutant SOD1 mice with muscle specific SOD1 expression | Dobrowolny et al., 2008 |
| Mitochondrial abnormalities | System & SOD1 targeted to mitochondria | References |
| Induction of apoptosis | N2A cells, SOD1 targeted to matrix | Takeuchi et al., 2002 |
| Axonal transport defect, mitochondrial depolarization | NSC34 cells, SOD1 targeted to IMS | Magrane et al., 2009 |
| Abnormal morphology, caspase-3 activation | NSC34 cells, SOD1 targeted to IMS | Cozzolino et al., 2009 |
Mitochondrial defects are also present in muscle, where weakness and atrophy develop as a result of MN denervation. In ALS patients, decreased respiratory chain activity in skeletal muscle was reported (Vielhaber et al., 2000; Vielhaber et al., 1999; Wiedemann et al., 1998). Up-regulation of uncoupling protein 3 in the skeletal muscle, but not spinal cord, implied a muscular mitochondria specific metabolic defect from human and SOD1-fALS mouse model (Dupuis et al., 2003). Oxidative damage and compensatory SOD1 enzyme upregulation was also observed in mutant SOD1 skeletal muscle (Mahoney et al., 2006). Moreover, transgenic mice expressing mutant SOD1 exclusively in the skeletal muscle develop progressive muscle weakness, pathologically characterized by alterations in the contractile apparatus and mitochondrial dysfunction (Dobrowolny et al., 2008). These reports indicate that defective muscle mitochondria play a direct role in muscle degeneration during ALS progression.
4. How does SOD1 affect mitochondria?
SOD1, which is a predominantly cytosolic protein, also localizes to mitochondria (Higgins et al., 2002; Higgins et al., 2001; Jaarsma et al., 2001; Mattiazzi et al., 2002; Okado-Matsumoto and Fridovich, 2001). The consensus on the sub-mitochondrial localization is that SOD1 is in the intermembrane space (IMS); however, mutant SOD1 accumulation on the outer membrane (OM) (Liu et al., 2004; Pasinelli et al., 2004; Vande Velde et al., 2008) and matrix (Vijayvergiya et al., 2005) has also been reported.
Recently, we reported that mammalian SOD1 localization in mitochondria is dependent on oxygen concentration and its interaction with its copper chaperone CCS (Kawamata and Manfredi, 2008). CCS is imported by a disulfide relay system in a respiratory chain dependent manner, and mitochondrial CCS levels modulate SOD1 levels in mitochondria (Kawamata and Manfredi, 2008; Reddehase et al., 2009). Mutant SOD1 is relatively insensitive to physiological regulation and dominated by misfolding and aggregation (Kawamata and Manfredi, 2008) that are likely to involve intermolecular disulfide linkages (Cozzolino et al., 2009; Deng et al., 2006; Ferri et al., 2006).
Wild type SOD1 in the IMS may provide antioxidant activity to remove superoxide produced as a result of respiratory chain activity on the IMS side, which is inaccessible to the matrix residing MnSOD. On the other hand, mitochondrial localization of mutant SOD1 has been correlated with detrimental consequences on mitochondrial function (Ferri et al., 2006; Son et al., 2007). Targeting mutant SOD1 specifically to the IMS also resulted in mitochondrial dysfunction in cultured neuronal cells (Cozzolino et al., 2009; Magrane et al., 2009), while targeting to the matrix caused apoptosis (Takeuchi et al., 2002). The targets of mutant SOD1 in mitochondria are not fully characterized, but defects in the activity of complex IV of the respiratory chain have been consistently described (Jung et al., 2002; Mattiazzi et al., 2002; Son et al., 2008; Son et al., 2007). These defects could be attributed to impaired association of cytochrome c with the inner membrane (Kirkinezos et al., 2005), competition for mitochondrial copper supply between mutant SOD1 and complex IV (Horn et al., 2008), or inhibition by excess NO production (Arciello et al., 2009).
Mutant SOD1 could also cause mitochondrial damage from the outside, because the OM houses many proteins important for mitochondrial morphology, dynamics, transport, and protein import. Recent studies showed impaired mitochondrial transport along axons causing mitochondrial dysfunction and mislocalization (De Vos et al., 2007; Magrane et al., 2009; Sotelo-Silveira et al., 2009). These defects could result from aberrant interactions between mutant SOD1 and motor and cargo adapter complexes, such as dynein (Zhang et al., 2007). Other examples of potential interference of mutant SOD1 on the OM include interactions with the anti-apoptotic protein Bcl2 (Pasinelli et al., 2004) and the precursor of mitochondrial lysyl-tRNA synthetase (Kawamata et al., 2008). It has also been proposed, but not yet demonstrated, that accumulation of mutant SOD1 on the OM might interfere with the import of mitochondrial proteins (Boillee et al., 2006a; Liu et al., 2004; Vande Velde et al., 2008). Figure 1 summarizes the putative sites and mechanisms of action of mutant SOD1 on mitochondria.
Figure 1.

Mitochondrial abnormalities in SOD1-fALS.
The potential toxic effects of mutant SOD1 to mitochondria (indicated by black jagged symbols) are numerous. See also table 1 for a detailed list of reported mitochondrial abnormalities. Mitochondrial morphological abnormalities, including fragmentation and swelling due to expansion of the IMS, may involve interactions with proteins involved in mitochondrial fusion and fission, protein import, or Bcl proteins. Abnormalities of axonal transport of mitochondria along microtubule axes may result from mutant SOD1 interference with motors and cargo adaptors. Mutant SOD1 accumulation in mitochondria can affect availability of copper necessary for cuproenzymes. Mutant SOD1 in mitochondria can induce excess ROS and NO production. Mitochondrial mutant SOD1 can reduce association of cytochrome c with the IM, and enhance its release thereby activating caspase-dependent cell death. Mutant SOD1 reduces mitochondrial ATP synthesis, respiration, and electron transport chain (ETC) complex activities, especially complex IV. Decreased mitochondrial membrane potential can impair calcium uptake through the mitochondrial uniporter. Mutant SOD1 can causes sensitivity to mPTP opening resulting in release of ions and solutes.
5. Mitochondrial calcium handling defects in ALS
Mitochondrial dysfunction in mutant SOD1 mice can affect bioenergetics in many ways. In addition to deficient ATP output and increased ROS production, a particularly severe consequence of defective mitochondrial function is impaired calcium handling (Fig. 1). There are several lines of evidence that mitochondrial calcium handling is impaired in fALS. Our group has demonstrated defective calcium uptake in isolated mitochondria from mutant SOD1 affected tissues, such as brain and spinal cord, but not in unaffected liver (Damiano et al., 2006). Recently, abnormal calcium-induced mitochondrial membrane depolarization has been observed in motor terminals of transgenic mice (Nguyen et al., 2009). In addition, mitochondrial calcium handling defects have been shown in organotypic brainstem slices (Jaiswal and Keller, 2009) and cultured neuronal cells (Jaiswal et al., 2009). Mitochondrial calcium handling impairment could contribute to abnormally high intracellular calcium levels (Carri et al., 1997; Kruman et al., 1999) and makes ALS MNs vulnerable to degeneration (Kim et al., 2007; Kim et al., 2002). Consistent with the involvement of muscle mitochondria in fALS, in skeletal muscle of mutant SOD1 mice, depolarized mitochondria were localized to neuromuscular junctions, and were accompanied by hyperactive calcium release from the sarcoplasmic reticulum (Zhou et al., 2009).
In the following sections, we will outline the role of mitochondria in intracellular calcium homeostasis and the potential detrimental effects of impaired mitochondrial calcium handling in ALS.
6. Role of mitochondria in intracellular calcium homeostasis
Calcium is a universal intracellular signaling molecule that regulates many pathways critical to cell survival. This versatile intracellular signal for control of physiological processes is generated by transient elevation of cytosolic calcium (Clapham, 1995). For this purpose, cells maintain a tightly controlled resting cytosolic free calcium concentration of ∼100nM by extruding excess calcium by plasma membrane pumps and exchangers, and by compartmentalizing calcium in the endoplasmic reticulum (ER; 100-800μM range) and mitochondria (Fig. 2).
Figure 2.
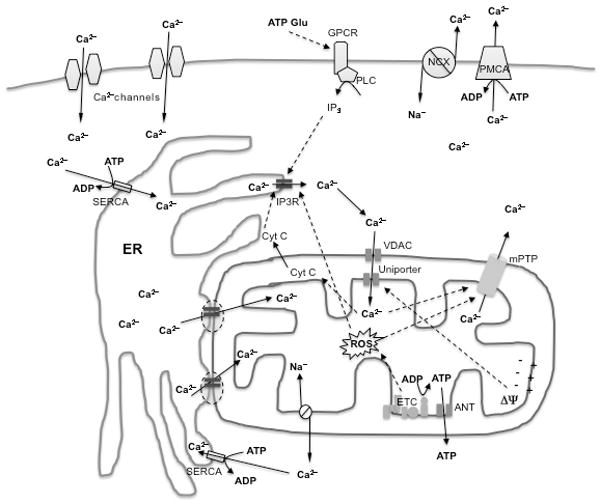
Mechanisms of intracellular calcium homeostasis and signaling.
Calcium enters cells from the extracellular space via calcium channels, including store-operated, ligand- and voltage-gated channels. In addition, intracellular calcium rises in response to metabotropic receptor stimulation, for example by glutamate or ATP, and production of IP3, which activates IP3 receptors (IP3R) or ryanodine receptors (not shown in figure) on the ER. IP3R activation induces calcium release from the ER stores, which contain high calcium concentrations because of the activity of SERCA pumps. Calcium is taken up by mitochondria through the membrane potential-dependent activity of the uniporter. Calcium transfer from ER to mitochondria can occur directly through “hotspots” localized on juxtaposed ER/mitochondrial membranes (indicated by dashed ovals). Mitochondrial calcium is extruded by the exchange with sodium by the mitochondrial Na/Ca exchanger. Under mitochondrial permeability transition conditions, stimulated by high calcium and reactive oxygen species (ROS), calcium, along with other solutes, is released through the mPTP. Cytosolic calcium is extruded by the Na/Ca exchanger (NCX) and the plasma membrane calcium ATPase (PMCA).
Mitochondria at rest have low free calcium (∼100nM) and at baseline cytosolic calcium concentration is below the setpoint for mitochondrial uptake. However, mitochondria can take up calcium up to millimolar concentrations through the activity of the calcium uniporter, when cytosolic calcium rises significantly in the micromolar range (Montero et al., 2000). Calcium uptake through the uniporter is dependent upon mitochondrial membrane potential, and it is accompanied by transient mitochondrial depolarization (Duchen, 2000).
Mitochondrial calcium uptake increases oxidative phosphorylation under physiological conditions (Jouaville et al., 1999) through activation of calcium-sensitive dehydrogenases of the Krebs cycle and pyruvate dehydrogenase (Hansford, 1991). Thus, it is speculated that calcium transfer from cytosol to mitochondria acts as a signal coupling ATP supply with demand. Calcium is also critical for the regulation of mitochondrial movement along axons. Neuronal mitochondria transition between mobile and stationary states in response to intracellular calcium (Yi et al., 2004). It is thought that this mechanism helps retaining mitochondria in presynaptic terminals and postsynaptic dendritic spines, where calcium influx is dynamic and homeostasis maintenance is heavily required. In these regions, mitochondria directly buffer calcium and provide ATP for plasma membrane calcium exchangers. It was recently determined that the EF-hand calcium binding domain of Miro1, a member of the subfamily of mitochondrial rho GTPases, is a calcium sensor for the regulation of mitochondrial motility that may serve to localizing mitochondria at sites of demand (Macaskill et al., 2009; Saotome et al., 2008; Wang and Schwarz, 2009). As discussed above, mutant SOD1 impedes correct mitochondrial transport. Whether mutant SOD1 hinders the normal calcium signaling to Miro1 and its interactions with other components of the transport machinery, such as kinesin or dynein motors and cargo adaptors, thereby disrupting the regulation of mitochondrial motility, remain to be further explored.
7. Mitochondrial calcium overload and cell death
Under physiological conditions, mitochondrial calcium levels are maintained through calcium cycling between cytosol and matrix through the uniporter and Na/Ca exchanger activities. In addition, large amounts of calcium can be found in the form of precipitates in the matrix (Chinopoulos et al., 2003). Calcium overload occurs when mitochondria are exposed to matrix calcium concentration exceeding their capacity, and may result in membrane de-energization, ROS production (Fig. 2), and ultimately permeability transition (mPT). mPT is a sudden increase of inner membrane permeability to ions and solutes, resulting in dissipation of membrane potential, loss of ATP synthesis and ion homeostasis, and diffusion of solutes (Hunter and Haworth, 1979).
Mitochondrial calcium overload is associated with activation of cell death pathways (Bernardi, 1999; Takuma et al., 2004) and is observed in many pathological conditions (Honda and Ping, 2006; Norenberg and Rao, 2007). The mechanisms of calcium overload are not entirely clear, but it is likely that defects in mitochondrial bioenergetics, such as those found in mutant SOD1 mitochondria, can decrease their ability to handle calcium and sensitize them to overload. Theoretically, defects of the mitochondrial Na/Ca exchanger could also be involved in causing calcium overload in ALS mitochondria, although this putative mechanism has not yet been directly explored. Another potential factor contributing to calcium overload could be the functional and physical link between mitochondria and ER. Transfer of calcium from the large stores in the ER to mitochondria depends on the position of the two organelles, and is thought to occur at calcium hotspots, which are sites where ER and mitochondrial membranes are in close contact (Rizzuto et al., 1999). It has been shown that shorter distance between the two organelles results in increased mitochondrial calcium accumulation upon ER calcium release and cell death (Csordas et al., 2006). Since mutant SOD1 accumulates in ER (Kikuchi et al., 2006; Urushitani et al., 2006) and mitochondrial (Liu et al., 2004) membranes, in mutant cells the structure of these calcium hotspots could be altered leading to abnormal calcium signaling.
Activation of cell death by mitochondrial calcium overload involves the opening of the mitochondrial permeability transition pore (mPTP), release of cytochrome c, and downstream activation of apoptosis. Cytochrome c released into the cytosol can propagate apoptotic signaling by binding to the inositol triphosphate receptor on the ER to desensitize the autoinhibition of the receptor by calcium, and cause further calcium release from ER stores (Boehning et al., 2003). Ablation of cyclophilin D (CypD) delays the occurrence of mPT (Basso et al., 2005) and has a protective effect against neuronal death in models of ischemia (Baines et al., 2005; Schinzel et al., 2005). In ALS, it was reported that the deletion of CypD in the G93A mutant SOD1 mice delays disease onset and extends lifespan (Martin et al., 2009). Furthermore, two studies in mutant SOD1 mice using the immunosuppressant cyclosporin A, which binds to CypD to inhibit mPTP, suggests that inhibition of mPTP may be of benefit to ALS (Keep et al., 2001; Kirkinezos et al., 2004).
Another mechanism whereby calcium can participate to the activation of cell death pathways is by stimulating mitochondrial reactive oxygen species (ROS) production. ROS can be damaging to proteins, lipids, and DNA, and oxidative stress is a common feature of aging-related diseases, including ALS (Lin and Beal, 2006). Mitochondrial dysfunction increases ROS production (Wei et al., 1998) and so does calcium influx in mitochondria (Petrosillo et al., 2004). High mitochondrial calcium can enhance cytochrome c release through a mechanism involving ROS-mediated oxidation of cardiolipin (Iverson and Orrenius, 2004). Oxidized cardiolipin changes its physical properties (Vercesi et al., 1997), resulting in cytochrome c release. Lipid peroxidation (Mattiazzi et al., 2002) and dissociation of cytochrome c from the mitochondrial inner membrane (Kirkinezos et al., 2005) have been reported in mutant SOD1 mice. Finally, intracellular calcium dysregulation may also be linked to necrotic death of neurons (Nicholls et al., 1999).
8. Calcium and excitotoxicity in ALS
ALS mitochondria are susceptible to calcium overload and calcium-induced damage that lead to MN death (Fig. 3). Conditions that favor elevated intracellular calcium levels contribute to mitochondrial toxicity. Intracellular calcium rises in MNs primarily through the activation of voltage gated channels or ionotropic receptors. Glutamate is the primary neurotransmitter involved in MN excitation. Glutamate receptor over-activation resulting in extended elevation of cytosolic calcium is one of the leading contenders as the triggering cause of ALS (Rothstein, 1996). In support of this hypothesis, increased glutamate levels were found in the cerebrospinal fluid of ALS patients (Rothstein et al., 1990), but other studies found it decreased in some areas of the CNS (Perry et al., 1987; Tsai et al., 1991).
Figure 3.
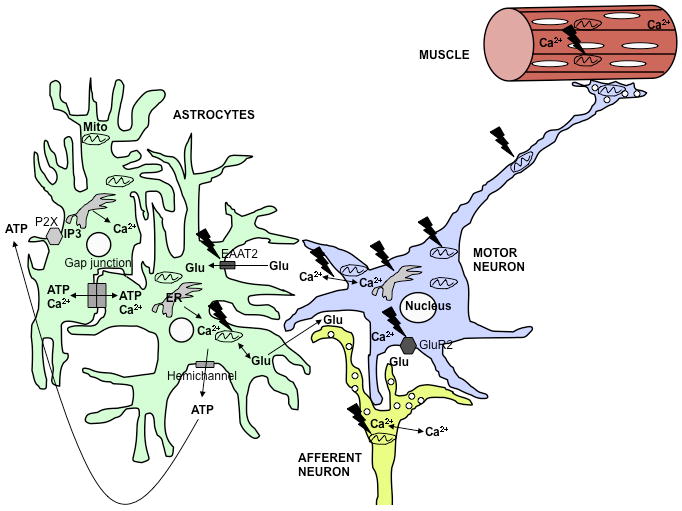
Involvement of calcium dysregulation in ALS.
In the ALS spinal cord, multiple cell types, including MNs and astrocytes, are involved in the disease. The black jagged symbols indicate potential sites of mutant SOD1 toxicity. MNs are particularly vulnerable to increased cytosolic calcium due to low levels of calcium binding proteins and enhanced permeability to calcium through AMPA receptors that contain few GluR2 subunits. Calcium uptake defects mitochondria can cause extended intracellular calcium exposure. In MNs, this may contribute to excitotoxicity, impaired mitochondrial transport, permeability transition, and apoptosis. Mitochondrial bioenergetic dysfunction contributes to increased intracellular calcium, thereby propagating a vicious cycle of calcium dysregulation and cellular damage. Additionally, defective mitochondrial calcium uptake in afferent neurons or astrocytes may result in excessive glutamate release in excitatory synapses. ALS astrocytes have low glutamate transporter (EAAT2) expression, which exacerbates glutamate excitotoxicity. Increased intracellular calcium in astrocytes can also result in excessive propagation of intercellular calcium waves, either through gap junctions or through opening of hemichannels. The resulting extracellular release of modulatory molecules, such as ATP, induces intracellular calcium rise through activation of metabotropic receptors on neighboring cells, and propagates the wave. Furthermore, muscle mitochondrial calcium handling defects can also play a role in muscle degeneration.
MNs express ionotropic (AMPA, NMDA, kainite) and metabotropic glutamate receptors, but are particularly sensitive to glutamate excitotoxicity exerted through AMPA receptors (Carriedo et al., 1996) with relatively few GluR2 subunits, which causes increased calcium permeability. Defective RNA editing of GluR2 has been shown to increase calcium permeability in MNs (Kawahara et al., 2004). Interestingly, GluR2 expression in MNs appears to be regulated by astrocytes and provides protection against glutamate/AMPA-mediated excitotoxicity. This protective effect is lost when mutant SOD1 is expressed in astrocytes (Van Damme et al., 2007).
Alternatively, mutant SOD1 may expose MNs to high intracellular calcium levels by impairing calcium extrusion from the cytosol. It was found that the influx of calcium through AMPA receptors or voltage-activated calcium channels in mutant SOD1 MNs did not differ from controls, but mutant SOD1 MNs had slower cytosolic calcium clearance resulting in higher intracellular calcium (Guatteo et al., 2007).
Mutant SOD1 expressing astrocytes also contribute to glutamate excitotoxicity (Fig. 3). Reduced levels of astrocytic glutamate transporters (GLT1 and EAAT2, for mouse and human, respectively) have been consistently reported, both in ALS patients and fALS mouse models (Bendotti et al., 2001; Howland et al., 2002; Rothstein et al., 1995). Reduced astrocytic glutamate transporters are thought to cause persistent glutamate presence in the ALS synapse, since these transporters partake in the majority of glutamate clearance in the CNS. Decreased levels of astrocytic glutamate transporters may be caused by oxidative damage of the protein or by caspase-mediated cleavage (Boston-Howes et al., 2006; Trotti et al., 1999).
9. Role of astrocytic calcium in ALS
Altered calcium handling in mutant SOD1 mitochondria has been demonstrated by biochemical methods in brain or spinal cord preparations, where cell type specific defects cannot be isolated. As mentioned earlier, some studies have shown mitochondrial calcium abnormalities in isolated MNs. However, the role of glial cells in SOD1-fALS is well established and it is necessary to consider the pathogenic potential of calcium dysregulation in these cells. Astrocyte-MN co-culture studies have shown toxic effects of mutant SOD1 expressing astrocytes on MN viability. This toxicity involved impaired calcium homeostasis maintenance in MN (Bilsland et al., 2008), as well as excessive productions of ROS (Cassina et al., 2008; Marchetto et al., 2008). However, it is not clear yet whether calcium signaling abnormalities in mutant SOD1 astrocytes are responsible for the changes observed in co-cultured MNs, because intracellular calcium homeostasis in general, and mitochondrial calcium handling in particular, have not been investigated in mutant SOD1 astrocytes.
Considering the fundamental role of astrocytes in the “tripartite” synapse in regulating neuronal transmission (Perea and Araque, 2005), there are several astrocytic functions that could be affected in ALS, resulting in MN toxicity. In the CNS, calcium can be transmitted across astrocytes, in the form of inter-cellular calcium waves. Recently, rare events of inter-cellular calcium waves originating from astrocytes near amyloid-β plaques were demonstrated in a model of Alzheimer's disease, but not in control mice. This was the first example of inter-cellular calcium wave recording in vivo, and suggested a pathogenic signaling role for these events (Kuchibhotla et al., 2009). This kind of novel neurophysiological studies has not yet been applied to ALS. By buffering calcium, mitochondria can regulate the speed of propagation of intracellular calcium waves (Boitier et al., 1999) and thereby modulate the release of signaling molecules, such as ATP, IP3, and calcium itself, that determine inter-cellular propagation of the stimulus. Furthermore, glutamate release from astrocytes by exocytosis is calcium-dependent and is directly regulated by mitochondrial function (Reyes and Parpura, 2008). Therefore, mitochondrial bioenergetic impairment in mutant SOD1 mitochondria (Cassina et al., 2008) might result in a defect in calcium wave buffering and accelerated waves that propagate among astrocytes as well as increased glutamate release, resulting in excitotoxicity to MNs (Fig. 3).
10. Perspectives and conclusions
Despite the wealth of evidence supporting the involvement of mitochondria in ALS, the question still remains of whether mitochondrial dysfunction represents a trigger or a target. There are several possibilities to start addressing this issue experimentally. One way to approach the direct role of mitochondrial dysfunction in the pathogenesis of SOD1-linked fALS is to direct the mutant protein specifically to mitochondria. These studies have been performed in cultured neuronal cells (Cozzolino et al., 2009; Magrane et al., 2009), as mentioned above, and they are also being currently performed by our group in transgenic mice. One limitation of this approach is that it only addresses the pathogenic role of mutant SOD1 localized in mitochondria and it may fail to detect the pathogenic contribution of mitochondrial defects originating outside of the organelles. For example, mitochondria may be affected by extra-mitochondrial mutant SOD1 interfering with axonal transport, mitochondrial protein import, or with intracellular signaling modulating mitochondrial biogenesis. Nevertheless, investigating ALS models initiating in mitochondria may help determine the need to include therapies that specifically target mitochondrial dysfunction.
Our understanding of mitochondrial involvement in ALS is currently limited mostly to SOD1-linked fALS, and it is not certain that the pathogenic mechanisms involving mutant SOD1 in mitochondria may be extended to other forms of fALS or sporadic ALS. The latter represent the overwhelming majority of ALS cases, but our ability to investigate the mitochondrial involvement in sporadic ALS is clearly hampered by the lack of understanding of the underlying causes and of appropriate disease models. Most of the available information originates from post-mortem studies, where mitochondrial abnormalities may represent a terminal feature of cell degeneration, without etiological significance. The fast developing human induced pluripotent stem (iPS) cell technology (Di Giorgio et al., 2007) will provide ALS researchers with valuable tools to investigate mitochondrial dysfunction in cells derived from patients affected by both sporadic and hereditary forms of the disease.
Even in the context of well-characterized disease models, such as mutant SOD1 mice, there are still several challenges that need to be faced to understand the contribution of mitochondrial dysfunction. We still don't know if cellular ATP depletion could arise from defective mitochondrial ATP synthesis, and whether this has any pathogenic significance, or whether other aspects of bioenergetic failure, such as defective calcium handling, have a more critical role. In this review, we have highlighted the potentially detrimental consequences of impaired mitochondrial calcium handling in neurons and other cells involved in ALS. More work is needed in the SOD1-fALS and other models of ALS to address the role of mitochondria in specific cell types. It is clear that in ALS glial cell toxicity affects mitochondria in MNs, but whether glial mitochondria are also defective and whether this causes toxicity to MNs remain to be investigated.
Understanding the molecular and biochemical basis of mitochondrial dysfunction in ALS is in tall order, but it is of great importance, because it may explain the mechanisms of mitochondrial toxicity and cell death. ATP depletion would suggest that cellular demise might depend on necrotic mechanisms, whereas calcium dysregulation would indicate that cells might degenerate through apoptotic processes. Clearly, identifying the different modalities of cell death would be useful for deciding therapeutic priorities and strategies.
Acknowledgments
This work is supported by grants R01-NS051419, R01-NS062055, Muscular Dystrophy Association, and the Robert Packard ALS Research Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arciello M, Capo CR, Cozzolino M, Ferri A, Nencini M, Carri MT, Rossi L. Inactivation of cytochrome c oxidase by mutant SOD1s in mouse motoneuronal NSC-34 cells is independent from copper availability but is because of nitric oxide. J Neurochem. 2009;112(1):183–192. doi: 10.1111/j.1471-4159.2009.06441.x. [DOI] [PubMed] [Google Scholar]
- Bacman SR, Bradley WG, Moraes CT. Mitochondrial involvement in amyotrophic lateral sclerosis: trigger or target? Mol Neurobiol. 2006;33(2):113–131. doi: 10.1385/MN:33:2:113. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434(7033):658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280(19):18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Tortarolo M, Suchak SK, Calvaresi N, Carvelli L, Bastone A, Rizzi M, Rattray M, Mennini T. Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J Neurochem. 2001;79(4):737–746. doi: 10.1046/j.1471-4159.2001.00572.x. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79(4):1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bilsland LG, Nirmalananthan N, Yip J, Greensmith L, Duchen MR. Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J Neurochem. 2008;107(5):1271–1283. doi: 10.1111/j.1471-4159.2008.05699.x. [DOI] [PubMed] [Google Scholar]
- Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5(12):1051–1061. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006a;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006b;312(5778):1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Boitier E, Rea R, Duchen MR. Mitochondria exert a negative feedback on the propagation of intracellular Ca2+ waves in rat cortical astrocytes. J Cell Biol. 1999;145(4):795–808. doi: 10.1083/jcb.145.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boston-Howes W, Gibb SL, Williams EO, Pasinelli P, Brown RH, Jr, Trotti D. Caspase-3 cleaves and inactivates the glutamate transporter EAAT2. J Biol Chem. 2006;281(20):14076–14084. doi: 10.1074/jbc.M600653200. [DOI] [PubMed] [Google Scholar]
- Carri MT, Ferri A, Battistoni A, Famhy L, Gabbianelli R, Poccia F, Rotilio G. Expression of a Cu,Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett. 1997;414(2):365–368. doi: 10.1016/s0014-5793(97)01051-x. [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996;16(13):4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de Leon A, Robinson KM, Mason RP, Beckman JS, Barbeito L, Radi R. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. 2008;28(16):4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Starkov AA, Fiskum G. Cyclosporin A-insensitive permeability transition in brain mitochondria: inhibition by 2-aminoethoxydiphenyl borate. J Biol Chem. 2003;278(30):27382–27389. doi: 10.1074/jbc.M303808200. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 1995;80(2):259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Cozzolino M, Pesaresi MG, Amori I, Crosio C, Ferri A, Nencini M, Carri MT. Oligomerization of mutant SOD1 in mitochondria of motoneuronal cells drives mitochondrial damage and cell toxicity. Antioxid Redox Signal. 2009;11(7):1547–1558. doi: 10.1089/ars.2009.2545. [DOI] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174(7):915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;145(6):1271–1279. [PMC free article] [PubMed] [Google Scholar]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Flint Beal M, Manfredi G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006;96(5):1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007;16(22):2720–2728. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, Lukas T, Dal Canto MC, O'Halloran TV, Siddique T. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A. 2006;103(18):7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10(5):608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, Belia S, Wannenes F, Nicoletti C, Del Prete Z, Rosenthal N, Molinaro M, Protasi F, Fano G, Sandri M, Musaro A. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008;8(5):425–436. doi: 10.1016/j.cmet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Doi K, Nakano T, Kitayama M, Watanabe Y, Yasui K, Fukada Y, Morino S, Kaidoh T, Nakashima K, Inoue T. Mitochondrial changes in motor neurons of homozygotes of leucine 126 TT deletion SOD1 transgenic mice. Neuropathology. 2008;28(3):269–276. doi: 10.1111/j.1440-1789.2007.00876.x. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and Ca(2+)in cell physiology and pathophysiology. Cell Calcium. 2000;28(5-6):339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- Dupuis L, di Scala F, Rene F, de Tapia M, Oudart H, Pradat PF, Meininger V, Loeffler JP. Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. FASEB J. 2003;17(14):2091–2093. doi: 10.1096/fj.02-1182fje. [DOI] [PubMed] [Google Scholar]
- Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, Rotilio G, Valentine JS, Carri MT. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A. 2006;103(37):13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander RM, Brown RH, Gagliardini V, Wang J, Yuan J. Inhibition of ICE slows ALS in mice. Nature. 1997;388(6637):31. doi: 10.1038/40299. [DOI] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26(34):8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guatteo E, Carunchio I, Pieri M, Albo F, Canu N, Mercuri NB, Zona C. Altered calcium homeostasis in motor neurons following AMPA receptor but not voltage-dependent calcium channels' activation in a genetic model of amyotrophic lateral sclerosis. Neurobiol Dis. 2007;28(1):90–100. doi: 10.1016/j.nbd.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Guegan C, Vila M, Teismann P, Chen C, Onteniente B, Li M, Friedlander RM, Przedborski S. Instrumental activation of bid by caspase-1 in a transgenic mouse model of ALS. Mol Cell Neurosci. 2002;20(4):553–562. doi: 10.1006/mcne.2002.1136. [DOI] [PubMed] [Google Scholar]
- Hansford RG. Dehydrogenase activation by Ca2+ in cells and tissues. J Bioenerg Biomembr. 1991;23(6):823–854. doi: 10.1007/BF00786004. [DOI] [PubMed] [Google Scholar]
- Hervias I, Beal MF, Manfredi G. Mitochondrial dysfunction and amyotrophic lateral sclerosis. Muscle Nerve. 2006;33(5):598–608. doi: 10.1002/mus.20489. [DOI] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Ding H, Xu Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22(6):RC215. doi: 10.1523/JNEUROSCI.22-06-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Xu Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003;4(1):16. doi: 10.1186/1471-2202-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CMJ, Jung C, Gatha N, Xu ZS. Mutant Cu, Zn superoxide dismutase that causes amyotrophic lateral sclerosis (ALS) is in mitochondria in the central nervous system. Soc Neuroscience Abstr. 2001;31:580–585. [Google Scholar]
- Hirano A, Donnenfeld H, Sasaki S, Nakano I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984;43(5):461–470. doi: 10.1097/00005072-198409000-00001. [DOI] [PubMed] [Google Scholar]
- Honda HM, Ping P. Mitochondrial permeability transition in cardiac cell injury and death. Cardiovasc Drugs Ther. 2006;20(6):425–432. doi: 10.1007/s10557-006-0642-0. [DOI] [PubMed] [Google Scholar]
- Horn D, Al-Ali H, Barrientos A. Cmc1p is a conserved mitochondrial twin CX9C protein involved in cytochrome c oxidase biogenesis. Mol Cell Biol. 2008;28(13):4354–4364. doi: 10.1128/MCB.01920-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99(3):1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys. 1979;195(2):453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- Iverson SL, Orrenius S. The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch Biochem Biophys. 2004;423(1):37–46. doi: 10.1016/j.abb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol (Berl) 2001;102(4):293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- Jaiswal MK, Keller BU. Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol Pharmacol. 2009;75(3):478–489. doi: 10.1124/mol.108.050831. [DOI] [PubMed] [Google Scholar]
- Jaiswal MK, Zech WD, Goos M, Leutbecher C, Ferri A, Zippelius A, Carri MT, Nau R, Keller BU. Impairment of mitochondrial calcium handling in a mtSOD1 cell culture model of motoneuron disease. BMC Neurosci. 2009;10(1):64. doi: 10.1186/1471-2202-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A. 1999;96(24):13807–13812. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C, Higgins CMJ, Xu Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J Neurochem. 2002;83(3):535–545. doi: 10.1046/j.1471-4159.2002.01112.x. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Sanchez I, Jing N, Yuan J. Dissociation between neurodegeneration and caspase-11-mediated activation of caspase-1 and caspase-3 in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2003;23(13):5455–5460. doi: 10.1523/JNEUROSCI.23-13-05455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. Glutamate receptors: RNA editing and death of motor neurons. Nature. 2004;427(6977):801. doi: 10.1038/427801a. [DOI] [PubMed] [Google Scholar]
- Kawamata H, Magrane J, Kunst C, King MP, Manfredi G. Lysyl-tRNA synthetase is a target for mutant SOD1 toxicity in mitochondria. J Biol Chem. 2008;283(42):28321–28328. doi: 10.1074/jbc.M805599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata H, Manfredi G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum Mol Genet. 2008;17(21):3303–3317. doi: 10.1093/hmg/ddn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keep M, Elmer E, Fong KS, Csiszar K. Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res. 2001;894(2):327–331. doi: 10.1016/s0006-8993(01)02012-1. [DOI] [PubMed] [Google Scholar]
- Kieran D, Woods I, Villunger A, Strasser A, Prehn JH. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc Natl Acad Sci U S A. 2007;104(51):20606–20611. doi: 10.1073/pnas.0707906105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi H, Almer G, Yamashita S, Guegan C, Nagai M, Xu Z, Sosunov AA, McKhann GM, Przedborski S., 2nd Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A. 2006;103(15):6025–6030. doi: 10.1073/pnas.0509227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Im W, Kim S, Kim SH, Sung JJ, Kim M, Lee KW. Calcium-influx increases SOD1 aggregates via nitric oxide in cultured motor neurons. Exp Mol Med. 2007;39(5):574–582. doi: 10.1038/emm.2007.63. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim M, Kim SH, Sung JJ, Lee KW. Alteration in intracellular calcium homeostasis reduces motor neuronal viability expressing mutated Cu/Zn superoxide dismutase through a nitric oxide/guanylyl cyclase cGMP cascade. Neuroreport. 2002;13(9):1131–1135. doi: 10.1097/00001756-200207020-00012. [DOI] [PubMed] [Google Scholar]
- Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, Bradley WG, Moraes CT. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci. 2005;25(1):164–172. doi: 10.1523/JNEUROSCI.3829-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkinezos IG, Hernandez D, Bradley WG, Moraes CT. An ALS mouse model with a permeable blood-brain barrier benefits from systemic cyclosporine A treatment. J Neurochem. 2004;88(4):821–826. doi: 10.1046/j.1471-4159.2003.02181.x. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18(9):3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Pedersen WA, Springer JE, Mattson MP. ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp Neurol. 1999;160(1):28–39. doi: 10.1006/exnr.1999.7190. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323(5918):1211–1215. doi: 10.1126/science.1169096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43(1):5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61(4):541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, Hervias I, Henning MS, Damiano M, Kawamata H, Manfredi G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum Mol Genet. 2009;18(23):4552–4564. doi: 10.1093/hmg/ddp421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, Manfredi G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11(7):1615–1626. doi: 10.1089/ars.2009.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney DJ, Kaczor JJ, Bourgeois J, Yasuda N, Tarnopolsky MA. Oxidative stress and antioxidant enzyme upregulation in SOD1-G93A mouse skeletal muscle. Muscle Nerve. 2006;33(6):809–816. doi: 10.1002/mus.20542. [DOI] [PubMed] [Google Scholar]
- Marchetto MC, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3(6):649–657. doi: 10.1016/j.stem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol. 2009;218(2):333–346. doi: 10.1016/j.expneurol.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA, Golden WC. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol. 2007;500(1):20–46. doi: 10.1002/cne.21160. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277(33):29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZM, Dong L, Figlewicz DA, Shaw PJ. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002;125(Pt 7):1522–1533. doi: 10.1093/brain/awf167. [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Carnicero E, Cuchillo-Ibanez I, Albillos A, Garcia AG, Garcia-Sancho J, Alvarez J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat Cell Biol. 2000;2(2):57–61. doi: 10.1038/35000001. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL, Ward MW, Castilho RF. Excitotoxicity and mitochondria. Biochem Soc Symp. 1999;66:55–67. doi: 10.1042/bss0660055. [DOI] [PubMed] [Google Scholar]
- Nguyen KT, Garcia-Chacon LE, Barrett JN, Barrett EF, David G. The Psi(m) depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals. Proc Natl Acad Sci U S A. 2009;106(6):2007–2011. doi: 10.1073/pnas.0810934106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norenberg MD, Rao KV. The mitochondrial permeability transition in neurologic disease. Neurochem Int. 2007;50(7-8):983–997. doi: 10.1016/j.neuint.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh YK, Shin KS, Kang SJ. AIF translocates to the nucleus in the spinal motor neurons in a mouse model of ALS. Neurosci Lett. 2006;406(3):205–210. doi: 10.1016/j.neulet.2006.07.044. [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276(42):38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43(1):19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7(9):710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Houseweart MK, Brown RH, Jr, Cleveland DW. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97(25):13901–13906. doi: 10.1073/pnas.240305897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Glial calcium signaling and neuron-glia communication. Cell Calcium. 2005;38(3-4):375–382. doi: 10.1016/j.ceca.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Perry TL, Hansen S, Jones K. Brain glutamate deficiency in amyotrophic lateral sclerosis. Neurology. 1987;37(12):1845–1848. doi: 10.1212/wnl.37.12.1845. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Ruggiero FM, Pistolese M, Paradies G. Ca2+-induced reactive oxygen species production promotes cytochrome c release from rat liver mitochondria via mitochondrial permeability transition (MPT)-dependent and MPT-independent mechanisms: role of cardiolipin. J Biol Chem. 2004;279(51):53103–53108. doi: 10.1074/jbc.M407500200. [DOI] [PubMed] [Google Scholar]
- Reddehase S, Grumbt B, Neupert W, Hell K. The disulfide relay system of mitochondria is required for the biogenesis of mitochondrial Ccs1 and Sod1. J Mol Biol. 2009;385(2):331–338. doi: 10.1016/j.jmb.2008.10.088. [DOI] [PubMed] [Google Scholar]
- Reyes RC, Parpura V. Mitochondria modulate Ca2+-dependent glutamate release from rat cortical astrocytes. J Neurosci. 2008;28(39):9682–9691. doi: 10.1523/JNEUROSCI.3484-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Brini M, Chiesa A, Filippin L, Pozzan T. Mitochondria as biosensors of calcium microdomains. Cell Calcium. 1999;26(5):193–199. doi: 10.1054/ceca.1999.0076. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Excitotoxicity hypothesis. Neurology. 1996;4747(4 Suppl 2):S19–25. doi: 10.1212/wnl.47.4_suppl_2.19s. discussion S26. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65 1:S3–9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28(1):18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38(1):73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105(52):20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Iwata M. Dendritic synapses of anterior horn neurons in amyotrophic lateral sclerosis: an ultrastructural study. Acta Neuropathol (Berl) 1996;91(3):278–283. doi: 10.1007/s004010050426. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007;66(1):10–16. doi: 10.1097/nen.0b013e31802c396b. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Warita H, Murakami T, Shibata N, Komori T, Abe K, Kobayashi M, Iwata M. Ultrastructural study of aggregates in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2005;109(3):247–255. doi: 10.1007/s00401-004-0939-7. [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102(34):12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Gal J, Kwinter DM, Liu X, Zhu H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2009 doi: 10.1016/j.bbadis.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M, Leary SC, Romain N, Pierrel F, Winge DR, Haller RG, Elliott JL. Isolated cytochrome c oxidase deficiency in G93A SOD1 mice over-expressing CCS protein. J Biol Chem. 2008;283(18):12267–12275. doi: 10.1074/jbc.M708523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M, Puttaparthi K, Kawamata H, Rajendran B, Boyer PJ, Manfredi G, Elliott JL. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proc Natl Acad Sci U S A. 2007;104(14):6072–6077. doi: 10.1073/pnas.0610923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo-Silveira JR, Lepanto P, Elizondo MV, Horjales S, Palacios F, Martinez Palma L, Marin M, Beckman JS, Barbeito L. Axonal mitochondrial clusters containing mutant SOD1 in transgenic models of ALS. Antioxid Redox Signal. 2009;11(7):1535–1545. doi: 10.1089/ars.2009.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Kobayashi Y, Ishigaki S, Doyu M, Sobue G. Mitochondrial Localization of Mutant Superoxide Dismutase 1 Triggers Caspase-dependent Cell Death in a Cellular Model of Familial Amyotrophic Lateral Sclerosis. J Biol Chem. 2002;277(52):50966–50972. doi: 10.1074/jbc.M209356200. [DOI] [PubMed] [Google Scholar]
- Takuma K, Baba A, Matsuda T. Astrocyte apoptosis: implications for neuroprotection. Prog Neurobiol. 2004;72(2):111–127. doi: 10.1016/j.pneurobio.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2(5):427–433. doi: 10.1038/8091. [DOI] [PubMed] [Google Scholar]
- Tsai GC, Stauch-Slusher B, Sim L, Hedreen JC, Rothstein JD, Kuncl R, Coyle JT. Reductions in acidic amino acids and N-acetylaspartylglutamate in amyotrophic lateral sclerosis CNS. Brain Res. 1991;556(1):151–156. doi: 10.1016/0006-8993(91)90560-i. [DOI] [PubMed] [Google Scholar]
- Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85(1):94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9(1):108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Bogaert E, Dewil M, Hersmus N, Kiraly D, Scheveneels W, Bockx I, Braeken D, Verpoorten N, Verhoeven K, Timmerman V, Herijgers P, Callewaert G, Carmeliet P, Van Den Bosch L, Robberecht W. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci U S A. 2007;104(37):14825–14830. doi: 10.1073/pnas.0705046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008;105(10):4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercesi AE, Kowaltowski AJ, Grijalba MT, Meinicke AR, Castilho RF. The role of reactive oxygen species in mitochondrial permeability transition. Biosci Rep. 1997;17(1):43–52. doi: 10.1023/a:1027335217774. [DOI] [PubMed] [Google Scholar]
- Vielhaber S, Kunz D, Winkler K, Wiedemann FR, Kirches E, Feistner H, Heinze HJ, Elger CE, Schubert W, Kunz WS. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain. 2000;123(Pt 7):1339–1348. doi: 10.1093/brain/123.7.1339. [DOI] [PubMed] [Google Scholar]
- Vielhaber S, Winkler K, Kirches E, Kunz D, Buchner M, Feistner H, Elger CE, Ludolph AC, Riepe MW, Kunz WS. Visualization of defective mitochondrial function in skeletal muscle fibers of patients with sporadic amyotrophic lateral sclerosis. J Neurol Sci. 1999;169(1-2):133–139. doi: 10.1016/s0022-510x(99)00236-1. [DOI] [PubMed] [Google Scholar]
- Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25(10):2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukosavic S, Stefanis L, Jackson-Lewis V, Guegan C, Romero N, Chen C, Dubois-Dauphin M, Przedborski S. Delaying caspase activation by Bcl-2: A clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2000;20(24):9119–9125. doi: 10.1523/JNEUROSCI.20-24-09119.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136(1):163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei YH, Lu CY, Lee HC, Pang CY, Ma YS. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann N Y Acad Sci. 1998;854:155–170. doi: 10.1111/j.1749-6632.1998.tb09899.x. [DOI] [PubMed] [Google Scholar]
- Wiedemann FR, Winkler K, Kuznetsov AV, Bartels C, Vielhaber S, Feistner H, Kunz WS. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci. 1998;156(1):65–72. doi: 10.1016/s0022-510x(98)00008-2. [DOI] [PubMed] [Google Scholar]
- Wootz H, Hansson I, Korhonen L, Lindholm D. XIAP decreases caspase-12 cleavage and calpain activity in spinal cord of ALS transgenic mice. Exp Cell Res. 2006;312(10):1890–1898. doi: 10.1016/j.yexcr.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Wootz H, Hansson I, Korhonen L, Napankangas U, Lindholm D. Caspase-12 cleavage and increased oxidative stress during motoneuron degeneration in transgenic mouse model of ALS. Biochem Biophys Res Commun. 2004;322(1):281–286. doi: 10.1016/j.bbrc.2004.07.118. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11(3):251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Weaver D, Hajnoczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol. 2004;167(4):661–672. doi: 10.1083/jcb.200406038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Strom AL, Fukada K, Lee S, Hayward LJ, Zhu H. Interaction between familial ALS-linked SOD1 mutants and the dynein complex: Implications of retrograde axonal transport in ALS. J Biol Chem. 2007;282(22):16691–16699. doi: 10.1074/jbc.M609743200. [DOI] [PubMed] [Google Scholar]
- Zhou J, Yi J, Fu R, Liu E, Siddique T, Rios E, Deng HX. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J Biol Chem. 2009;285(1):705–712. doi: 10.1074/jbc.M109.041319. [DOI] [PMC free article] [PubMed] [Google Scholar]