

Short Summary
Xeroderma pigmentosum (XP) is a genetic disease that results in poor genomic defense from endogenous and exogenous stressors. Patients with mutations in the XPC and XPA genes exhibit cochlear hearing loss and to date, the underlying molecular mechanism is unknown. However, recent evidence suggests that the cochlea experiences persistent oxidative stress under normal conditions. In the current study, XPC and XPA gene products were purified from the cochlea and quantitative polymerase chain reaction was used to study the kinetics and magnitude of their expression. Additionally, immunohistochemistry was used to locate their respective polypeptides in the cochlea. The results revealed a significant demand for XP genes in the mammalian cochlea, which suggest that XP genomic defenses contribute in counterbalancing endogenous stress in the peripheral end-organ under normal conditions.
Introduction
Xeroderma pigmentosum (XP) is a rare autosomal recessive disease characterized by severe sensitivity of the skin and eyes to sunlight and a 1000-fold increase in sunlight-induced melanomas and cutaneous basal and squamous cell carcinomas (Rappin et al. 2000). These XP phenotypes result from loss-of-function mutations in any of eight genes, termed XPA-G and V. Their gene products regulate the multi-enzymatic process called nucleotide excision repair (NER), which is the primary molecular pathway for repairing bulky helix-distorting sunlight induced DNA lesions, such as cyclobutane pyrimidine dimmers and (6-4) photoproducts (Sugasawa 2008). Twenty to thirty percent of patients with XP world-wide belong to a subgroup called XP-neurologic disease which occurs from mutations in the XPD, XPC or XPA genes (Reardon et al. 1997; Rapin et al. 2000). In addition to the typical XP phenotypes, these patients exhibit neuronal loss that results in brain atrophy and hearing loss (Robbins et al. 1991). However, hearing loss may occur in the absence of brain atrophy, which is frequent among XP patients (Kenyon et al. 1985; Robbin et al. 1991; Oh et al. 2006). Audiologic assessments have revealed that the hearing loss is localized to the cochlea (Kenyon et al. 1985; Robbins et al. 1991). The sunlight induced skin-cancers among patients with XP-neurologic disease is consistent with the fact that XP gene products are directly responsible for removing ultraviolet (UV) DNA lesions from the genome. However, brain atrophy and cochlear hearing loss is less obvious since both the brain and cochlea are shielded from light. Therefore, in addition to repairing UV DNA lesions, the genes (XPD, XPC and XPA) that underlie XP neurologic disease may harbor other cell survival activities.
The XPD gene product is a 5′→3′ helicase that serves as a vital subunit of the general transcription factor, TFIIH (Lehmann 2001; Guthrie 2009). XPD maintains the stability of the TFIIH complex during transcription initiation and promoter escape (Lehmann 2001). Additionally, its helicase activity is needed to unwind DNA around sites of UV lesions to facilitate the docking of DNA repair factors such as XPA during NER (Evans et al. 1997; Winkler et al. 2000). Therefore, in addition to sunlight induced DNA repair, XPD plays a role in transcriptional regulation. A loss-of-function mutation in the XPD gene is expected to manifest as brain atrophy and cochlear hearing loss through faulty transcriptional events coupled with poor DNA repair. However, beyond DNA repair, XPC and XPA have no other known cellular function (Guthrie, 2008b). The XPC gene product scans transcriptionally inactive (silent genes) regions of the genome and localize chemical and/or physical aberrations to the normal Watson-Crick structure of DNA. Once XPC identifies the lesion, it initiates NER (Sugasawa et al. 1998). Although, XPC may initially identify a DNA lesion, NER only proceeds after the XPA gene product verifies that the lesion is cytotoxic (Riedl et al. 2003). This DNA damage verification role of XPA is required for at least two genetically distinct sub-divisions of the NER pathway that operates on transcriptionally active and inactive regions of the genome (Sugasawa et al. 1998).
XPC and XPA have evolved across phylogeny to protect the genome from UV induced DNA lesions (Thoma and Vasquez 2003). However, recent research has revealed that they also exhibit specificity for a broad range of endogenous and exogenous DNA lesions (Brooks 2007; Riedl et al. 2003; Thoma and Vasquez 2003). Research on the molecular basis of XP-neurologic disease have revealed that both XPC and XPA are involved in repairing various types of oxidative DNA lesions (Brooks et al. 2000; Kuraoka et al. 2000; Kassam et al. 2007). Indeed, cells from patients with XPC and XPA mutations reveal elevated levels of oxidative DNA lesions (Reardon et al. 1997). The high oxygen metabolism of the brain makes it particularly susceptible to oxidative DNA damage in an XP mutant background which explains the comorbidity with skin cancers (Brooks 2007; Kuraoka et al. 2000). Unlike the brain, the molecular basis of cochlear hearing loss has remained unresolved.
We are interested in the role of XP genes in the cochlea because they may help us understand the hyper-vulnerability of the cochlea to oxidative stressors such as, cisplatin, aminoglycosides and acoustic over-exposure. A mechanistic understanding of the hyper-vulnerability of the cochlea to oxidative stressors is a prerequisite to the development of targeted therapeutic strategies. Our previous experiments revealed for the first time, the existence and distribution of post-translational products from XP genes in the cochlea (Guthrie 2008c, 2009). Additionally, we demonstrated that these post-translational products are expressed at high levels in the cochlear neurosensory epithelia. These finding are important because recent research has revealed that the cochlea experiences high levels of oxidative stress under normal conditions (Takumida and Anniko 2001; Bánfi et al. 2004; Tiede et al. 2007; García-Berrocal et al. 2007), therefore high expression levels of XP genes would be needed for protection. However, if XP genes are busy with endogenous protection, then the cochlea would be unprotected from exogenous oxidative stressors, which provides a basis to interpret cochlear hyper-vulnerability to oxidative stressors. The current study contributes to this line of thinking by providing quantitative evidence through real-time quantitative reverse-transcription polymerase chain reaction (rt-qRT-PCR) to support a high basal demand for XPC and XPA gene products in the cochlea. Additionally, the current study supports previous studies by revealing the localization of XPC and XPA gene products in the cochlea through immunohistochemistry.
Materials and Methods
Animals and tissue preparation
The animals used in the current experiment have been described previously (Guthrie et al. 2008). Briefly, thirty female Fischer344 CDF rats were acquired from Charles River Laboratories, Malvern, PA, USA. The animals were housed at 23 ± 2° C on a 12-hr light/dark cycle and allowed free access to food and water. After the animals acclimated to the rat facility, fifteen were euthanized with a lethal dose of pentobarbital (100 mg/kg, i.p.) and decapitated. The heads were rapidly skinned and the skull resected to allow removal of the brain and access to the osseous labyrinth. Cochlear tissues were dissected immediately from the osseous labyrinth in ice-cold phosphate buffered saline (PBS; pH 7.4) under a stereomicroscope and flash frozen on a dry-ice aluminum block then stored at -80° C in a monophasic lysis reagent (TRIzol; Invitrogen, Carlsbad, CA, USA). Kidney tissues were also harvested in a similar manner for use as a control organ. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh, Pittsburgh, PA. The committee ensured that all protocols were consistent with the United States Department of Agriculture and NIH guidelines for the ethical treatment of animals and that all attempts were made to minimize both animal use and suffering.
RNA purification
Frozen tissues (50-100 mg) were homogenized in 1 ml of monophasic lysis reagent. The homogenate was centrifuged after adding chloroform (0.2 ml) to separate the RNA phase from the DNA phase. Isopropyl alcohol (0.25 ml) was used to precipitate RNA which was rinsed with 75% ethanol and solubilized in diethyl pyrocarbonate treated double distilled water. To remove DNA contamination, the RNA was treated with DNase I (Ambion Inc., Austin, TX, USA) according to the manufacturer's protocol.
Reverse transcription
Reverse transcription produced complementary DNA (cDNA) from DNA-free RNA. The reverse transcription reaction included 10 μl of 10× PCR Taq Gold Buffer II (Amplified Biosystems Inc., Foster City, CA), 30 μl of 25 mM MgCl2, 4 μl of 25 mM of each dNTP, 5 μl of 100 μM of random primer (GIBCO BRL, Gaithersburgh, MD, USA), 40 units of RNasin (Applied Biosystems Inc.), 250 units of Super-Script-II (GIBCO) and 200 ng of total apo-RNA. The reaction was incubated for 10 min at 25° C, 30 min at 48° C and 5 min at 95° C in a 9600 thermocycler (Applied Biosystems Inc., Foster City, CA, USA).
Real-time quantification
Real time quantification was accomplished with SYBR Green chemistry. The reaction consisted of 5 μl of 10× SYBR green PCR buffer (Applied Biosystems, Inc.), 6 μl of 25 mM MgCl2, 4 μl of each dNTPs (blended with 2.5 mM dATP, dGTP and dCTP and 5 mM dUTP), 2.5 μl of specific gene primers (5 μM), 0.5 units of AmpErase UNG, 1.25 units of AmpliTaq Gold and 5 μl of cDNA in a final volume of 50 μl. The rubric for the thermo-cycling was 2 min at 50° C, 12 min at 95° C, 40 cycles at 95° C for 15 seconds and 1 min at 60° C in an ABI PRISM 7700 sequence Detection system (Applied Biosystems Inc). The gene-specific primers for XPC, XPA and 18S rRNA (internal control gene) were reported previously (Guthrie et al. 2008).
Analysis
The ABI Prism 7700 Sequence Detector Software (Amplified Biosystems, Inc.) was used to measure the net fluorescent spectra of the thermal cycler continuously during PCR amplification. Changes in the emission spectra (ΔRn) were calculated as, ΔRn= (Rn+) − (Rn-) where, (Rn+) is the SYBR Green fluorescent signal at any given time after the start of PCR and (Rn-) is baseline fluorescence (ROX; 6-carboxy-X-rhodamine) before the PCR reaction. ROX does not participate in the PCR amplification therefore it provides an internal reference to normalize the fluorescent signal that results from the SBR Green-dsDNA complex. The ΔRn as a function of PCR cycle number or ΔRn–cycle function was derived in real-time during the PCR reaction. The cycle number at which the fluorescence signal crossed the mid-linear portion of the ΔRn–cycle function is the cycle threshold denoted, CT (Schmittgen et al. 2000). For statistical and computational analyses, the CT was converted to 2-CT or 2-ΔΔCT (Schmittgen and Livak 2008). The maximum 2-CT for a particular gene was determined by monitoring CT levels for the gene over 22 days (maximum historic expression). Therefore, the percent expression for any gene was relative to its maximum historic expression under the current experimental conditions. To determine fold-change in cochlear gene expression relative to the kidney, the 2-ΔΔCT method was used (Schmittgen et al. 2000; Schmittgen and Livak, 2008), where ΔΔCT = (CTtarget gene − CTinternal control gene)cochlea − (CTtarget gene − CT internal controlgene)kidney. For example the fold-change for the cochlear XPA target gene relative to that of the kidney was quantified as: 2-ΔΔCT (ΔΔCT = CTXPA − CT18S rRNA)cochlea − CTXPA − CT18S rRNA)kidney. The rat endogenous 18S rRNA (internal control) is used to normalize CT across organs (Guthrie et al. 2008). To determine fold-change in cochlear gene expression relative to another cochlear gene, the 2-ΔΔCT method was used (Schmittgen et al., 2000; Schmittgen and Livak, 2008), where ΔΔCT = (CTtarget gene − CTinternal control gene)cochlea − (CTreference gene − CTinternal controlgene)cochlea. For example the fold-change for the cochlear XPA target gene relative to the cochlear XPC gene was quantified as: 2-ΔΔCT (ΔΔCT = CTXPA − CT18S rRNA)cochlea − CTXPC − CT18S rRNA)cochlea. Differences in mRNA expression were examined with repeated measures analysis of variance (ANOVA) and/or t-test.
Immunohistochemistry
The immunohistochemical procedure has been described previously (Guthrie 2008b). Briefly, fifteen rats received intraperitoneal administration of 100 mg of pentobarbital per kilogram of body weight. After a negative response to a paw pinch, the animals were transcardially perfused with phosphate-buffered saline (PBS) then periodate-lysine-paraformaldehyde fixative. The heads were removed, skinned and post-fixed for 24 hours at room temperature. They were then decalcified in 10% formic acid, trimmed and paraffin embedded. These embedded specimens were then sectioned at 8 μm with a rotary microtome and mounted on subbed slides. The sections were de-paraffinized in xylene and a graded series of ethanol then hydrated. They were then exposed to 30% hydrogen peroxide for 10 minutes then heat treated for 20 minutes at 90-98° C. The sections were then pre-treated with a blocking solution of normal goat or horse serum for 1 hour at room temperature. The primary antibodies anti-XPC and anti-XPA (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) have been characterized previously (Guthrie et al., 2008; Guthrie 2008b, 2009). The sections were treated with the antibodies at a concentration of 1:200 for 24 hours at room temperature. For negative controls the antibodies were omitted (Guthrie et al., 2008; Guthrie 2008b). Following treatment with the antibody, the sections were incubated with biotinylated secondary goat or rabbit antibodies for 1 hour at room temperature and reacted with preformed avidin-biotinylated enzyme complex. The reaction product was then used to oxidize 3,3′-diaminobenzidine tetrahydrochloride which served as chromogen. The sections were then de-hydrated and cover-slipped.
Results
XP DNA repair activity in the kidney is among the highest of all the major organs such as brain, heart, lung, spleen and muscle (Gospodivov et al. 2003). Therefore, the kinetics and magnitude of XP mRNA expression from the cochlea was compared to that of the kidney on postnatal day (p) 83, 97 and 101. These postnatal days correspond to survival times from a previous study (Guthrie et al. 2008) and are arbitrary to the current study. They provide a convenient means of tracking gene expression beyond one time point. Both the kidney and cochlea are composed of several types of cells; therefore, expression refers to the pooled mRNA expression of cells/tissues in the organs. Such pooled expression provides a profile that is specific to each organ and allows for monitoring the kinetics of each gene. Figure 1 is an organ profile of the level and pattern of XPA and XPC gene expression from the kidney and cochlea. The level of expression is shown relative to the maximum expression derived for each organ under the current experimental conditions. In the kidney, XPA gene expression ranged from 42-92% of its maximum expression level and a repeated measures ANOVA revealed significant differences in the level of expression between different time points (F(2,8) = 54.848, p < 0.01). For instance, follow-up paired samples t–test revealed significant differences between p83 and p101 (t(4) = 13.939, p < 0.01) and between p97 and p101 (t(4) = 7.587, p < 0.01). However, there were no significant differences between p83 and p97 (t(4) = 2.405, p > 0.05). Additionally, XPC gene expression ranged from 55-78% of its maximum expression level and a repeated measures ANOVA revealed significant differences in the level of expression between the different time points (F(2,8) = 8.835, p < 0.05). For instance, follow-up paired samples t –test revealed a significant difference between p83 and p101 (t(4) = 5.768, p < 0.01). However, there were no significant differences between p83 and p97 (t(4) = 1.546, p >0.05) and p97 and p101 (t(4) = 2.150, p > 0.05). The high expression of XPA (up to 92% of maximum capacity) and XPC (up to 78% of maximum capacity) is expected, since XP DNA repair activity in the kidney is higher than most organs (Gospodivov et al. 2003).
Figure 1.

Organ profiling of the level and pattern of XPA/C mRNA expression. The percent expression (2−CT: y-axis) relative to the maximum expression level for each organ (kidney or cochlea) was tracked overtime (p = postnatal day). Each bar (N = 5 animals) represents the mean±S.E. for triplicate trials.
The cochlea revealed high levels of XP gene expression, similar to the kidney. Cochlear XPA gene expression ranged from 45-95% of its maximum expression level and a repeated measures ANOVA revealed significant differences in the level of expression between different time points (F(2,8) = 20.938, p < 0.05). For instance, follow-up paired samples t-test revealed a significant difference between p97 and p101 (t(4) = -6.178, p < 0.01). However, there were no significant differences between p83 and p97 (t(4) = 4.322, p > 0.05) and p83 and p101 (t(4) = -1.222, p > 0.05). Additionally, XPC expression ranged from 42-69% of its maximum expression level and a repeated measures ANOVA revealed no significant differences in the level of expression between different time points (F(2,8) = 2.686, p > 0.05). The expression level of XPA from both organs (kidney and cochlea) was closer to saturation (maximum level) than XPC. This suggests that there is a greater basal demand for XPA over XPC.
The pattern of gene expression from each organ is illustrated in Figure 1. In the kidney, both XPC and XPA showed a similar pattern of expression. For instance, a graded (down-ward) expression pattern was observed for both genes at serial time points. In the cochlea, both XPC and XPA showed a similar pattern of expression. But unlike the kidney, the cochlear pattern of expression revealed an alternating morphology, characterized as high expression followed by low expression and again by high expression. Therefore, the expression kinetics is organ specific. The combined results reveal that although, XPC and XPA revealed similar kinetics, there is a greater demand for XPA in both kidney and cochlea.
The results in Figure 2 reveal that XP gene expression is higher in the cochlea than in the kidney. For instance, cochlear XPA expression may be equal to or 3-fold greater than that of the kidney. Interestingly, when cochlear XPA is expressed at 45% of its maximum capacity on p97, it is equal to that of the kidney which is expressing XPA at 77% of its maximum capacity. Additionally, when the kidney is expressing XPA at 92% of its maximum capacity on p83, the cochlea is expressing XPA at 86% (equal to the kidney) of its maximum capacity. Cochlear XPC expression may be 3-6 folds greater than that of the kidney. Interestingly, when cochlear XPC is expressed at only 42% of its maximum capacity on p97, it is 3-fold greater than XPC expression in the kidney which is expressed at 70% of its maximum capacity. Additionally, when the kidney is expressing XPC at 78% of its maximum capacity on p83, the cochlea is expressing XPC at 63% of its maximum capacity which is 4-fold greater than the kidney. Figure 3 shows the high expression level of XPA. For instance, in the cochlea XPA may be up to 160-fold higher than XPC, which further illustrates a high basal demand for XPA.
Figure 2.

Fold-change in cochlear XPA and XPC mRNA expression relative to the kidney (p = postnatal day). Each bar (N = 5 animals) represents the mean±S.E. for triplicate trials.
Figure 3.

Abundance of XPA mRNA relative to that of XPC (p = postnatal day). Each bar (N = 5 animals) represents the mean±S.E. for triplicate trials.
The immunohistochemical staining pattern of XP polypeptides over time, under normal conditions, is consistent. However, several previous experiments have revealed prominent changes in the staining pattern of XP polypeptides in the cochlea under ototoxic stress (Guthrie et al. 2008; Guthrie 2008c, 2009). Figures 4-7 provides representative photomicrographs of XP immunostaining for the three postnatal days (p83, p97 and p101). Unlike the quantitative nature and high resolution of RT-qPCR, changes in XPC and XPA are less obvious with immunostaining under normal conditions. Figure 4 reveals that spiral ganglion cells are XP immunopositive and the staining is predominantly cytoplasmic. This cytoplasmic compartmentalization is consistent with translational events but indicates demobilization of the polypeptides (Guthrie et al., 2008). However, this cytoplasmic pattern of expression becomes primarily nuclear after genomic stress from the mutagen, cisplatin (Guthrie et al., 2008). A nuclear pattern of expression of XP polypeptides indicates mobilization due to damaged DNA (Rademakers et al., 2003; Moné et al., 2004; Politi et al., 2005; Wu et al., 2007). Figure 5 reveals that hair cells and supporting cells of the cochlear neurosensory epithelium are XP immunopositive and the expression is predominantly nuclear with residual cytoplasmic staining. This pattern of expression is consistent with or without genomic stress from the mutagen, cisplatin and indicates persistent mobilization of XP polypeptides (Guthrie 2008c).
Figure 4.
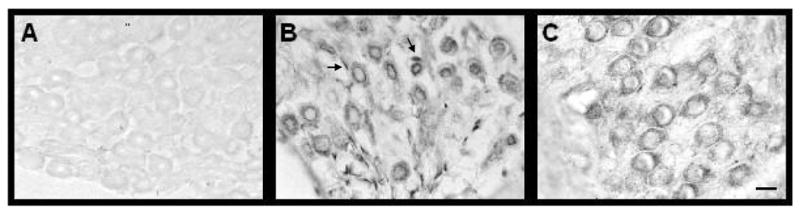
Spiral ganglion cells are immunopositive for XPA and XPC polypeptides. Panel A reveals that omitting the antibody during the immunohistochemical procedure results in negative staining. Panel B shows immunostaining for the XPA polypeptide while panel C shows immunostaining for the XPC polypeptide. Both polypeptides were predominantly localized in the cytoplasm of spiral ganglion cells. The arrows point to satellite cells. Scale bar (10 μm) in panel C, also applies to panels A and B.
Figure 7.
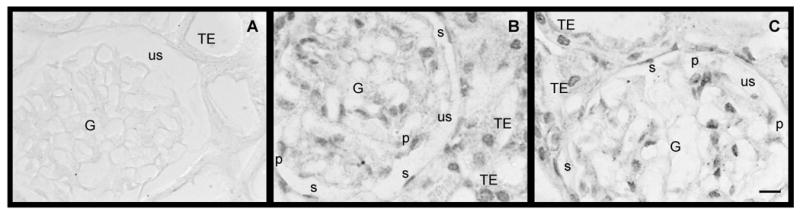
Several types of kidney cells are immunopositive for XPA and XPC polypepetides. Panel A reveals that omitting the antibody during the immunohistochemical procedure results in negative staining. Panel B shows immunostaining for the XPA polypeptide while panel C shows immunostaining for the XPC polypeptide. Both polypeptides were predominantly localized in the nucleus with residual cytoplasmic staining. Abbreviations: G, glumerulus; us, urinary space; TE, tubular epithelium cells; P, podocytes; s, simple squamous epithelial cells. Scale bars (10 μm) in panel C also applies to panels A and B.
Figure 5.
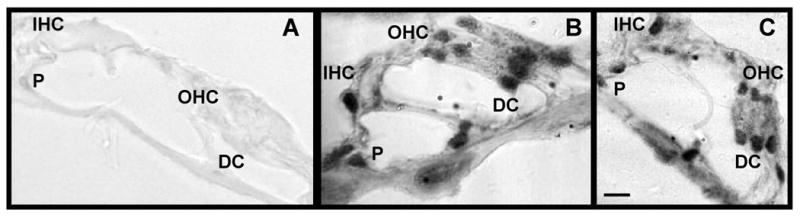
Hair cells and supporting cells are immunopositive for XPA and XPC polypeptides. Panel A reveals that omitting the antibody during the immunohistochemical procedure results in negative staining. Panel B shows immunostaining for the XPA polypeptide while panel C shows immunostaining for the XPC polypeptide. Both polypeptides were predominantly localized in the nucleus with residual cytoplasmic staining for hair cells and supporting cells. Abbreviations: IHC, inner hair cells; OHC, outer hair cells; DC, Dieter's cells; P, pillar cells. Scale bar (10 μm) is panel C, applied to panels A and B as well.
Fibrocytes of the spiral limbus and lateral wall constitute a significant portion of the cells in the cochlea. It is known that fibrocytes and their progenitors are proficient at mobilizing genomic defenses (Brammer et al. 2004). Figure 6 reveals that cochlear fibrocytes from different tissues (spiral limbus and spiral ligament) are XP immunopositive and the expression is predominantly diffused in the cytoplasm with residual nuclear staining. This diffused cytoplasmic pattern of expression changes to prominent reaction products in the nucleus after genomic stress from the mutagen, cisplatin (Guthrie 2009). Figure 7 reveals that several kidney cells are XP immunopositive and the expression is predominantly nuclear with residual cytoplasmic staining. This kidney pattern of expression is consistent with its high intrinsic level of genomic stress which underlies its high susceptibility to exogenous ROS stressors (Dmitrieva et al. 2005; Guthrie 2008c). Table 1 provides a summary of the immunohistochemical findings.
Figure 6.
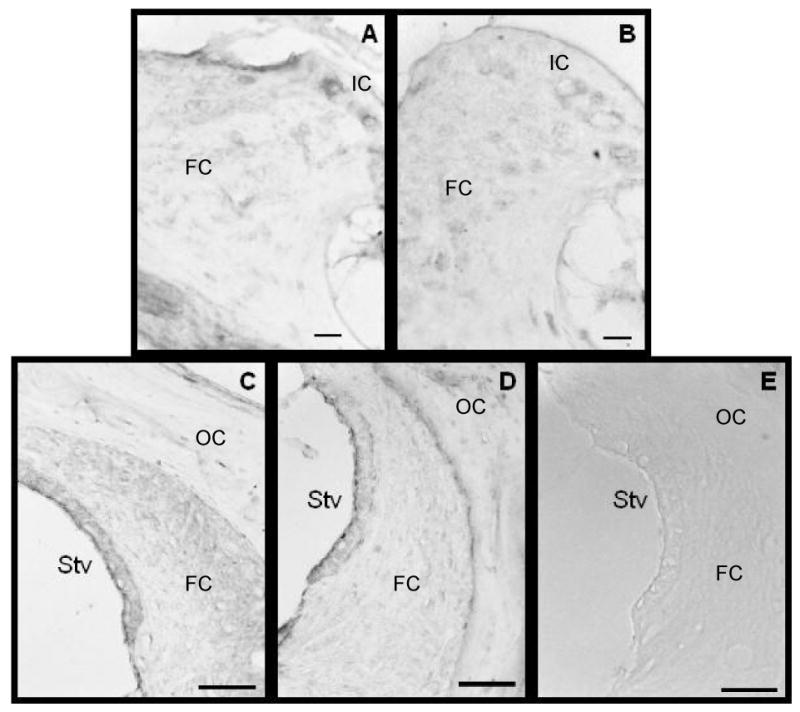
Fibrocytes is the spiral limbus and lateral wall are immunopositive for XPA and XPC polypeptides. Fibrocytes constitute a significant proportion of cochlear cells and are known to be proficient at mobilizing genomic defenses. Panels A (XPA) and B (XPC) shows immunopositive staining in the spiral limbus and panels C (XPA) and D (XPC) shows immunopositive staining in the lateral wall. The staining is predominantly diffused in the cytoplasm with residual nuclear staining. Panel E reveals that omitting the antibody during the immunohistochemical procedure results in negative staining. Scale bars in panels A and B = 10 μm and those of panels C-E = 50 μm. Abreviations: IC, interdental cells; FC, fibrocytes; OC, osteoclasts; Stv, stria vascularis.
Table 1.
Summary of XPA and XPC immunopositive cells/tissues from the cochlea and kidney.
| Cell/tissue-types | XPC | XPA |
|---|---|---|
| Cochlea | ||
| Spiral ganglion cells | + | + |
| Satellite cells | + | |
| Inner hair cells | + | + |
| Outer hair cells | + | + |
| Dieter's cells | + | + |
| Pillar cells | + | + |
| Spiral limbus fibrocytes | + | + |
| Interdental cells | + | + |
| Stria vascularis | + | + |
| Spiral ligament fibrocytes | + | + |
| Osteoclasts | + | + |
| Kidney | ||
| Glumerulus | + | + |
| Tubular epithelium cells | + | + |
| Simple squamous epithelium cells | + | + |
| Podocytes | + | + |
Discussion
The diagnostic criterion for XP-neurologic disease is brain atrophy and cochlear hearing loss (Brooks 2007; Rapin et al. 2000). While genomic stress from high oxidative metabolism compels the need for XP genes in the brain, the basis of cochlear hearing loss has remained unresolved. It is possible that similar to the brain, the cochlea is experiencing genomic stress from high oxidative metabolism. Indeed, the high metabolic activity of the cochlea promotes high reactive oxygen species (ROS; Kopke et al. 1999; García-Berrocal et al. 2007). For instance, the balance between reduced-nicotinamide adenine dinucleotide coenzyme (NADH) and oxidized-flavoproteins (Fp) have revealed significant levels of oxidative redox-activity in the cochlea (Tiede et al. 2007). A prominent source of ROS in the cochlea is based on the activity of NADPH oxidase (NOX)-3. NOX-3 enzymes are single electron transporters whose primary role is the production of ROS (Bedard and Krause 2007). In the cochlea, NOX-3 mRNA is expressed 50-870 times higher than other organs in the body (Bánfi et al. 2004). Additionally, there are several sources of reactive nitrogen species (RNS) production in the cochlea. For instance, nitric oxide (NO) has been directly localized among cochlear cells and isoforms of NO producing enzymes (NO synthase I, II and II) are abundant in the cochlea (Takumida and Anniko 2001). Antioxidants are generally required to combat ROS/RNS mediated damage to biomolecules. However, endogenous cochlear antioxidants have been measured at levels lower than other organs (El Barbary et al. 1993). For instance, the level of cytosolic antioxidants such as glutathione, glutathione-S-transferase, glutathione reductase, selenium-dependent glutathione peroxidase and selenium-independent glutathione peroxidase are several folds lower in the cochlea compared to other organs (Lautermann et al. 1997). Therefore, high production of ROS/RNS coupled with low levels of cytosolic antioxidants suggest that the cochlea is experiencing persistent genomic stress that would require high levels of genome defense molecules to maintain genome integrity and cell survival (Guthrie 2008c).
XP gene products are important for defending the genome against ROS (Brooks et al. 2000; Brooks 2007). Our experimental results showed that cochlear XP gene expression may be as high as 95% of maximum expression capacity, suggesting that under normal conditions XP genes operate close to maximum expression levels. Human mutations in XP genes are known to result in cochlear hearing loss which implies that XP gene products may be vital genome defense molecules in the cochlea (Kenyon et al. 1985; Robbins et al. 1991). Furthermore, cochlear XP gene expression may be up-to 6-fold greater than that of the kidney, which is particularly significant because XP DNA repair activity in the kidney is among the highest of all the major organs. XP DNA repair operates on genetically active (used) and inactive (unused) regions of the genome. Removal of lesions among genetically active regions is faster and more efficient than genetically inactive regions (Balajee and Bohr 2000). This difference is based on the notion that the defense of actively transcribed genes, has greater priority than inactive genes (Guthrie 2008b). XPA plays a critical role in protecting both active and inactive genes, while XPC only protects inactive genes (Sugasawa 2008; Thoma and Vasquez 2003). Our data revealed that the expression level of XPA from both kidney and cochlea was closer to saturation (maximum levels) than XPC. This suggests that there is a greater demand for XPA over XPC. Additionally, in the cochlea, XPA could be up to 160-fold greater than XPC. This high expression may reflect the role of XPA in protecting the total genome. High levels of either XPA or XPC suggest that the cochlea is experiencing persistent genomic stress (possibly from endogenous ROS) that obligates XP genome defenses.
Genome defense research coupled with cancer biology has demonstrated that XP factors are not only important for maintaining genome integrity from endogenous ROS but they are also implicated in cell survival from exogenous mutagens (Guthrie et al. 2008; Thoma and Vasquez 2003). For instance, cancer cells that are resistant to cisplatin (an inorganic mutagen) are proficient at up-regulating XP mRNA levels as a function of treatment (States and Reed 1996; Weaver et al. 2005). Our current data reveal that the normal (non-malignant) cochlea expresses high levels of XP mRNA. However, it is known from both human and animal research that cochlear damage is a frequent side effect of cisplatin chemotherapy (van den Berg 2006; Rybak et al., 2007). Cisplatin chemotherapy results in DNA damage among the various cochlear epithelia yet the neurosensory epithelium is significantly more susceptible to degeneration than the nonsensory epithelia (Hoistad et al. 1998; Cheng et al. 2001; van Ruijven et al. 2005; Thomas et al. 2006). This suggests that there is a difference in XP DNA repair capacity between cochlear epithelia.
Recent experiments have revealed that under normal conditions the cochlear neurosensory epithelium expresses XP translational products at high levels relative to the nonsensory epithelia (Guthrie 2008c, 2009). Additionally, under conditions of genomic stress from cisplatin, the neurosensory epithelium is unable to mobilize XP translational products beyond basal demand while the nonsensory epithelia which is much more resistant to cisplatin, actively mobilized XP translational products beyond basal demand. These observations have led to the hypothesis of basal demand interference, where basal demand for high levels of genome defenses precludes a substantive response to exogenous stress (Guthrie 2008c). This line of thinking is particularly significant because it may help to elucidate the intrinsic vulnerability of the cochlea to exogenous auditory stressors, such as acoustic-overexposure and ototoxic xenobiotics (Guthrie 2008a). Both of these exogenous auditory stressors perpetuate additional ROS production in the cochlea (Rybak et al. 2007; Kopke et al. 1999). If cochlear XP defenses are operating close to saturation under normal conditions (due to high endogenous ROS), then a limited reservoir would be available for mobilization under conditions of exogenous stress. Therefore, the current data supplements previous experiments by providing a basis to interpret and further investigate the intrinsic susceptibility of the cochlea to exogenous auditory stressors. Implicit in the data, is the notion that turning down cochlear levels of oxidative stress under normal conditions might relieve XP DNA repair enzymes to defend the cochlea under stressful conditions. Therefore, future clinical strategies aimed at protecting the cochlea from oxidative stress might benefit from freeing up endogenous defense enzymes prior to exposure to an ROS inducing condition. The combined line of research for the first time, implicates genomic stress and XP DNA repair factors as mechanisms underlying cochlear pathophysiology.
In conclusion, the current work revealed that XP genes are expressed at high levels in the cochlea under normal conditions, which suggest a high demand for XP genome defense in the cochlea and provides a basis to further explore why mutations in XP genes result in cochlear hearing loss. Additionally, the data may be audiologically/otologically important because it supports the hypothesis that basal demand interference may underlie the intrinsic susceptibility of the cochlea to exogenous ROS inducing stressors. Therefore, future therapeutic strategies may yield clinical success by reducing endogenous stress, so that endogenous defenses are free to manage stress from exogenous sources.
Acknowledgments
The authors would like to thank Dr. Ha-Sheng Li-Korotky for helpful discussions and technical assistance and Chia-Yi Lo for expert technical assistance. This work was supported by NIH-NIDCD, the K. Leroy Irvis Award and the SHRS Research Development Fund. F.A.C.-M. is an ASCB MAC visiting scholar at Duke. This award is supported by a MARC grant from the NIH-NIGMS to the American Society for Cell Biology Minorities Affairs Committee.
Sources of Support: This work was supported by NIH-NIDCD, the K. Leroy Irvis Award and the SHRS Research Development Fund. F.A.C.-M. is an ASCB MAC visiting scholar at Duke. This award is supported by a MARC grant from the NIH-NIGMS to the American Society for Cell Biology Minorities Affairs Committee.
References
- Balajee A, Bohr V. Genomic heterogeneity of nucleotide excision repair. Gene. 2000;250:15–30. doi: 10.1016/s0378-1119(00)00172-4. [DOI] [PubMed] [Google Scholar]
- Bánfi B, Malgrange B, Knisz J, et al. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Brammer I, Herskind C, Haase O, Rodemann HP, Dikomey E. Induction and repair of radiation-induced DNA double-strand breaks in human fibroblasts are not affected by terminal differentiation. DNA Repair (Amst) 2004;3:113–120. doi: 10.1016/j.dnarep.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Brooks PJ. The case for 8,5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience. 2007;145:1407–1417. doi: 10.1016/j.neuroscience.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ, Wise DS, Berry DA. The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Bio Chem. 2000;275:22355–22362. doi: 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
- Cheng PW, Kaga K, Koyama S. Temporal bone histopathology after treatment by a large amount of cisplatin: a case study. Otolaryngol Head Neck Surg. 2001;125:411–413. doi: 10.1067/mhn.2001.117408. [DOI] [PubMed] [Google Scholar]
- Dmitrieva NI, Burg MB, Ferraris JD. DNA damage and osmotic regulation in the kidney. Am J Physiol Renal Physiol. 2005;289:2–7. doi: 10.1152/ajprenal.00041.2005. [DOI] [PubMed] [Google Scholar]
- el Barbary A, Altschuler RA, Schacht J. Glutathione S-transferases in the organ of Corti of the rat: Enzymatic activity, subunit composition and immunohistochemical localization. Hear Res. 1993;71:80–90. doi: 10.1016/0378-5955(93)90023-t. [DOI] [PubMed] [Google Scholar]
- Evans E, Moggs JG, Hwang JR. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997;16:6559–6573. doi: 10.1093/emboj/16.21.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Berroca JR, Nevado J, Ramírez-Camacho R, et al. The anticancer drug cisplatin induces an intrinsic apoptotic pathway inside the inner ear. Br J Pharmacol. 2007;152:1012–1020. doi: 10.1038/sj.bjp.0707405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gospodinov A, Ivanov R, Anachova B. Nucleotide excision repair in rat tissues. Eur J Biochem. 2003;270:1000–1005. doi: 10.1046/j.1432-1033.2003.03473.x. [DOI] [PubMed] [Google Scholar]
- Guthrie OW. Aminoglycoside induced ototoxicity. Toxicology. 2008a;249:91–96. doi: 10.1016/j.tox.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Guthrie OW. Dys-synchronous regulation of XPC and XPA among trigeminal ganglion neurons following cisplatin treatment cycles. Anticancer Res. 2008b;28:2637–2640. [PubMed] [Google Scholar]
- Guthrie OW. Preincision complex-I from the excision nuclease reaction among cochlear spiral limbus and outer hair cells. J Mol Histol. 2008c;39:617–625. doi: 10.1007/s10735-008-9202-1. [DOI] [PubMed] [Google Scholar]
- Guthrie OW. DNA repair proteins and telomerase reverse transcriptase in the cochlear lateral wall of cisplatin treated rats. J Chemother. 2009;21:74–79. doi: 10.1179/joc.2009.21.1.74. [DOI] [PubMed] [Google Scholar]
- Guthrie OW, Li-Korotki HS, Durrant JD, et al. Cisplatin induces cytoplasmic to nuclear translocation of nucleotide excision repair factors among spiral ganglion neurons. Hear Res. 2008;239:79–91. doi: 10.1016/j.heares.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Hoistad DL, Ondrey FG, Mutlu C. Histopathology of human temporal bone after cis-platinum, radiation, or both. Otolaryngol Head Neck Surg. 1998;118:825–832. doi: 10.1016/S0194-5998(98)70276-1. [DOI] [PubMed] [Google Scholar]
- Kassam SN, Rainbow AJ. Deficient base excision repair of oxidative DNA damage induced by methylene blue plus visible light in xeroderma pigmentosum group C fibroblasts. Biochem Biophys Res Commun. 2007;359:1004–1009. doi: 10.1016/j.bbrc.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Kenyon GS, Booth JB, Prasher DK. Neuro-otological abnormalities in xeroderma pigmentosum with particular reference to deafness. Brain. 1985;108:771–784. doi: 10.1093/brain/108.3.771. [DOI] [PubMed] [Google Scholar]
- Kopke R, Allen KA, Henderson D. A radical demise: Toxins and trauma share common pathways in hair cell death. Anal NY Acad Sci. 1999;884:171–191. doi: 10.1111/j.1749-6632.1999.tb08641.x. [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Bender C, Romieu A, et al. Removal of oxygen free-radical-induced 5′8-purine cyclodeoxynucleoside from DNA by the nucleotide excision-repair pathway in human cells. PNAS. 2000;97:3832–3837. doi: 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautermann J, Crann SA, McLaren J, et al. Glutathione-dependent antioxidant systems in the mammalian inner ear: Effects of aging, ototoxic drugs and noise. Hear Res. 1997;114:75–82. doi: 10.1016/s0378-5955(97)00154-8. [DOI] [PubMed] [Google Scholar]
- Lehmann AL. The xeroderma pigmentosum group D (XPD) gene: One gene, two functions three diseases. Genes and Development. 2001;15:15–23. doi: 10.1101/gad.859501. [DOI] [PubMed] [Google Scholar]
- Moné MJ, Bernas T, Dinant C, Goedvree FA, Manders EMM, Volker M, Houtsmuller AB, Hoeijmakers JHJ, Vermeulen W, van Driel R. In vivo dynamics of chromatin-associated complex formation in mammalian nucleotide excision repair. PNAS. 2004;101:15933–15937. doi: 10.1073/pnas.0403664101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi A, Moné MJ, Houtsmuller AB, Hoogstraten D, Vermeulen W, Heinrich R, van Driel R. Mathematical modeling of nucleotide excision repair reveals efficiency of sequential assembly strategies. Molecular Cell. 2005;19:679–690. doi: 10.1016/j.molcel.2005.06.036. [DOI] [PubMed] [Google Scholar]
- Oh KS, Khan SG, Jaspers NG. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Hum Mutat. 2006;27:1092–103. doi: 10.1002/humu.20392. [DOI] [PubMed] [Google Scholar]
- Rademakers S, Volker M, Hoogstraten D, Nigg AL, Moné MJ, van Zeeland AA, Hoeijmakers JHJ, Houtsmuller ABD, Vermeulen W. Xeroderma pigmentosum group A protein loads as a separate factor onto DNA lesions. Molecular and Cellular Biology. 2003;23:5755–5767. doi: 10.1128/MCB.23.16.5755-5767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH, et al. Cockayne syndrome and xeroderma pigmentosum: DNA repair disorders with overlaps and paradoxes. Neurology. 2000;55:1442–1449. doi: 10.1212/wnl.55.10.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Bessho T, Kung HC, et al. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: Possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc Natl Acad Sci USA. 1997;95:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl T, Hanaoka F, Egly JM. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 2003;22:5293–5303. doi: 10.1093/emboj/cdg489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins H, Brumback RA, Mendiones M, et al. Neurologic disease in xeroderma pigmentosum: Documentation of a late onset type of the juvenile onset form. Brain. 1991;114:1335–1361. doi: 10.1093/brain/114.3.1335. [DOI] [PubMed] [Google Scholar]
- Rybak LP, Whitworth CA, Mukherjea D, et al. Mechanisms of cisplatin-induced ototoxicity and prevention. Hear Res. 2007;226:157–167. doi: 10.1016/j.heares.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Zakrajsek BA, Mills AG. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- States JC, Reed E. Enhanced XPA mRNA levels in cisplatin-resistant human ovarian cancer are not associated with XPA mutations or gene amplifications. Cancer Lett. 1996;108:233–237. doi: 10.1016/s0304-3835(96)04428-x. [DOI] [PubMed] [Google Scholar]
- Sugasawa K, Ng JM, Masutani C, et al. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- Sugasawa K. Xeroderma pigmentosum genes: Functions inside and outside DNA repair. Carcinogenesis. 2008;29:455–465. doi: 10.1093/carcin/bgm282. [DOI] [PubMed] [Google Scholar]
- Takumida M, Anniko M. Direct evidence of nitric oxide production in the guinea pig organ of Corti. Acta Otolaryngol. 2001;121:342–345. doi: 10.1080/000164801300102716. [DOI] [PubMed] [Google Scholar]
- Thoma BS, Vasquez KM. Critical DNA damage recognition functions of XPC-hHR23b and XPA-RPA in nucleotide excision repair. Molecular Carcinogenesis. 2003;38:1–13. doi: 10.1002/mc.10143. [DOI] [PubMed] [Google Scholar]
- Thomas JP, Lautermann J, Liedert B, et al. High accumulation of platinum-DNA adducts in strial marginal cells of the cochlea is an early event in cisplatin but not carboplatin ototoxicity. Mol Pharmacol. 2006;70:23–29. doi: 10.1124/mol.106.022244. [DOI] [PubMed] [Google Scholar]
- Tiede LM, Rocha-Sanchez SM, Hallworth R. Determination of hair cell metabolic state in isolated cochlear preparations by two-photon microscopy. J Biomed Opt. 2007;12:1–15. doi: 10.1117/1.2714777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg JH, Beijnen JH, Balm AJ. Future opportunities in preventing cisplatin induced ototoxicity. Cancer Treat Rev. 2006;32:390–397. doi: 10.1016/j.ctrv.2006.04.011. [DOI] [PubMed] [Google Scholar]
- van Ruijven MWM, de Groot JCMJ, Hendriksen F. Immunohistochemical detection of platinated DNA in the cochlea of cisplatin-treated guinea pigs. Hear Res. 2005;203:112–121. doi: 10.1016/j.heares.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Weaver DA, Crawford EL, Warner KA, et al. ABCC5, ERCC2, XPA and XRCC1 transcript abundance levels correlate with cisplatin chemoresistance in non-small cell lung cancer cell lines. Mol Cancer. 2005;4:18–26. doi: 10.1186/1476-4598-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler GS, Araújo SJ, Fiedler U, et al. TFIIH with inactive XPD helicase functions in transcription initiation but is defective in DNA repair. J Biol Chem. 2000;275:4258–4266. doi: 10.1074/jbc.275.6.4258. [DOI] [PubMed] [Google Scholar]
- Wu X, Shell SM, Liu Y, Zou Y. ATR-dependent checkpoint modulates XPA nuclear import in response to UV irradiation. Oncogene. 2007;26:757–764. doi: 10.1038/sj.onc.1209828. [DOI] [PMC free article] [PubMed] [Google Scholar]