

Abstract
Endogenous cardiotonic steroids (CTS), also called digitalis like factors, have been postulated to play important roles in pathogenesis of hypertension for nearly half of a century. For the past 50 years biomedical scientists have been in quest of an unidentified factor or hormone that both increases blood pressure and renal sodium excretion; this “natriuretic hormone” was, in fact, postulated to interact with the Na/K-ATPase. Recent discoveries have led to the identification of steroid molecules which are present in humans, rodents and amphibians, and which, in a complex manner, interact with each other and with the other systems that regulate renal salt handling and contribute to the salt-sensitivity of blood pressure.
Recent findings include the specific identification of endogenous cardenolide (endogenous ouabain) and bufadienolide (marinobufagenin) CTS in humans along with the delineation of mechanisms by which CTS can signal through the Na/K-ATPase. Although CTS were first considered important in the regulation of renal sodium transport and arterial pressure, more recent work implicates these hormones in the central regulation of blood pressure and regulation of cell growth, and development of cardiovascular and renal fibrosis in particular.
1. Concept of natriuretic hormone
The NaCl-sensitivity of blood pressure is thought to be due, at least in part, to a compromised ability of the kidneys to excrete sodium, which is mediated by variety of factors, both genetic and environmental [1]. One of such factors are endogenous digitalis-like inhibitors of the Na/K-ATPase, or cardiotonic steroids (CTS) [2].
Lewis Dahl was one of the first to hypothesize that salt-induced hypertension may be mediated by a humoral factor, which raises the blood pressure [3]. Based on numerous observations made in human subjects and in experimental animals under the conditions of NaCl loading and volume expansion, deWardener and others postulated that a humoral prohypertensive factor implicated in the pathogenesis of NaCl-sensitive hypertension is an endogenous natriuretic [2]. Because Na/K-ATPase comprises a major sodium transporting mechanism in the kidney, and because digitalis glycosides are specific ligands of the Na/K-ATPase, it has been further postulated that a putative natriuretic hormone has digitalis-like properties. According to the “concept of natriuretic hormone”, the primary role of endogenous digitalis is to promote natriuresis via inhibition of the Na/K-ATPase and sodium reabsorption in the renal proximal tubules [2]. The increased plasma levels of digitalis-like CTS could also contribute to vasoconstriction via inhibition of the Na,K-pump, coupled with activation of Na+/Ca2+ exchange in vascular smooth muscle [4]. In accord with these views, Na/K-ATPase activity in the cardiovascular tissues from dogs and rats with low renin hypertension was found to be decreased [5,6], and plasma saline volume expansion of dogs was shown to raise levels of circulating digoxin-like immunoreactive material [7]. Subsequently, Hamlyn et al. demonstrated that plasma Na/K-ATPase inhibitory activity is positively associated with blood pressure in hypertensive patients [8]. Kojima et al [9] showed that anti-digoxin antibody lowers blood pressure in rats with deoxycorticosterone-salt-induced hypertension. These findings prompted search for the identity of “endogenous digitalis”.
2. Endogenous ouabain
Endogenous ouabain (EO) was the first CTS to be identified in human plasma [10,11], but in humans EO did not appear to be natriuretic, and peripheral levels of this hormone were not stimulated by chronic high NaCl intake [12]. In rats, ouabain exhibits high affinity for the α2/α3 isoforms of Na/K-ATPase [13], while tubular cells of the rat kidney, the target for a “natriuretic hormone”, express mainly the α1 isoform, which is relatively insensitive to ouabain [14]. Although EO does not fulfill the criteria for a putative natriuretic hormone, experimental data indicate that EO plays an important role in the pathogenesis of NaCl-sensitive hypertension.
Experimental studies performed by Takahashi’s group demonstrated that in normotensive Wistar rats and in rats with DOCA-NaCl hypertension central administration of low concentrations of ouabain elicits pressor and natriuretic which are dependent on the activation of renin-angiotensin system [15,16]. Subsequently, the same group demonstrated that development of DOCA-salt hypertension is associated with increases in the levels of two different CTS, in the brain and in the adrenal cortex [17]. Later, in Leenen’s laboratory, in three strains of rats, in Dahl salt-sensitive (DS), Wistar, and spontaneously hypertensive rats, brain EO was found to be responsive to acute and chronic NaCl loading [18]. Notably, pressor responses to central administration of ouabain and to NaCl were shown to stimulate brain EO levels via activation of central RAAS [19,20]. In DS, NaCl-induced activation of brain EO was accompanied by an increase in plasma Na/K-ATPase inhibitory activity in presence of unchanged plasma levels of ouabain-like immunoreactivity, suggesting that CTS other than EO were involved in these events [21,22].
3. Endogenous bufadienolides
For centuries it had been known that the skin and parotid gland of several amphibian species contain substantial amounts of bufadienolides, CTS which differ from cardenolides, such as digoxin and ouabain, in having a doubly unsaturated six-membered lactone ring [23]. Since antiquity bufadienolides from amphibian skin have been used in traditional Oriental medicine to treat cardiac failure [23]. Considering that in amphibia, skin participates in the regulation of water/electrolyte homeostasis, it was hypothesized that the Na/K-ATPase and bufadienolides represent a system which regulates water/electrolyte balance, i.e. that bufadienolides function as the putative mammalian “natriuretic hormone” [24]. Accordingly, the highest levels of bufadienolides were found in those amphibian species which migrate from dry to aquatic environment [25]. Later, in agreement with this hypothesis, brain and skin levels of bufadienolides in the toads were shown to change according to the changes in environmental salinity [26].
Shortly thereafter, several groups demonstrated that human fluids contain material which cross-react with antibodies against one of the bufadienolides, bufalin [27–29]. Later, Lichtstein et al. demonstrated the presence of bufalin derivatives in the eye lenses of several mammalian species and proposed a role for these compounds in cataract formation [30]. Sich et al. reported that human plasma and bovine adrenals contain material which cross-reacts with an antibody against a plant-derived bufadienolide, proscillaridin A [31]. In 1996 Hilton and co-workers mass-spectrometrically identified a bufadienolide compound in human placentae [32].
4. Marinobufagenin
At least one of the circulating mammalian CTS is a bufadienolide, marinobufagenin (MBG) [23]. MBG emerged as a candidate CTS based on the in vitro studies which demonstrated that this steroid at low concentrations induces vasoconstriction in isolated human blood vessels and exhibits higher, as compared to ouabain, affinity to α1 isoform of the Na/K-ATPase, an exclusive sodium pump isoform in renotubular epithelium [14,33,34]. Normotensive human subjects on a high NaCl diet exhibit elevations in plasma levels and renal excretion of MBG [35]. In normotensive rats and dogs plasma MBG was stimulated by acute plasma volume expansion and by chronic administration of a high NaCl diet [36–39]. Increased production of MBG occurs in volume-expanded states, such as essential hypertension, primary aldosteronism, chronic renal failure [40], congestive heart failure [41], and pregnancy [42,43]. Elevated levels of MBG in plasma and placenta are observed in preeclampsia [42,44]. In hypertensive DS and in pregnant rats with hypertension, induced by NaCl supplementation, monoclonal anti-MBG antibody reduced blood pressure and increased activity of the sodium pump in the vasculature [43]. Recently, Komiyama et al. purified MBG and telocinobufagin, a possible MBG precursor from human plasma and found that levels of TCB and MBG were significantly elevated in plasma of uremic patients [45]. Subsequently, the same group demonstrated that murine adrenocortical Y1 cells elaborate MBG and its conjugated form, marinobufotoxin [46].
5. Cardiotonic steroids and regulation of sodium excretion
Before identification of individual CTS in the mammalian tissues, it had been demonstrated that the endogenous CTS are capable of substantially inhibiting renal Na/K-ATPase activity [47]. Three potential mechanisms may be offered to describe how low nanomolar or subnanomolar concentrations of CTS present in mammalian plasma promote natriuresis via interaction with renotubular Na/K-ATPase. First, circulating concentrations of MBG have been shown to significantly inhibit Na/K-ATPase activity in rat renal tissue despite the rodent a1 subunit being known to have decreased sensitivity to most cardiotonic steroids [39,48]. In DS and in the normotensive Sprague-Dawley rats, the natriuretic responses to acute and chronic NaCl loading were associated with substantial elevation in plasma concentration and renal excretion of MBG, and in vivo administration of anti-MBG antibody reduced renal sodium excretion and increased activity of renal Na/K-ATPase [49,50]. Some of this may be related to the particular intracellular architecture of the Na/K-ATPase in renal tissues. Na/K-ATPase partially purified from renal membranes under “gentle” conditions, including pretreatment with moderate concentration of SDS [50], as well as a preparation of membrane-bound Na/K-ATPase without detergent, but with further application of ionophore alamethicin [48] displays a biphasic response to MBG inhibition despite the exclusive presence of α1 Na/K-ATPase in this preparation [48,50]. Importantly, under these conditions, rat renal Na/K-ATPase exhibits high affinity Ki for the interaction with MBG and with endogenous MBG-immunoreactive material purified from urine of NaCl-loaded DS [48]. Moreover, when intact renal membrane vesicles were pretreated with gradually increasing concentrations of SDS, the biphasic nature of MBG inhibitory curve gradually changed to a monophasic exhibiting a single Ki (Fedorova, unpublished observation) This suggests that a degree of polymerization of Na/K-ATPase α-1 subunits (dimers or tetramers) or some other membrane proteins (e.g., the γ subunit of the Na/K-ATPase) can modulate the Na/K-ATPase responsiveness to some CTS, including MBG.
CTS may interact with the other endogenous natriuretic hormones, such as natriuretic peptides. Thus, low concentrations of α-hANP, via cGMP/PKG-dependent mechanism, increase sensitivity of renal Na/K-ATPase to MBG, but reduce MBG-sensitivity of vascular sodium pump [51]. The potential importance of such interaction could be illustrated by the results of an experiment in which we compared blood pressure, renal sodium excretion, activity of the sodium pump in aorta and renal medulla, and levels of MBG, ANP, and cGMP in salt-loaded DS and Sprague-Dawley rats [52]. In this experiment, NaCl loading elicited increases in plasma ANP and in renal cGMP excretion in normotensive Sprague-Dawley rats, but not in DS. NaCl loading produced sustained elevations in renal MBG excretion in both DS and Sprague-Dawley rats. While in NaCl-loaded DS SBP rose and aortic sodium pump was inhibited, in Sprague-Dawley rat blood pressure and activity of aortic sodium pump did not change. NaCl-loaded Sprague-Dawley rats excreted twice as much sodium as DS, and in Sprague-Dawley rats renal sodium pump was inhibited more than that in DS. Thus, in NaCl-loaded normotensive and salt-sensitive rats, a comparable MBG response is associated with preferential inhibition of the sodium pump in the kidney and in vascular smooth muscle, respectively, resulting in an adaptive natriuresis in Sprague-Dawley rats, but sodium retention and pressor response in DS [52]. The lack of ANP response to NaCl loading in DS appears to be one likely factor underlying different patterns of Na/K-ATPase inhibition in two strains of rats.
Another mechanism of CTS-induced natriuresis may involve CTS-induced endocytosis of the Na/K-ATPase in kidney tissues. In chronically NaCl-loaded Sprague-Dawley rats, renal MBG excretion was significantly elevated and anti-MBG antibody reduced natriuresis and restored sodium pump activity in the renal cortex [39]. The same study demonstrated that in addition to the direct inhibition of the Na/K-ATPase, MBG is capable to exhibit its natriuretic effects via internalization of the sodium pump in the proximal tubule [39]. The endocytosis of the proximal tubular Na/K-ATPase induced by CTS has been shown to proceed through clathrin coated pits and require PI(3)K activation as well as the plasmalemmal pump being in the context of caveolae, as well as signaling through the Src–EGFR pathway [53,54]. Further work demonstrated that CTS can induce decreases in the apical expression of one of the plasma membrane Na+/H+ exchanger, NHE3 [55,56]. Taken together, these data suggest that increases in the circulating levels of MBG accompany salt loading which, in turn, may induce decreases in both basolateral and apical sodium transport in the proximal tubule through both the classical or ionic signaling mechanism and the more recently described Na/K-ATPase-Src-EGFR pathway.
6. Endogenous ouabain and marinobufagenin in Dahl salt-sensitive rats
Lewis Dahl proposed the existence of a prohypertensive natriuretic hormone based on experiments with an inbred strain of rats which develop progressive hypertension on a high NaCl intake [57], i.e., in DS. Our laboratory studied CTS in this model, and demonstrated that on a high NaCl diet (8% NaCl) DS exhibited a transient rise in the levels of EO which was followed by a progressive increase in plasma concentration and renal excretion of MBG which paralleled elevation of the blood pressure [50]. When DS with established hypertension were administered anti-MBG antibody, blood pressure markedly decreased, while anti-ouabain antibody did not exert such an effect [50]. Therefore, elevated levels of MBG contributed to the maintenance of high blood pressure in this genetic form of NaCl sensitive hypertension.
When central and peripheral levels of EO and MBG were compared in both chronically and acutely NaCl-loaded DS the responses of both substances exhibited similar patterns, and transient EO response preceded a more sustained increase in MBG excretion [13,50](Fig. 1 A,B). Following acute NaCl loading, levels of EO in the amygdala, hippocampus, pituitary, adrenals and plasma exhibited a transient peak response which was followed by a sustained rise in plasma levels and renal excretion of MBG [58]. Such a pattern suggested that a causative relationship might exist between an acute EO response and a sustained elevation in MBG, i.e., that brain EO could trigger MBG production. In accord with this hypothesis, pretreatment of the acutely NaCl-loaded with anti-MBG antibodies did not attenuate the EO response, but prevented NaCl-induced blood pressure increase [58]. In the same experiment, pretreatment of NaCl-loaded DS with anti-ouabain antibody markedly decreased NaCl-induced renal MBG excretion [58].
Figure 1. Transient response of endogenous ouabain precedes sustained increase in MBG excretion during high NaCl intake.
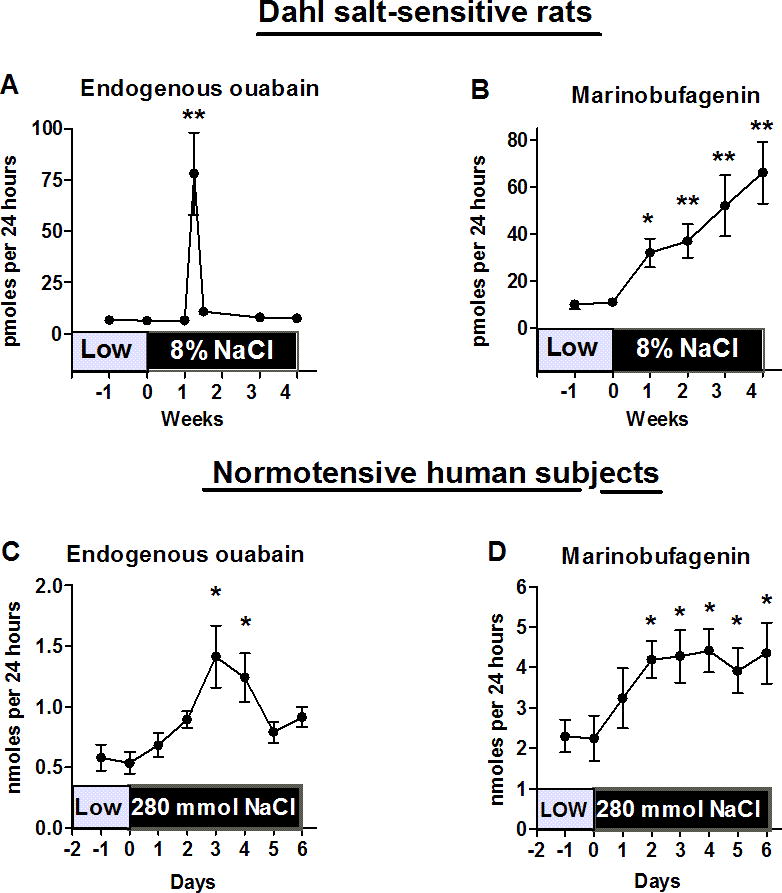
Similar patterns of renal excretion of endogenous cardiotonic steroids during chronic NaCl loading of Dahl-S rats (administration of 8% NaCl diet) (A and B) and subacute dietry NaCl supplementation of normotensive human subjects (280 vs. 50 mmol NaCl per day) (C and D). By one-way ANOVA and Newman-Keuls test: *-P<0.05; **-P<0.01 vs. day 0. Adapted from references 50 and 35.
Considering that, in previous studies from the laboratories of Takahashi and Leenen, activation of RAAS was shown to be implicated in the response of brain EO to high NaCl [17,18], we hypothesized that brain RAAS may be a factor linking central (EO) and peripheral (MBG) CTS. The effects of acute NaCl loading on blood pressure, central and peripheral EO, on central and adrenal ATII, on plasma norepinephrine and MBG, and Na/K-pump in renal medulla were investigated in 10 week old DS [49]. NaCl loading induced transient peak increases of EO in the hippocampus and pituitary followed by a transient increase in pituitary ATII, increases in plasma NE and adrenocortical ATII levels, a sustained increase in MBG excretion, a 45% inhibition of the renal Na/K-pump, and a 35 mmHg rise in arterial pressure [49,58]. In these experiments, pretreatment of rats with anti-ouabain or anti-MBG polyclonal antibodies prevented the NaCl-induced pressor response and renal Na/K-pump inhibition. Anti-ouabain antibody pretreatment also prevented the increases in pituitary and adrenal ATII, and reduced MBG production. Anti-MBG antibody, in contrast, did not affect levels of central EO or ATII. In the primary culture of adrenocortical cells from DS, 1 nmol/L ATII, in a losartan-sensitive manner, doubled MBG production [49]. Thus, in response to NaCl loading, brain EO stimulated adrenocortical MBG via AT1 receptor signaling, and MBG inhibited the renal Na/K-pump and elevated blood pressure, i.e., it behaved like a natriuretic hormone. This sequence of events is schematically presented in Figure 1.
Subsequently, it has been demonstrated that intrahippocampal administration of an extremely low dose of ouabain to DS mimics effects of NaCl loading including activation RAAS in the hypothalamus and adrenal cortex, increase in the adrenocortical levels of MBG, inhibition of the sodium pump in vasculature and in the kidney, and pressor and natriuretic responses [58]. Importantly, in this experiment, in vivo peripheral administration of anti-MBG antibody reversed the above effects induced by central administration of ouabain [58].
Although relevance of this scenario to pathogenesis of human hypertension remains to be proven, in NaCl-loaded normotensive human subjects [35] renal excretion of CTS exhibits a pattern similar to that observed in NaCl-loaded DS [50], i.e., a transient increase in renal excretion of EO precedes a more sustained increase in the excretion of MBG (Figure 2).
Figure 2. Interactions between brain endogenous ouabain, renin-angiotensin system, and marinobufagenin in pathogenesis of NaCl-sensitive hypertension.
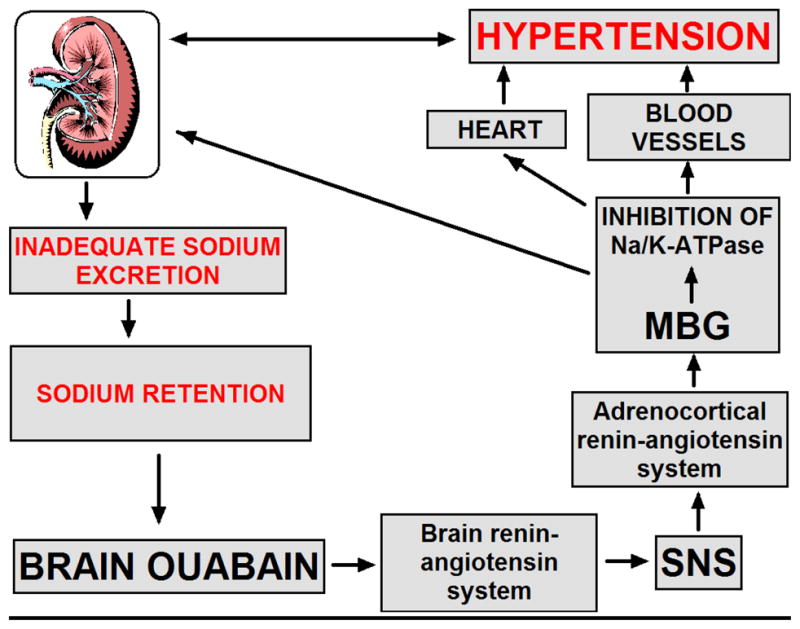
In NaCl-loaded Dahl salt-sensitive rats, in the presence of impaired renal sodium transport, sodium retention stimulates endogenous ouabain in the hippocampus, hypothalamus and pituitary. Brain endogenous ouabain stimulates renin-angiotensin system in the hypothalamus and pituitary, and activates sympathetic nervous system. These events stimulate renin-angiotensin system in the adrenal cortex, and activate adrenocortical production of marinobufagenin (MBG). MBG, a natriuretic and a vasoconstrictor, is produced with a primarily adaptive aim, to induce natriuresis via inhibition of renotubular Na/K-ATPase. An excessive MBG elaboration, however, induces a maladaptive effect and leads to the inhibition of the Na/K-ATPase in vascular smooth muscle cells and potentiates vasoconstriction.
Several studies analyzed mechanisms which could link NaCl loading/sodium retention to activation of brain EO. Leenen and coworkers demonstrated in rats that an increase in cerebrospinal fluid NaCl precedes the development of hypertension [59] and that sodium ions in the brain enter the intracellular space via epithelial sodium channels [60], which is modulated by central mineralocorticoid receptors [61]. Accordingly, Gabor and Leenen demonstrated in Wistar rats that immunoneutralization of brain EO prevented the potentiating effect of exogenously administered aldosterone on pressor response induced by central administration of sodium chloride [62]. Some data, however, suggest that brain-specific Nax rather than epithelial sodium channels are involved in this process [63]. Importantly, in DS, NaCl-induced increase in the concentration of sodium in cerebrospinal fluid may be dependent on the altered properties of the Na/K-ATPase in the chorioid plexus [64]. Thus, in DS an abnormal function of the sodium pump is important not only for renal sodium retention [65], but also for triggering of NaCl-induced EO-dependent central mechanisms of establishment of hypertension.
7. Endogenous ouabain and adducin paradigm
Despite lack of evidence for natriuretic activity of EO, several lines of evidence suggest prohypertensive role of EO, including induction of hypertension in ouabain-treated rodents [66,67] and elevation of EO levels in patients with essential hypertension [68]. In addition to EO contributing to hypertension acting as a central mediator, two other mechanisms, the “Adducin paradigm”, and the existence of highly sensitive ouabain binding sites in vascular smooth muscle were proposed to link the peripheral effects of EO to vasoconstriction in hypertension.
In Milan hypertensive rats increased levels of EO in the presence of a mutation of a cytoskeletal protein, adducin, are associated with heightened expression and activity of the Na/K-ATPase in the renotubular epithelium [69,70]. The increase in renal sodium pump activity in Milan hypertensive rats occurs due to an increase of the resident time of the Na/K-ATPase in the cellular membrane [70–72]. This effect of EO has been mimicked by chronic infusion of low dose of ouabain to normotensive rats which doubled plasma ouabain levels, rose the blood pressure and increased levels of α1/β1 Na-K-ATPase subunits, Src and EGFr in isolated caveolae membranes along with activation of ERK1/2 [70]. It is believed that EO exhibits these effects via interaction with a fraction of ouabain-sensitive sodium pumps localized in the caveolae of renotubular cells [70]. Accordingly, administration of an ouabain antagonist Rostafuroxin (PST2238), a digitoxin derivative, to Milan hypertensive rats antagonized the interaction of EO and adducin on the renal sodium pump, lowered the blood pressure and normalized EO-induced up-regulation of Na/K-ATPase in the renal medulla [70,73]. In agreement with the above scenario, hypertensive patients with polymorphisms of the adducin gene also exhibit altered renal sodium reabsoption [74]. Recently, Manunta et al. [75] have demonstrated that saline loading produces renal sodium retention in the hypertensives with elevated plasma EO levels and mutant adducin.
Elevated levels of EO can also elevate blood pressure via inhibition of ouabain-sensitive α2 Na/K-ATPase and promoting Ca2+ entry via Na+/Ca2+ exchanger in vascular smooth muscle [67,76]. Thus, the genetically engineered mice that express ouabain-resistant α2 Na/K-ATPase, unlike control mice with more ouabain-sensitive sodium pumps, do not increase blood pressure following chronic administration of ouabain [67]. Vascular smooth muscle from these mice with ouabain-insensitive α2 sodium pumps is insensitive to the pressor effect of ouabain [77]. Moreover, genetically engineered mice which have reduced expression of α2 (but not α1) Na/K-ATPase become hypertensive and their arteries exhibit enhanced vascular tone in vitro [76].
8. Marinobufagenin and fibrosis
Growing evidence suggests that contribution of CTS to pathogenesis of hypertension is not limited to involvement of these hormones in the mechanisms of vasoconstriction and impaired renal sodium excretion. Discovery of signaling functions of the sodium pump and identification of specific mechanisms underlying effects of low concentrations of CTS on growth, development, and apoptosis add a new dimension to understanding of pathophysiological roles of these hormones [23]. Most recent findings indicate that CTS are also implicated in the mechanisms of development of fibrosis. Several lines of experimental evidence suggest that the classical i.e. “ionic mechanism” might not be sufficient to explain the effects of CTS on the Na/K-ATPase both in vitro and in vivo. Thus, the effects of CTS on the phosphorylation of key signaling proteins such as the EGFR can be observed in cell free systems where changes in [Na] are essentially excluded [78]. The positive evidence supporting the alternative or “signaling” function for the Na/K-ATPase derived from several studies can be summarized as follows. The Na/K-ATPase involved in signaling through Src and the EGFR resides in caveolae and does not appear to actively pump sodium or potassium [79]. Exposure of several different cell types to CTS induce rapid phosphorylation of the EGFR in a Src dependent manner. The phosphorylation pattern for the EGFR is distinctly different than that observed when EGFR induces autophosphorylation of this protein [80,81]. In addition to the EGFR, other signaling proteins appear to be recruited including phospholipase C (PLC), TRP proteins, PI(3)K and several isoforms of protein kinase C (PKC) [54,78,82–87]. The caveolar Na/K-ATPase binds closely with Src and maintains Src in an inactive form, and binding of CTS to the Na/K-ATPase induces a conformation change which alters the relationship between the Na/K-ATPase and Src and allows Src to become activated. This active Src is then able to phosphorylate other proteins [87]. Notably, the binding of CTS to the plasmalemmal Na/K-ATPase induces the endocytosis of the CTS-Na/K-ATPase-Src-EGFR complex [39,53,54,88]. CTS binding to the Na/K-ATPase produces elevation in cellular ROS which is dependent on the function of RAS [89]. The downstream biochemical (e.g., activation of ERK) and physiological (e.g., increases in cytosolic Ca) consequences of CTS binding to the Na/K-ATPase can be prevented by ROS scavenging [83,89–91].
The possibility that CTS signaling may lead to fibrosis is derived from a group of experiments performed in our laboratories. First, we noted that experimental renal failure produced a tremendous amount of cardiac fibrosis in both rat and mouse [91–93]. The cardiomyopathy associated with human renal failure is also complicated by considerable fibrosis, although fibrosis, in general, does appear to develop much faster in the rodent models. Active immunization against an MBG-albumin conjugate which resulted in a high antibody titer, as well as reduction of circulating levels of MBG by adrenalectomy, prevented cardiac fibrosis seen with experimental renal failure and treatment of animals with an infusion of MBG which achieved similar plasma levels of MBG as seen with experimental renal failure caused a similar degree of cardiac fibrosis [91,92]. Activation of Na/K-ATPase signaling as evidenced by increases in both Src and MAPK phosphorylation in cardiac tissue was also seen along with the cardiac fibrosis [91,92].
Based on these in vivo results, we examined whether MBG and other CTS had effects on fibroblasts grown in culture. First, we saw that both MBG and other CTS (e.g., ouabain, digoxin) caused fibroblasts grown to confluence to increase proline incorporation as well as collagen production, the latter measured with Western blot. This was, again, coincident with evidence of Na/K-ATPase signaling in that Src and MAPK activation could be observed as well as the effectiveness of ROS scavenging and Src inhibition in preventing the increases in proline incorporation and collagen production [92]. Increases in mRNA for collagen following exposure to MBG was also noted [92]. Interestingly, we did not note increases in TGF beta or SMAD proteins, but antagonism of the TGF beta system with SB421542 did block MBG induced stimulation of collagen production [92]. Based on previous work in dermal fibroblasts demonstrating that Fli-1 is a negative regulator of collagen synthesis [94], we chose to examine whether MBG signaling altered the expression of Fli-1. We noted that MBG induced decreases in Fli-1 expression in several types of fibroblasts (cardiac, renal and dermal), and we further noted that the decreases in Fli-1 appeared to be necessary for MBG to induce increases in collagen [95]. Further studies demonstrated that MBG appears to induce translocation of PKCdelta from the cytosol to the nucleus in a PLC-dependant manner, and that the translocation of PKCdelta appeared to result in the phosphorylation and subsequent degradation of Fli-1 [95].
Because of these results as well as controversy as to how mineralocorticoid antagonists ameliorated cardiac fibrosis, we examined the effects of spironolactone and its major metabolite, canrenone, in a series of in vitro and in vivo studies. We observed that both spironolactone and canrenone could attenuate MBG induced increases in collagen production by cardiac fibroblasts, a finding which was corroborated in vivo by marked attenuation of the cardiac fibrosis caused by experimental renal failure with spironolactone treatment [96]. However, in vitro, we could not see a substantial effect of aldosterone on cardiac collagen production. That said, we did observe that both spironolactone and canrenone both prevented MBG signaling. Moreover, both spironolactone and canrenone appeared to act as competitive inhibitors of CTS binding to the Na/K-ATPase [96], a finding first proposed by Finnotti and Palatini [97–99]. Based on these findings, it appears that CTS signaling may be a fertile area for therapeutic drug development.
As mentioned above, the effects of MBG (and other CTS) are not specific for cardiac fibroblasts. In fact, renal fibroblasts have a very similar response as cardiac fibroblasts [100]. This suggested that MBG might potentially be involved in renal fibrosis. Moving back to the in vivo model of MBG infusion, we observed that MBG administration was associated with increases in renal collagen content. We also observed that Snail, a transcription factor known to be involved in epithelial-mesenchymal transformation (EMT), appeared to be up-regulated with MBG infusion. In cell culture, LLC-PK1 cells could be induced to undergo EMT by exposure to MBG in a dose and time dependent fashion [100]. This suggests the following “trade off” which might explain why high salt diet appears to lead to hypertension and renal functional impairment over a long period of time in humans (Figure 3).
Figure 3. Salt, hypertension and renal failure. Trade off of CTS effects?
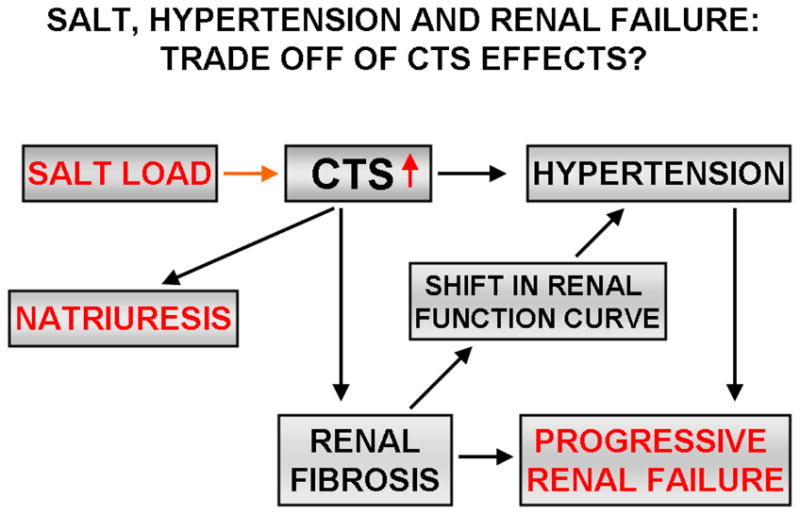
The schema illustrates how CTS may be the molecular link explaining how long exposure to a high salt diet may produce hypertension and cardiovascular disease. Specifically, we propose that the profibrotic effects of CTS may, as a trade off for maintaining sodium homeostasis, initially sub-clinically injure the renal parenchyma and in this manner, shift the relationship between blood pressure and sodium. Because of this, patients might be expected to develop hypertension which may, in and of itself, increase circulating CTS levels and synergistically injure organs including the heart and kidney.
In addition to their potential role in uremic cardiomyopathy and renal fibrosis, CTS-induced synthesis of collagen may be one of the factors contributing to vascular dysfunction in preeclampsia. Recently, we demonstrated that development of preeclampsia is associated with elevated levels of MBG in placentae, and decreased levels of Fli-1 and markedly increased levels of collagen-1 in umbilical arteries [101]. Notably, in the in vitro experiments, rings of umbilical arteries from patients with preeclampsia, in the presence of unaltered responsiveness to endothelin-1, exhibited a marked reduction in the responsiveness to vasorelaxant action of sodium nitroprusside, as compared to that in the vessels obtained from subjects with uncomplicated pregnancies [101].
Although much of our discussion has revolved around the “newer” signaling function of the pump, i.e., the signal transduction mediated through its caveolar association with Src, the EGFR and other signaling partners, the classical concept that CTS signal by inhibiting the pumping function of the Na/K-ATPase is still relevant. For example, it is well known that red blood cells of patients with end stage renal disease or preeclampsia, as well as NaCl-loaded DS, have decreases in Na/K-ATPase activity which can be reversed by incubation, ex vivo, with antibodies to MBG or with Digibind, an ovine digoxin antibody which interacts with CTS [13,43,102]. From these and other data which have been summarized in several recent reviews, we would propose that both the classical and alternative pathways may work both in parallel as well as synergistically to effect physiological consequences of CTS binding to the Na/K-ATPase.
9. Conclusions
A number of studies have now validated the initial intuition of deWardener, Blaustein and the others [2–7], and possibly extended it beyond what was initially conceived. In fact, it is becoming clear that several endogenous factors including EO, MBG, natriuretic peptides interact with each other on multiple levels and in complex manner and together account for the effects of the putative natriuretic hormone. The role of CTS viewed only as natriuretic hormones is an incomplete view. The recently described “signaling” function of the Na/K-ATPase has presented a number of potential therapeutic targets for existing medications as well as for pharmacological development addressing diseases ranging from hypertension to progressive renal failure.
Acknowledgments
This work was supported by the Intramural Research Program, National Institute on Aging, National Institutes of Health (OVF and AYB), and by NIH grant HL071556 (JIS).
References
- 1.Folkow B. Physiological aspects of primary hypertension. Physiol Rev. 1992;62:347–504. doi: 10.1152/physrev.1982.62.2.347. [DOI] [PubMed] [Google Scholar]
- 2.DeWardener HE, Clarkson EM. Concept of natriuretic hormone. Physiol Rev. 1985;65:658–759. doi: 10.1152/physrev.1985.65.3.658. [DOI] [PubMed] [Google Scholar]
- 3.Dahl LK, Knudsen KD, Iwai J. Humoral transmission of hypertension: Evidence from parabiosis. Circ Res. 1969;24(5Suppl):21–33. [PubMed] [Google Scholar]
- 4.Blaustein MP. Sodium ions, calcium ions, blood pressure regulation and hypertension: A reassessment and a hypothesis. Am J Physiol. 1977;232:C167–C173. doi: 10.1152/ajpcell.1977.232.5.C165. [DOI] [PubMed] [Google Scholar]
- 5.Haddy FJ, Overbeck HW. The role of humoral agents in volume expanded hypertension. Life Sci. 1976;19:935–947. doi: 10.1016/0024-3205(76)90284-8. [DOI] [PubMed] [Google Scholar]
- 6.Overbeck HW, Pamnani MB, Akera T, Brody TM, Haddy FJ. Depressed function of a ouabain-sensitive sodium-potassium pump in blood vessels from renal hypertensive dogs. Circ Res. 1976;38(6 Suppl 2):48–52. doi: 10.1161/01.res.38.6.48. [DOI] [PubMed] [Google Scholar]
- 7.Gruber KA, Whitaker JM, Buckalew VM. Endogenous digitalis-like substance in plasma of volume-expanded dogs. Nature. 1980;287:743–745. doi: 10.1038/287743a0. [DOI] [PubMed] [Google Scholar]
- 8.Hamlyn JM, Ringel R, Schaeffer J, Levinson PD, Hamilton BP, Kowarski AA, Blaustein MP. A circulating inhibitor of (Na,K)ATPase associated with essential hypertension. Nature. 1982;300:650–652. doi: 10.1038/300650a0. [DOI] [PubMed] [Google Scholar]
- 9.Kojima I, Yoshihara S, Ogata E. Involvement of endogenous digitalis-like substance in genesis of deoxycorticosterone-salt hypertension. Life Sci. 1982;30:1775–1781. doi: 10.1016/0024-3205(82)90313-7. [DOI] [PubMed] [Google Scholar]
- 10.Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, Mathews WR, Ludens JH. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci USA. 1991;88:6259–6563. doi: 10.1073/pnas.88.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider R, Wray V, Nimtz M, Lehmann WD, Kirch U, Antolovic R, Schoner W. Bovine adrenals contain, in addition to ouabain, a second inhibitor of the sodium pump. J Biol Chem. 1998;273:784–792. doi: 10.1074/jbc.273.2.784. [DOI] [PubMed] [Google Scholar]
- 12.Manunta P, Messaggio E, Ballabeni C, Sciarrone MT, Lanzani C, Ferrandi M, Hamlyn JM, Cusi D, Galletti F, Bianchi G. Salt Sensitivity Study Group of the Italian Society of Hypertension, Plasma ouabain-like factor during acute and chronic changes in sodium balance in essential hypertension. Hypertension. 2001;38:198–203. doi: 10.1161/01.hyp.38.2.198. [DOI] [PubMed] [Google Scholar]
- 13.Blanco G, Mercer RW. Isozymes of the Na/K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1988;275:F633–F650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- 14.Fedorova OV, Bagrov AY. Endogenous cardenolide and bufadienolides Na/K-ATPase inhibitors. How they work together in NaCl-sensitive hypertension. Front Biosci. 2005;10:2250–2256. doi: 10.2741/1694. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi H, Iyoda I, Takeda K, Okajima H, Sasaki S, Yoshimura M, Nakagawa M, Ijichi H. Centrally-induced vasopressor responses to ouabain are augmented in spontaneously hypertensive rats. Clin Exp Hypertens. 1984;A6:1499–1515. doi: 10.3109/10641968409044065. [DOI] [PubMed] [Google Scholar]
- 16.Iyoda I, Takahashi H, Lee LC, Okajima H, Inoue A, Sasaki S, Takeda K, Yoshimura M, Ijichi H. Cardiovascular and sympathetic responses to ouabain injected into the hypothalamus in rats. Cardiovasc Res. 1986;20:294–298. doi: 10.1093/cvr/20.4.294. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi H, Matsusawa M, Ikegaki I, Nishimura M, Yoshimura M, Yamada H, Sano Y. Brain renin-angiotensin system and the hypothalamic, digitalis-like Na,K-ATPase inhibitor in rats. Clin Exp Hypertens. 1988;A10:1285–1287. doi: 10.1080/07300077.1988.11878920. [DOI] [PubMed] [Google Scholar]
- 18.Leenen FHH, Ruzicka M, Huang BS. The brain and salt-sensitive hypertension. Curr Hypertens Rep. 2002;4:129–135. doi: 10.1007/s11906-002-0037-y. [DOI] [PubMed] [Google Scholar]
- 19.Huang BS, Leenen FHH. Sympathoexcitatory and pressor responses to increased brain sodium and ouabain are mediated via brain ANGII. Am J Physiol. 1996;270:H275–H280. doi: 10.1152/ajpheart.1996.270.1.H275. [DOI] [PubMed] [Google Scholar]
- 20.Huang BS, Leenen FH. Both brain angiotensin II and “ouabain” contribute to sympathoexcitation and hypertension in Dahl S rats on high salt intake. Hypertension. 1998;32:1028–1033. doi: 10.1161/01.hyp.32.6.1028. [DOI] [PubMed] [Google Scholar]
- 21.Leenen FHH, Harmsen E, Yu H. Dietary sodium and central vs. peripheral ouabain-like activity in Dahl salt-sensitive vs. salt-resistant rats. Am J Physiol. 1994;267:H1969–H1920. doi: 10.1152/ajpheart.1994.267.5.H1916. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Leenen FHH. Brain sodium channels and central sodium-induced increases in brain ouabain-like compound and blood pressure. J Hypertens. 2003;21:1519–1524. doi: 10.1097/00004872-200308000-00016. [DOI] [PubMed] [Google Scholar]
- 23.Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev. 2009;61:9–38. doi: 10.1124/pr.108.000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flier JS, Maratos-Flier E, Pallota JA, McIsaac D. Endogenous digitalis-like activity in the plasma of the toad Bufo marinus. Nature. 1979;279:341–343. doi: 10.1038/279341a0. [DOI] [PubMed] [Google Scholar]
- 25.Flier JS, Edwards MW, Daly JW, Myers CW. Widespread occurrence in frogs and toads of skin compounds interacting with the ouabain site of Na,K-ATPase. Science. 1980;208:503–505. doi: 10.1126/science.6245447. [DOI] [PubMed] [Google Scholar]
- 26.Lichtstein D, Gati I, Babila T, Katz U. Effect of salt acclimation on digitalis-like compounds in the toad. Biochim Biophys Acta. 1991;1073:65–68. doi: 10.1016/0304-4165(91)90183-h. [DOI] [PubMed] [Google Scholar]
- 27.Kieval RS, Butler VP, Derguini F, Bruening RC, Rosen MR. Cellular electrophysiologic effects of vertebrate digitalis-like substances. J Am Coll Cardiol. 1988;11:637–643. doi: 10.1016/0735-1097(88)91543-4. [DOI] [PubMed] [Google Scholar]
- 28.Goto A, Yamada K, Ishii M, Sugimoto T, Yoshioka M. Immunoreactivity of endogenous digitalislike factors. Biochem Pharmacol. 1991;41:1261–1263. doi: 10.1016/0006-2952(91)90668-u. [DOI] [PubMed] [Google Scholar]
- 29.Oda M, Kurosawa M, Numazawa S, Tanaka S, Akizawa T, Ito K, Maeda M, Yoshida T. Determination of bufalin-like immunoreactivity in serum of humans and rats by time-resolved fluoroimmunoassay for using a monoclonal antibody. Life Sci. 2001;68:1107–1117. doi: 10.1016/s0024-3205(00)01013-4. [DOI] [PubMed] [Google Scholar]
- 30.Lichtstein D, Gati I, Samuelov S, Berson D, Rosenman Y, Landau L, Deutsch J. Identification of digitalis-like compounds in human cataractous lenses. Eur J Biochem. 1993;216:261–268. doi: 10.1111/j.1432-1033.1993.tb18141.x. [DOI] [PubMed] [Google Scholar]
- 31.Sich B, Kirch U, Tepel M, Zidek W, Schoner W. Pulse pressure correlates in humans with a proscillaridin-A immunoreactive compound. Hypertension. 1996;27:1073–1077. doi: 10.1161/01.hyp.27.5.1073. [DOI] [PubMed] [Google Scholar]
- 32.Hilton PJ, White RW, Lord GA, Garner GV, Gordon DB, Hilton MJ, Forni LG, McKinnon W, Ismail FM, Keenan M, Jones K, Morden WE. An inhibitor of the sodium pump obtained from human placenta. Lancet. 1996;348:303–305. doi: 10.1016/s0140-6736(96)02257-x. [DOI] [PubMed] [Google Scholar]
- 33.Bagrov AY, Dmitrieva RI, Fedorova OV, Kazakov GP, Roukoyatkina NI, Shpen VM. Endogenous marinobufagenin-like immunoreactive substance. A possible endogenous Na,K-ATPase inhibitor with vasoconstrictor activity. Am J Hypertens. 1996;9:982–990. doi: 10.1016/0895-7061(96)00148-3. [DOI] [PubMed] [Google Scholar]
- 34.Fedorova OV, Bagrov AY. Inhibition of Na/K ATPase from rat aorta by two endogenous Na/K pump inhibitors, ouabain and marinobufagenin. Evidence of interaction with different alpha-subunit isoforms. Am J Hypertens. 1997;10:929–935. doi: 10.1016/s0895-7061(97)00096-4. [DOI] [PubMed] [Google Scholar]
- 35.Anderson DE, Fedorova OV, Morrell CH, Longo DL, Kashkin VA, Metzler JD, Bagrov AY, Lakatta EG. Endogenous sodium pump inhibitors and age-associated increases in salt sensitivity of blood pressure in normotensives. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1248–R1254. doi: 10.1152/ajpregu.00782.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bagrov AY, Fedorova OV, Dmitrieva RI, French AW, Anderson DE. Plasma marinobufagenin-like and ouabain-like immunoreactivity during saline volume expansion in anesthetized dogs. Cardiovasc Res. 1996;31:296–305. [PubMed] [Google Scholar]
- 37.Fedorova OV, Doris PA, Bagrov AY. Endogenous marinobufagenin-like factor in acute plasma volume expansion. Clin Exp Hypertens. 1998;20:581–591. doi: 10.3109/10641969809053236. [DOI] [PubMed] [Google Scholar]
- 38.Fedorova OV, Anderson DE, Lakatta EG, Bagrov AY. Interaction of high sodium chloride intake and psychosocial stress on endogenous ligands of the sodium pump and blood pressure in normotensive rats. Am J Physiol. 2001;281:R352–R358. doi: 10.1152/ajpregu.2001.281.1.R352. [DOI] [PubMed] [Google Scholar]
- 39.Periyasamy SM, Liu J, Tanta F, Kabak B, Wakefield B, Malhotra D, Kennedy DJ, Nadoor A, Fedorova OV, Gunning W, Xie Z, Bagrov AY, Shapiro JI. Salt loading induces redistribution of the plasmalemmal Na/K-ATPase in proximal tubule cells. Kidney Int. 2005;67:1868–1877. doi: 10.1111/j.1523-1755.2005.00285.x. [DOI] [PubMed] [Google Scholar]
- 40.Gonick HC, Ding Y, Vaziri ND, Bagrov AY, Fedorova OV. Simultaneous measurement of marinobufagenin, ouabain and hypertension-associated protein in various disease state. Clin Exp Hypertens. 1998;20:617–627. doi: 10.3109/10641969809053240. [DOI] [PubMed] [Google Scholar]
- 41.Fridman AI, Matveev SA, Agalakova NI, Fedorova OV, Lakatta EG, Bagrov AY. Marinobufagenin, an endogenous ligand of α-1 Na/K-ATPase, is a marker of congestive heart failure severity. J Hypertens. 2002;20:1189–1194. doi: 10.1097/00004872-200206000-00032. [DOI] [PubMed] [Google Scholar]
- 42.Lopatin DA, Ailamazian EK, Dmitrieva RI, Shpen VM, Fedorova OV, Doris PA, Bagrov AY. Circulating bufodienolide and cardenolide sodium pump inhibitors in preeclampsia. J Hypertens. 1999;17:1179–1187. doi: 10.1097/00004872-199917080-00018. [DOI] [PubMed] [Google Scholar]
- 43.Fedorova OV, Simbirtsev AS, Kolodkin NI, Kotov AY, Agalakova NI, Kashkin VA, Tapilskaya NI, Bzhelyansky AM, Reznik VA, Nikitina ER, Frolova EV, Budny GV, Longo DL, Lakatta EG, Bagrov AY. Monoclonal antibody to an endogenous bufadienolide, marinobufagenin, reverses preeclampsia-induced Na/K-ATPase inhibition in lowers blood pressure in NaCl-sensitive hypertension. J Hypertens. 2008;26:2414–2425. doi: 10.1097/HJH.0b013e328312c86a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fedorova OV, Tapilskaya NI, Bzhelyansky AM, Frolova EV, Nikitina ER, Reznik VA, Kashkin VA, Bagrov AY. Interaction of Digibind with endogenous cardiotonic steroids from preeclamptic placentae. J Hypertens. 2010;28:361–366. doi: 10.1097/HJH.0b013e328333226c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Komiyama Y, Dong XH, Nishimura N, Masaki H, Yoshika M, Masuda M, Takahashi H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem. 2005;38:36–45. doi: 10.1016/j.clinbiochem.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 46.Yoshika M, Komiyama Y, Konishi M, Akizawa T, Kobayashi T, Date M, Kobatake S, Masuda M, Masaki H, Takahashi H. Novel digitalis-like factor, marinobufotoxin, isolated from cultured Y-1 cells, and its hypertensive effect in rats. Hypertension. 2007;49:209–214. doi: 10.1161/01.HYP.0000250433.64202.78. [DOI] [PubMed] [Google Scholar]
- 47.Hillyard SD, Lu E, Gonick HC. Further characterization of the natriuretic factor derived from kidney tissue of volume-expanded rats. Effects on short-circuit current and sodium-potassium-adenosine triphosphatase activity. Circ Res. 1976;38:250–255. doi: 10.1161/01.res.38.4.250. [DOI] [PubMed] [Google Scholar]
- 48.Fedorova OV, Kolodkin NI, Agalakova NI, Lakatta EG, Bagrov AY. Marinobufagenin, an endogenous alpha-1 sodium pump ligand, in hypertensive Dahl salt-sensitive rats. Hypertension. 2001;37:462–466. doi: 10.1161/01.hyp.37.2.462. [DOI] [PubMed] [Google Scholar]
- 49.Fedorova OV, Agalakova NI, Talan MI, Lakatta EG, Bagrov AY. Brain ouabain stimulates peripheral marinobufagenin via angiotensin II signalling in NaCl loaded Dahl-S rats. J Hypertens. 2005;23:1515–1523. doi: 10.1097/01.hjh.0000174969.79836.8b. [DOI] [PubMed] [Google Scholar]
- 50.Fedorova OV, Lakatta EG, Bagrov AY. Differential effects of acute NaCl loading on endogenous ouabain-like and marinobufagenin-like ligands of the sodium pump in Dahl hypertensive rats. Circulation. 2000;102:3009–3014. doi: 10.1161/01.cir.102.24.3009. [DOI] [PubMed] [Google Scholar]
- 51.Fedorova OV, Agalakova NI, Morrell CH, Lakatta EG, Bagrov AY. ANP differentially modulates marinobufagenin-induced sodium pump inhibition in kidney and aorta. Hypertension. 2006;48:1160–1168. doi: 10.1161/01.HYP.0000248129.20524.d0. [DOI] [PubMed] [Google Scholar]
- 52.Bagrov AY, Agalakova NI, Kashkin VA, Fedorova OV. Endogenous cardiotonic steroids and differential patterns of sodium pump inhibition in NaCl-loaded salt-sensitive and normotensive rats. Am J Hypertens. 2009;22:559–563. doi: 10.1038/ajh.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu J, Liang M, Liu L, Malhotra D, Xie Z, Shapiro JI. Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 2005;67:1844–1854. doi: 10.1111/j.1523-1755.2005.00283.x. [DOI] [PubMed] [Google Scholar]
- 54.Liu J, Kesiry R, Periyasamy SM, Malhotra D, Xie Z, Shapiro JI. Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int. 2004;66:227–241. doi: 10.1111/j.1523-1755.2004.00723.x. [DOI] [PubMed] [Google Scholar]
- 55.Oweis S, Wu L, Kiela PR, Zhao H, Malhotra D, Ghishan FK, Xie Z, Shapiro JI, Liu J. Cardiac glycoside downregulates NHE3 activity and expression in LLC-PK1 cells. Am J Physiol Renal Physiol. 2006;290:F997–F1008. doi: 10.1152/ajprenal.00322.2005. [DOI] [PubMed] [Google Scholar]
- 56.Liu J, Shapiro JI. Regulation of sodium pump endocytosis by cardiotonic steroids: Molecular mechanisms and physiological implications. Pathophysiology. 2007;14:171–181. doi: 10.1016/j.pathophys.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iwai J, Knudsen KD, Dahl LK, Heine M, Leitl G. Genetic influence on the development of renal hypertension in parabiotic rats. Evidence for a humoral factor. J Exp Med. 1969;129:507–522. doi: 10.1084/jem.129.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fedorova OV, Zhuravin IA, Agalakova NI, Yamova LA, Talan MI, Lakatta EG, Bagrov AY. Intrahippocampal microinjection of an exquisitely low dose of ouabain mimics NaCl loading and stimulates a bufadienolide Na/K-ATPase inhibitor. J Hypertens. 2007;25:1834–1844. doi: 10.1097/HJH.0b013e328200497a. [DOI] [PubMed] [Google Scholar]
- 59.Huang BS, Van Vliet BN, Leenen FH. Increases in CSF [Na+] precede the increases in blood pressure in Dahl S rats and SHR on a high-salt diet. Am J Physiol Heart Circ Physiol. 2004;287:H1160–H1166. doi: 10.1152/ajpheart.00126.2004. [DOI] [PubMed] [Google Scholar]
- 60.Wang H, Leenen FH. Brain sodium channels and central sodium-induced increases in brain ouabain-like compound and blood pressure. J Hypertens. 2003;21:1519–1524. doi: 10.1097/00004872-200308000-00016. [DOI] [PubMed] [Google Scholar]
- 61.Amin MS, Wang HW, Reza E, Whitman SC, Tuana BS, Leenen FH. Distribution of epithelial sodium channels and mineralocorticoid receptors in cardiovascular regulatory centers in rat brain. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1787–R1797. doi: 10.1152/ajpregu.00063.2005. [DOI] [PubMed] [Google Scholar]
- 62.Gabor A, Leenen FH. Mechanisms in the PVN mediating local and central sodium-induced hypertension in Wistar rats. Am J Physiol Regul Integr Comp Physiol. 2009;296:R618–R630. doi: 10.1152/ajpregu.90417.2008. [DOI] [PubMed] [Google Scholar]
- 63.Orlov SN, Mongin AA. Salt-sensing mechanisms in blood pressure regulation and hypertension. Am J Physiol Heart Circ Physiol. 2007;293:H2039–H2053. doi: 10.1152/ajpheart.00325.2007. [DOI] [PubMed] [Google Scholar]
- 64.Amin MS, Reza E, Wang H, Leenen FH. Sodium transport in the choroid plexus and salt-sensitive hypertension. Hypertension. 2009;54:860–867. doi: 10.1161/HYPERTENSIONAHA.108.125807. [DOI] [PubMed] [Google Scholar]
- 65.Orosz DE, Hopfer U. Pathophysiological consequences of changes in the coupling ration of Na,K-ATPase for renal sodium reabsorption and its implications for hypertension. Hypertension. 1996;27:219–227. doi: 10.1161/01.hyp.27.2.219. [DOI] [PubMed] [Google Scholar]
- 66.Briones AM, Xavier FE, Arribas SM, Gonzalez MC, Rossoni LV, Alonso MJ, Salaices M. Alterations in structure and mechanics of resistance arteries from ouabain-induced hypertensive rats. Am J Physiol Heart Circ Physiol. 2006;291:H193–H201. doi: 10.1152/ajpheart.00802.2005. [DOI] [PubMed] [Google Scholar]
- 67.Dostanic-Larson I, Lorenz JN, Van Huysse JW, Neumann JC, Moseley AE, Lingrel JB. Physiological role of the α1- and α2-isoforms of the Na-K-ATPase and biological significance of their cardiac glycoside binding site. Am J Physiol Regul Integr Comp Physiol. 2006;290:R524–R528. doi: 10.1152/ajpregu.00838.2005. [DOI] [PubMed] [Google Scholar]
- 68.Hamlyn JM, Manunta P. Ouabain, digitalis-like factors and hypertension. J Hypertens Suppl. 1992;10:S99–S111. [PubMed] [Google Scholar]
- 69.Bianchi G, Tripodi G, Casari G, Salardi S, Barber BR, Garcia R, Leoni P, Torielli L, Cusi D, Ferrandi M, et al. Two point mutations within the adducin genes are involved in blood pressure variation. Proc Natl Acad Sci USA. 1994;26:3999–4003. doi: 10.1073/pnas.91.9.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferrandi M, Molinari I, Barassi P, Minotti E, Bianchi G, Ferrari P. Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J Biol Chem. 2004;279:33306–33314. doi: 10.1074/jbc.M402187200. [DOI] [PubMed] [Google Scholar]
- 71.Efendiev R, Krmar RT, Ogimoto G, Zwiller J, Tripodi G, Katz AI, Bianchi G, Pedemonte CH, Bertorello AM. Hypertension-linked mutation in the adducin alpha-subunit leads to higher AP2-mu2 phosphorylation and impaired Na,K-ATPase trafficking in response to GPCR signals and intracellular sodium. Circ Res. 1994;95:1100–1108. doi: 10.1161/01.RES.0000149570.20845.89. [DOI] [PubMed] [Google Scholar]
- 72.Torielli L, Tivodar S, Montella RC, Iacone R, Padoani G, Tarsini P, Russo O, Sarnataro D, Strazzullo P, Ferrari P, Bianchi G, Zurzolo C. alpha-Adducin mutations increase Na/K pump activity in renal cells by affecting constitutive endocytosis: implications for tubular Na reabsorption. Am J Physiol Renal Physiol. 2008;295:F478–F487. doi: 10.1152/ajprenal.90226.2008. [DOI] [PubMed] [Google Scholar]
- 73.Ferrari P, Ferrandi M, Tripodi G, Torielli L, Padoani G, Minotti E, Melloni P, Bianchi G. PST 2238: a new antihypertensive compound that modulates Na,K-ATPase in genetic hypertension. J Pharmacol Exp Ther. 1999;288:1074–1083. [PubMed] [Google Scholar]
- 74.Wang JG, Staessen JA, Messaggio E, Nawrot T, Fagard R, Hamlyn JM, Bianchi G, Manunta P. Salt, endogenous ouabain and blood pressure interactions in the general population. J Hypertens. 2003;21:1475–1481. doi: 10.1097/00004872-200308000-00010. [DOI] [PubMed] [Google Scholar]
- 75.Manunta P, Maillard M, Tantardini C, Simonini M, Lanzani C, Citterio L, Stella P, Casamassima N, Burnier M, Hamlyn JM, et al. Relationships among endogenous ouabain, alpha-adducin polymorphisms and renal sodium handling in primary hypertension. J Hypertens. 2008;26:914–920. doi: 10.1097/HJH.0b013e3282f5315f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, et al. Sodium pump alpha2 subunits control myogenic tone and blood pressure in mice. J Physiol. 2005;569:243–256. doi: 10.1113/jphysiol.2005.091801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dostanic-Larson I, Van Huysse JW, Lorenz JN, Lingrel JB. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci U S A. 2005;102:15845–15850. doi: 10.1073/pnas.0507358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang H, Haas M, Liang M, Cai T, Tian J, Li S, Xie Z. Ouabain assembles signaling cascades through the caveolar Na+/K+-ATPase. J Biol Chem. 2004;279:17250–17529. doi: 10.1074/jbc.M313239200. [DOI] [PubMed] [Google Scholar]
- 79.Liang M, Tian J, Liu L, Pierre S, Liu J, Shapiro JI, Xie ZJ. Identification of a pool of non-pumping Na/K-ATPase. J Biol Chem. 2007;282:10585–10593. doi: 10.1074/jbc.M609181200. [DOI] [PubMed] [Google Scholar]
- 80.Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem. 2000;275:27832–27837. doi: 10.1074/jbc.M002951200. [DOI] [PubMed] [Google Scholar]
- 81.Haas M, Wang H, Tian J, Xie Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J Biol Chem. 2002;277:18694–18702. doi: 10.1074/jbc.M111357200. [DOI] [PubMed] [Google Scholar]
- 82.Kometiani P, Li J, Gnudi L, Kahn BB, Askari A, Xie Z. Multiple signal transduction pathways link Na+/K+-ATPase to growth-related genes in cardiac myocytes. The roles of Ras and mitogen-activated protein kinases. J Biol Chem. 1998;273:15249–15256. doi: 10.1074/jbc.273.24.15249. [DOI] [PubMed] [Google Scholar]
- 83.Liu J, Tian J, Haas M, Shapiro JI, Askari A, Xie Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem. 2000;275:27838–27844. doi: 10.1074/jbc.M002950200. [DOI] [PubMed] [Google Scholar]
- 84.Tian J, Liu J, Garlid KD, Shapiro JI, Xie Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. Mol Cell Biochem. 2003;242:181–187. [PubMed] [Google Scholar]
- 85.Liu L, Mohammadi K, Aynafshar B, Wang H, Li D, Liu J, Ivanov AV, Xie Z, Askari A. Role of caveolae in signal-transducing function of cardiac Na+/K+-ATPase. Am J Physiol Cell Physiol. 2003;284:C1550–C1560. doi: 10.1152/ajpcell.00555.2002. [DOI] [PubMed] [Google Scholar]
- 86.Mohammadi K, Kometiani P, Xie Z, Askari A. Role of protein kinase C in the signal pathways that link Na+/K+-ATPase to ERK1/2. J Biol Chem. 2001;276:42050–42056. doi: 10.1074/jbc.M107892200. [DOI] [PubMed] [Google Scholar]
- 87.Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, Maksimova E, Huang XY, Xie ZJ. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell. 2006;17:317–326. doi: 10.1091/mbc.E05-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu J, Periyasamy SM, Gunning W, Fedorova OV, Bagrov AY, Malhotra D, Xie Z, Shapiro JI. Effects of cardiac glycosides on sodium pump expression and function in LLC-PK1 and MDCK cells. Kidney Int. 2002;62:2118–21125. doi: 10.1046/j.1523-1755.2002.00672.x. [DOI] [PubMed] [Google Scholar]
- 89.Priyadarshi S, Valentine B, Han C, Fedorova OV, Bagrov AY, Liu J, Periyasamy SM, Kennedy D, Malhotra D, Xie Z, Shapiro JI. Effect of green tea extract on cardiac hypertrophy following 5/6 nephrectomy in the rat. Kidney Int. 2003;63:1785–1790. doi: 10.1046/j.1523-1755.2003.00914.x. [DOI] [PubMed] [Google Scholar]
- 90.Xie Z, Cai T. Na+-K+--ATPase-mediated signal transduction: from protein interaction to cellular function. Mol Interv. 2003;3:157–168. doi: 10.1124/mi.3.3.157. [DOI] [PubMed] [Google Scholar]
- 91.Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB, Xie Z, Malhotra D, Kolodkin NI, Lakatta EG, Fedorova OV, Bagrov AY, Shapiro JI. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension. 2006;47:488–495. doi: 10.1161/01.HYP.0000202594.82271.92. [DOI] [PubMed] [Google Scholar]
- 92.Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, Smaili S, Periyasamy SM, Hariri IM, Fedorova L, Liu J, Wu L, Kahaleh MB, Xie Z, Malhotra D, Fedorova OV, Kashkin VA, Bagrov AY, Shapiro JI. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension. 2007;49:215–224. doi: 10.1161/01.HYP.0000252409.36927.05. [DOI] [PubMed] [Google Scholar]
- 93.Kennedy DJ, Elkareh J, Shidyak A, Shapiro AP, Smaili S, Mutgi K, Gupta S, Tian J, Morgan E, Khouri S, Cooper CJ, Periyasamy SM, Xie Z, Malhotra D, Fedorova OV, Bagrov AY, Shapiro JI. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am J Physiol Renal Physiol. 2008;294:F450–F454. doi: 10.1152/ajprenal.00472.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kubo M, Czuwara-Ladykowska J, Moussa O, Markiewicz M, Smith E, Silver RM, Jablonska S, Blaszczyk M, Watson DK, Trojanowska M. Persistent down-regulation of Fli1, a suppressor of collagen transcription, in fibrotic scleroderma skin. Am J Pathol. 2003;163:571–581. doi: 10.1016/S0002-9440(10)63685-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Elkareh J, Periyasamy SM, Shidyak A, Vetteth S, Schroeder J, Raju V, Hariri IM, El-Okdi N, Gupta S, Fedorova L, Liu J, Fedorova OV, Kahaleh MB, Xie Z, Malhotra D, Watson DK, Bagrov AY, Shapiro JI. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli-1: implications for uremic cardiomyopathy. Am J Physiol Renal Physiol. 2009;296:F1219–F1226. doi: 10.1152/ajprenal.90710.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tian J, Shidyak A, Periyasamy SM, Haller S, Taleb M, El-Okdi N, Elkareh J, Gupta S, Gohara S, Fedorova OV, Cooper CJ, Xie Z, Malhotra D, Bagrov AY, Shapiro JI. Spironolactone attenuates experimental uremic cardiomyopathy by antagonizing marinobufagenin. Hypertension. 2009;54:1313–1320. doi: 10.1161/HYPERTENSIONAHA.109.140038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Finotti P, Palatini P. Canrenone as a partial agonist at the digitalis receptor site of sodium-potassium-activated adenosine triphosphatase. J Pharmacol Exp Ther. 1981;217:784–790. [PubMed] [Google Scholar]
- 98.de Mendonça M, Grichois ML, Pernollet MG, Wauquier I, Trouillet-Thormann B, Meyer P, Devynck MA, Garay R. Antihypertensive effect of canrenone in a model where endogenous ouabain-like factors are present. J Cardiovasc Pharmacol. 1988;11:75–83. doi: 10.1097/00005344-198801000-00012. [DOI] [PubMed] [Google Scholar]
- 99.Semplicini A, Serena L, Valle R, Ceolotto G, Felice M, Fontebasso A, Pessina AC. Ouabain-inhibiting activity of aldosterone antagonists. Steroids. 1995;60:110–113. doi: 10.1016/0039-128x(94)00005-w. [DOI] [PubMed] [Google Scholar]
- 100.Fedorova LV, Raju V, El-Okdi N, Shidyak A, Kennedy DJ, Vetteth S, Giovannucci DR, Bagrov AY, Fedorova OV, Shapiro JI, Malhotra D. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: implication of epithelial-to-mesenchymal transition. Am J Physiol Renal Physiol. 2009;296:F922–F934. doi: 10.1152/ajprenal.90605.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nikitina ER, Nikandrova ES, Frolova EV, Fadeev AV, Shman VV, Fedorova OV, Mikhailov AV, Bagrov AY. Endogenous digitalis and impairment of vasorelaxation in preeclampsia. Arterial Hypertension (in Russian) 2009;15:454–457. [Google Scholar]
- 102.Periyasamy SM, Chen J, Cooney D, Carter P, Omran E, Tian J, Priyadarshi S, Bagrov A, Fedorova O, Malhotra D, Xie Z, Shapiro JI. Effects of uremic serum on isolated cardiac myocyte calcium cycling and contractile function. Kidney Int. 2001;60:2367–2376. doi: 10.1046/j.1523-1755.2001.00053.x. [DOI] [PubMed] [Google Scholar]