

Abstract
Idiopathic epilepsies (IEs) are a group of disorders characterized by recurrent seizures in the absence of detectable brain lesions or metabolic abnormalities. IEs include common disorders with a complex mode of inheritance and rare Mendelian traits suggesting the occurrence of several alleles with variable penetrance. We previously described a large family with a recessive form of idiopathic epilepsy, named familial infantile myoclonic epilepsy (FIME), and mapped the disease locus on chromosome 16p13.3 by linkage analysis. In the present study, we found that two compound heterozygous missense mutations (D147H and A509V) in TBC1D24, a gene of unknown function, are responsible for FIME. In situ hybridization analysis revealed that Tbc1d24 is mainly expressed at the level of the cerebral cortex and the hippocampus. By coimmunoprecipitation assay we found that TBC1D24 binds ARF6, a Ras-related family of small GTPases regulating exo-endocytosis dynamics. The main recognized function of ARF6 in the nervous system is the regulation of dendritic branching, spine formation, and axonal extension. TBC1D24 overexpression resulted in a significant increase in neurite length and arborization and the FIME mutations significantly reverted this phenotype. In this study we identified a gene mutation involved in autosomal-recessive idiopathic epilepsy, unveiled the involvement of ARF6-dependent molecular pathway in brain hyperexcitability and seizures, and confirmed the emerging role of subtle cytoarchitectural alterations in the etiology of this group of common epileptic disorders.
Results and Discussion
Idiopathic epilepsies (IEs) are a group of disorders characterized by recurrent seizures in the absence of detectable brain lesions or metabolic abnormalities and affecting about 0.4% of the general population. Epidemiological studies highlighted the pivotal role of genetic factors in the etiology of these conditions.1 IEs include common disorders with a complex mode of inheritance and rare Mendelian traits suggesting the occurrence of several alleles with variable penetrance. The dissection of the complex genetics underlying IEs represented so far a challenging task, and alleles conferring susceptibility to seizures have not been identified yet. On the other hand, the investigation of rare Mendelian traits highlighted the critical role of genes encoding different ion channel subunits, including voltage-gated and ligand-gated channels, and shed light into epileptogenic mechanisms behind IEs.2,3 In addition, the identification of mutations in LGI14 (ADLTE [MIM 600512]) and EFHC15 (EJM [MIM 254770]) in familial forms of IE underlying subtle defects in embryonic or postnatal brain development provided evidence that pathogenesis of IE is more composite. In 2001, we described an autosomal-recessive early-onset idiopathic generalized epilepsy in a large family from Southern Italy characterized by myoclonic and generalized tonic-clonic seizures, photosensitivity, normal neurological and mental development, and good response to treatment (familial infantile myoclonic epilepsy, FIME [MIM 605021]) and mapped the FIME locus within a 3.4 cM interval on chromosome 16p13.3 between markers D16S3024 and D16S423.6,7
Toward the identification of the causative mutation, we performed high-density SNP genotyping by pyrosequencing and refined the FIME critical region to 2 Mb between rs35856 and rs9936111. The local Ethics Committee approved the study and a signed informed consent was obtained from family members participating to the study. Haplotype analysis indicated the occurrence of two distinct disease chromosomes suggestive of compound heterozygous mutations and confirmed initial mapping data (data not shown). The critical region contains 54 RefSeq genes, none of them with a definite role in neuronal excitability or epileptogenesis (Table S1 available online). The systematic mutational screening of 34 genes in two affected family members (III-1 and III-10) by Sanger sequencing of amplified exonic sequences and flanking intronic segments led to the identification of two compound heterozygous missense mutations in TBC1D24 (c.439G>C [p.D147H]; c.1526C>T [p.A509V]) (Figures 1A and 1B). These variants are not included in the SNP database and were not identified in 300 Italian controls. No other candidate mutations emerged from the remaining genes. We extended the mutational analysis to all available family members and confirmed that mutations segregate from different branches and that all patients affected by FIME are compound heterozygous carriers. Moreover, none of the unaffected family members carry both mutations. TBC1D24 encodes for a putative protein of 553 amino acids of unknown function (accession IDs: NM_020705.1 and NP_065756). BLASTP alignments indicated that TBC1D24 has no significant homology with other human proteins but is evolutionary conserved till lower vertebrates (HomoloGene:27469).
Figure 1.

Genetic Analysis of TBC1D24
(A) Pedigree of the family with FIME and segregation analysis of TBC1D24 mutations. Haplotypes are shown in colored bars. The red and green haplotypes cosegregate with c.1526C>T and c.439G>C TBC1D24 mutations, respectively. Affected patients are indicated by filled symbols.
(B) Electropherograms of c.439G>C and c.1526C>T mutations.
(C) Genomic organization and functional domains of human TBC1D24. Affected amino acids are highly conserved throughout evolution.
TBC1D24 is characterized by a Tre2/Bub2/Cdc16 (TBC) domain, shared by Rab GTPase-activating proteins (RabGAPs) and a TLDc domain with no reported putative function, despite occurring in four additional human genes.8,9 The identified mutations affect two highly conserved amino acids in TBC (D147H) and TLDc (A509V) domains (Figure 1C). The combination of TBC and TLDc domains is a unique feature among human proteins but it is found in about 30 proteins of different species.
The expression profile of TBC1D24 was evaluated in various human tissues by real-time PCR assay on an ABI Prism 7500 Real-Time PCR Systems (Applied Biosystems) with a TaqManMGB probe (assay ID; Hs00324855_m1, Applied Biosystems) specific for human TBC1D24. Each assay was carried out in triplicate, normalized to an endogenous reference (GAPDH [MIM 138400]), and expressed relative to a calibrator sample as previously described.10 TBC1D24 is expressed in several human tissues, with the highest level of expression in the brain (Figure 2A). To further analyze the distribution of TBC1D24 within the brain, RNA in situ hybridization was performed on coronal sections from 12-week-old mouse brain via an anti-digoxygenin Tbc1d24 antisense oligonucleotide probe, as previously described.11 The Tbc1d24 signal was mainly detected at the level of the cortex and the hippocampus (Figure 2B; Figure S1). In the cerebral cortex, Tbc1d24 was expressed through all layers, although more abundantly in layers V/VI (Figure 2C); in the hippocampus, Tbc1d24 was markedly expressed in the CA3 region and to a lower extent in the CA1 region and dentate gyrus (Figure 2D). Moreover, we evaluated the cortical expression of Tbc1d24 at different embryonic stages (days 15.5 and 18.5) and found that its expression increased during cortical development, particularly in the internal part of the cortical plate and in the subventricular zone (Figure S2).
Figure 2.
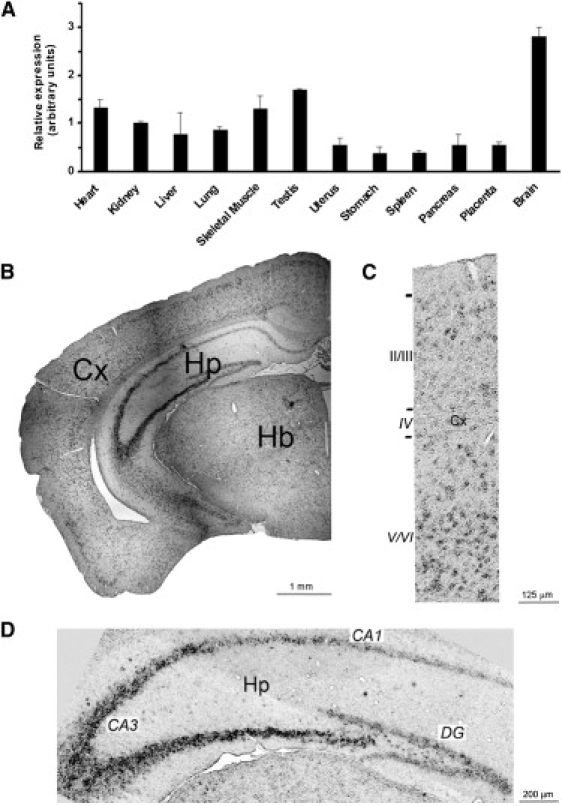
Expression Analysis of TBC1D24
(A) RT-PCR showing the expression profile of TBC1D24 in various human tissues. TBC1D24 is preferentially expressed in brain. Data are represented as means ± SEM.
(B) RNA in situ hybridization on brain sections from 12-week-old mouse shows that Tbc1d24 is abundantly expressed in the neocortex (Cx) and hippocampus (Hp).
(C) High-magnification image of the cerebral cortex revealed a higher expression of Tbc1d24 in deep cortical layers (V/VI) compared to superficial layers (II-III).
(D) High-magnification image of hippocampus shows the highest expression of Tbc1d24 in the CA3 region compared to the CA1 region and dentate gyrus (DG). Hb, hindbrain.
Sequence analysis showed that the TBC domain of TBC1D24 lacks critical residues conferring GAP properties to most RabGAPs, particularly the “arginine finger” at position 56 of the TBC domain consensus sequence (PF00566).12 Three human TBC proteins missing the arginine finger have been investigated and were all proven to lack RabGAP activity (Table S2). Among these, USP6/TRE17 and TBC1D3 have been shown to interact with the small GTPase ADP ribosylation factor 6 (ARF6 [MIM 600464]).13,14 ARF6 is implicated in the regulation of membrane trafficking between the plasma membrane and the endocytic compartment through the activation of the lipid-modifying enzymes phospholipase D and phosphotidyl-inositol-4-phosphate 5-kinase.15
We therefore investigated the possible interaction between TBC1D24 and ARF6 in COS7 cells (ATCC number CRL-1651) overexpressing GFP-tagged TBC1D24 and HA-tagged ARF6, in either its wild-type (WT), GDP-locked (T27N), or GTP-locked (Q67L) form.16 When transiently overexpressed in COS-7 cells and analyzed 36 hours after transfection, GFP-TBC1D24 showed a predominant cytoplasmatic localization (Figure 3A) and was partially expressed at the plasma membrane where it colocalized with coexpressed ARF6-HA (Figure 3B). For coimmunoprecipitation experiments, COS-7 cells were cotransfected with GFP-TBC1D24 and ARF6-HA and lysed 36 hr after transfection (lysis buffer: 50 mM Tris [pH 7.5], 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% Nonidet P40, 0.2 mM phenylmethylsulfonyl fluoride, 2 μg/ml pepstatin, and 1 μg/ml leupeptin). GFP-TBC1D24 or ARF6-HA were immunoprecipitated with anti-GFP or anti-HA polyclonal antibodies (Invitrogen), respectively, and interacting ARF6 or TBC1D24 were detected by retrospective immunoblotting with anti-HA or anti-GFP monoclonal antibodies (Millipore). ARF-6 coimmunoprecipitated with TBC1D24 (Figure 3C) as well as TBC1D24 coimmunoprecipitated with ARF6 (Figure S2). The intensity of the coimmunoprecipitated band increased in the presence of T27N-ARF6 and decreased in the presence of Q67L-ARF6, suggesting a GDP-dependent interaction (Figure 3C). When expressing TBC1D24 mutant forms, the interaction with ARF6 was significantly impaired by the D147H mutation and preserved in the A509V mutant (Figure 3C). These findings suggest an involvement of the TBC domain in the interaction with ARF6, as described for USP6,13 and reveal an essential role of the aspartic acid 147 in mediating TBC1D24-ARF6 interaction (Figure 3C).
Figure 3.
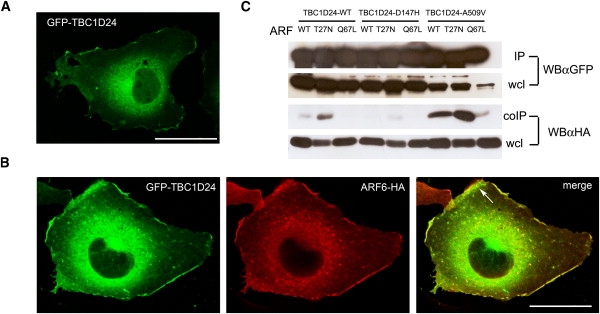
TBC1D24 Is an ARF6-Interacting Protein
(A) Representative image of a COS-7 cell transfected with GFP-TBC1D24 and visualized 36 hr after transfection.
(B) Representative images of a COS-7 cell cotransfected with GFP-TBC1D24 and ARF6-HA and visualized 36 hr after transfection. HA immunoreactivity was detected by HA antibody and Alexa546 conjugated secondary antibodies. The arrow in the merge panel shows colocalization of overexpressed ARF6 and TBC1D24 at the plasma membrane.
Scale bars represent 20 μm.
(C) TBC1D24-WT, D147H, and A509V were cotransfected with HA-tagged wild-type (WT), T27N, or Q67L ARF6 in COS-7 cells and immunoprecipitated (IP) with GFP antibody. Associated ARF6 (coIP) was detected by HA antibody (αHA) in retrospective western blotting (WB). Wcl, whole cell lysate.
The main function of ARF6 in the nervous system is the regulation of dendritic branching, spine formation, and axonal extension.17 To investigate the effect of TBC1D24 and FIME mutations in neuritogenesis, GFP-tagged wild-type and mutant forms of TBC1D24 were transiently expressed in primary cortical neurons prepared from C57Bl6J mouse embryos as previously described18 and neurite outgrowth and arborization were evaluated. Neurons were plated at low density (100 cells/mm2), transfected after 7 days in vitro (DIV) with lipofectamine transfection reagent (Invitrogen), and analyzed at 9 DIV. For analysis, neurons were fixed in 4% PFA, 4% sucrose in PBS and decorated with MAP2 antibody (Millipore) followed by Alexa 594 secondary antibody to distinguish dendrites from axons. For quantitative analysis of total neurite length, neurites were traced and their total length was measured. The analysis of neurite arborization was performed on 33 (GFP), 70 (WT), 43 (D147H), and 33 (A509V) neurons from three independent preparations via the Sholl analysis.19 Concentric circles with radii increasing at regular steps of 10 μm were centered to the cell body and the number of intersections was automatically evaluated with the Image J Sholl analysis plug-in. TBC1D24 overexpression resulted in a marked increase in neurite length and arborization compared to control, and the effect was evident at the level of both axonal and dendritic compartments (Figures 4A–4D). As the inactive ARF6 mutant (T27N) was shown to increase dendritic branching20 and axonal elongation,21 our results are consistent with a negative modulation of ARF6 function by TBC1D24. The FIME mutations significantly reverted this phenotype indicating a partial (D147H) or complete (A509V) loss of function (Figures 4A–4D). Notably, A509V mutation in the TLDc domain severely affected the ARF6-dependent TBC1D24 function. The TLDc domain is highly conserved through evolution from yeast to man, suggesting a key biological function. In mammals this domain is shared by genes involved in the response to oxidative stress, such as OXR1 and Ncoa7b, but its specific role has not been yet assessed.9
Figure 4.
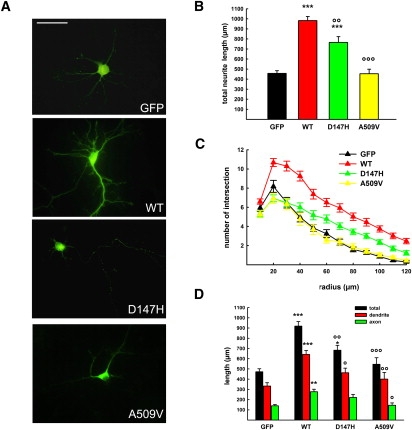
Functional Analysis of WT and Mutant TBC1D24
(A) Representative images of mouse cortical neurons transfected at 6 DIV with either GFP, GFP-TBC1D24-wt (WT), GFP-TBC1D24-D147H (D147H), or GFP-TBC1D24-A509V (A509V) and visualized 36 hr after transfection. Scale bar represents 50 μm.
(B and C) Overexpression of TBC1D24 provoked a massive increase in neurite length (B) and branching (C), whereas overexpression of its epileptogenic mutants (i.e., D147H and A509V) reverted this phenotype.
(D) Quantitative analysis of neurite (total), dendrite, or axon length in 33 cells for each genotype. Dendrites and axons were distinguished by MAP2 labeling and/or morphology. Data expressed as means ± SEM and compared via one-way analysis of variance (ANOVA, B and C) or repeated-measures ANOVA (D) followed by the Bonferroni's multiple comparison test. ∗∗∗p < 0.0001, ∗∗p < 0.001 versus GFP; °°°p < 0.0001, °°p < 0.001, °p < 0.01 versus GFP-TBC1D24-wt.
The identification of TBC1D24 mutations in patients with epilepsy provides evidence of the involvement of the ARF6-dependent molecular pathway in the generation of brain hyperexcitability and seizures. ARF6 has been shown to participate in several processes of neuronal development and plasticity by regulating axonal elongation and branching,20 dendrite arborization,21 and exo-endocytic cycling of synaptic vesicles.22 In addition, recent evidence points out a role for ARF6 in spine maturation and stability23 and in the control of AMPA receptor internalization during long-term depression,24 both processes implicated in the modulation of brain excitability.
The identification of TBC1D24 mutations in epileptic patients together with its predominant expression in critical epileptogenic brain areas also highlights a fundamental role of this protein in the regulation of neuronal network excitability. Moreover, the evidence that epileptogenic mutations affected neurite outgrowth and arborization suggests a critical role of TBC1D24 in developmentally regulated events essential for the morphological and functional maturation of neuronal circuitry. The increasing expression of TBC1D24 during embryogenesis in the cortex further strengthens this hypothesis.
Compelling evidence indicates that morpho-functional changes underlie epileptogenesis in different epileptic disorders with congenital or acquired gross structural brain abnormalities, such as defects in neuronal migration (e.g., lissencephaly or epileptic heterotopias), proliferation (e.g., tuberous sclerosis), and degeneration (e.g., progressive myoclonus epilepsies).25
However, recent data highlight the role of developmental dynamics also in idiopathic epilepsy and disclose novel mechanisms underlying brain hyperexcitability. LGI1,4 mutated in the autosomal-dominant lateral temporal lobe epilepsy, regulates the functional maturation and structural pruning of glutamatergic synapses during postnatal development, and its impairment markedly increases excitatory synaptic trasmission. EFHC1,5 which is involved in juvenile myoclonic epilepsy, one of the most frequent forms of idiopathic generalized epilepsy, interacts with microtubules to regulate cell division and cortical development and its loss disrupts radial migration of projection neurons in the developing rat neocortex.
In this study, we identified TBC1D24 as a gene involved in autosomal-recessive idiopathic epilepsy and showed that its protein product is a binding partner of ARF6 involved in neurite outgrowth. We unveiled the involvement of ARF6-dependent molecular pathway in brain hyperexcitability and seizures and confirmed the emerging role of subtle cytoarchitectural alterations in the etiology of this group of common epileptic disorders.
Acknowledgments
The authors wish to thank the family members for their participation in this study. We also thank Philippe Chavrier and Guillaume Montagnac (Institut Curie, Paris, France) for the kind gift of ARF6-HA constructs; Michele Zoli (University of Modena and Reggio Emilia, Modena, Italy) for statistical analysis and invaluable discussions, and Sonia Congia, Yoanne Clovis, and Franco Onofri for precious technical help. This work was supported by grants from the Italian Telethon Foundation (GGP02179 and GGP06180 to F.Z., GGP09134 to F.B.), the Italian Ministry of Health (GR-2007-657156 to A.F. and F.Z.), and the Italian Ministry of Research (to F.B.). The support of Compagnia di San Paolo- Torino (to F.B. and A.F.) and of the Italian League against Epilepsy is also acknowledged. The authors declare no conflict of interest.
Contributor Information
Anna Fassio, Email: afassio@unige.it.
Federico Zara, Email: federicozara@ospedale-gaslini.ge.it.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation
NCBI UniGene, http://www.ncbi.nlm.nih.gov/unigene
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.org/Omim
PFAM Protein Families Database, http://pfam.sanger.ac.uk/
References
- 1.Hauser W.A., Annegers J.F., Kurland L.T. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935-1984. Epilepsia. 1993;34:453–468. doi: 10.1111/j.1528-1157.1993.tb02586.x. [DOI] [PubMed] [Google Scholar]
- 2.Turnbull J., Lohi H., Kearny J.A., Rouleau G.A., Delgado-Escueta M.H., Cossette P., Minassian B.A. Sacred disease secrets revealed: The genetics of human epilepsy. Hum. Mol. Genet. 2005;14:2491–2500. doi: 10.1093/hmg/ddi250. [DOI] [PubMed] [Google Scholar]
- 3.Avanzini G., Franceschetti S. Cellular biology of epileptogenesis. Lancet Neurol. 2003;2:33–42. doi: 10.1016/s1474-4422(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 4.Zhou Y.D., Lee S., Jin Z., Wright M., Smith S.E., Anderson M.P. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat. Med. 2009;15:1208–1214. doi: 10.1038/nm.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Nijs L., Léon C., Nguyen L., Loturco J.J., Delgado-Escueta A.V., Grisar T., Lakaye B. EFHC1 interacts with microtubules to regulate cell division and cortical development. Nat. Neurosci. 2009;12:1266–1274. doi: 10.1038/nn.2390. [DOI] [PubMed] [Google Scholar]
- 6.Zara F., Gennaro E., Stabile M., Carbone I., Malacarne M., Majello L., Santangelo R., de Falco F.A., Bricarelli F.D. Mapping of a locus for a familial autosomal recessive idiopathic myoclonic epilepsy of infancy to chromosome 16p13. Am. J. Hum. Genet. 2000;66:1552–1557. doi: 10.1086/302876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Falco F.A., Majello L., Santangelo R., Stabile M., Bricarelli F.D., Zara F. Familial infantile myoclonic epilepsy: Clinical features in a large kindred with autosomal recessive inheritance. Epilepsia. 2001;42:1541–1548. doi: 10.1046/j.1528-1157.2001.26701.x. [DOI] [PubMed] [Google Scholar]
- 8.Fischer H., Zhang X.U., O'Brien K.P., Kylsten P., Engvall E. C7, a novel nucleolar protein, is the mouse homologue of the Drosophila late puff product L82 and an isoform of human OXR1. Biochem. Biophys. Res. Commun. 2001;281:795–803. doi: 10.1006/bbrc.2001.4345. [DOI] [PubMed] [Google Scholar]
- 9.Shkolnik K., Ben-Dor S., Galiani D., Hourvitz A., Dekel N. Molecular characterization and bioinformatics analysis of Ncoa7B, a novel ovulation-associated and reproduction system-specific Ncoa7 isoform. Reproduction. 2008;135:321–333. doi: 10.1530/REP-07-0402. [DOI] [PubMed] [Google Scholar]
- 10.Traverso M., Malnati M., Minetti C., Regis S., Tedeschi S., Pedemonte M., Bruno C., Biassoni R., Zara F. Multiplex real-time PCR for detection of deletions and duplications in dystrophin gene. Biochem. Biophys. Res. Commun. 2006;339:145–150. doi: 10.1016/j.bbrc.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 11.De Pietri Tonelli D., Calegari F., Fei J.F., Nomura T., Osumi N., Heisenberg C.P., Huttner W.B. Single-cell detection of microRNAs in developing vertebrate embryos after acute administration of a dual-fluorescence reporter/sensor plasmid. Biotechniques. 2006;41:727–732. doi: 10.2144/000112296. [DOI] [PubMed] [Google Scholar]
- 12.Pan X., Eathiraj S., Munson M., Lambright D.G. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature. 2006;442:303–306. doi: 10.1038/nature04847. [DOI] [PubMed] [Google Scholar]
- 13.Martinu L., Masuda-Robens J.M., Robertson S.E., Santy L.C., Casanova J.E., Chou M.M. The TBC (Tre-2/Bub2/Cdc16) domain protein TRE17 regulates plasma membrane-endosomal trafficking through activation of Arf6. Mol. Cell. Biol. 2004;24:9752–9762. doi: 10.1128/MCB.24.22.9752-9762.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frittoli E., Palamidessi A., Pizzigoni A., Lanzetti L., Garrè M., Troglio F., Troilo A., Fukuda M., Di Fiore P.P., Scita G., Confalonieri S. The primate-specific protein TBC1D3 is required for optimal macropinocytosis in a novel ARF6-dependent pathway. Mol. Biol. Cell. 2008;19:1304–1316. doi: 10.1091/mbc.E07-06-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Souza-Schorey C., Chavrier P. ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006;7:347–358. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- 16.Radhakrishna H., Donaldson J.G. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J. Cell Biol. 1997;139:49–61. doi: 10.1083/jcb.139.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaworski J. ARF6 in the nervous system. Eur. J. Cell Biol. 2007;86:513–524. doi: 10.1016/j.ejcb.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Chiappalone M., Casagrande S., Tedesco M., Valtorta F., Baldelli P., Martinoia S., Benfenati F. Opposite changes in glutamatergic and GABAergic transmission underlie the diffuse hyperexcitability of synapsin I-deficient cortical networks. Cereb. Cortex. 2009;19:1422–1439. doi: 10.1093/cercor/bhn182. [DOI] [PubMed] [Google Scholar]
- 19.Caserta F., Eldred W.D., Fernandez E., Hausman R.E., Stanford L.R., Bulderev S.V., Schwarzer S., Stanley H.E. Determination of fractal dimension of physiologically characterized neurons in two and three dimensions. J. Neurosci. Methods. 1995;56:133–144. doi: 10.1016/0165-0270(94)00115-w. [DOI] [PubMed] [Google Scholar]
- 20.Hernández-Deviez D.J., Casanova J.E., Wilson J.M. Regulation of dendritic development by the ARF exchange factor ARNO. Nat. Neurosci. 2002;5:623–624. doi: 10.1038/nn865. [DOI] [PubMed] [Google Scholar]
- 21.Hernández-Deviez D.J., Roth M.G., Casanova J.E., Wilson J.M. ARNO and ARF6 regulate axonal elongation and branching through downstream activation of phosphatidylinositol 4-phosphate 5-kinase alpha. Mol. Biol. Cell. 2004;15:111–120. doi: 10.1091/mbc.E03-06-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krauss M., Kinuta M., Wenk M.R., De Camilli P., Takei K., Haucke V. ARF6 stimulates clathrin/AP-2 recruitment to synaptic membranes by activating phosphatidylinositol phosphate kinase type Igamma. J. Cell Biol. 2003;162:113–124. doi: 10.1083/jcb.200301006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi S., Ko J., Lee J.R., Lee H.W., Kim K., Chung H.S., Kim H., Kim E. ARF6 and EFA6A regulate the development and maintenance of dendritic spines. J. Neurosci. 2006;26:4811–4819. doi: 10.1523/JNEUROSCI.4182-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scholz R., Berberich S., Rathgeber L., Kolleker A., Köhr G., Kornau H.C. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron. 2010;66:768–780. doi: 10.1016/j.neuron.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Andrade D.M. Genetic basis in epilepsies caused by malformations of cortical development and in those with structurally normal brain. Hum. Genet. 2009;126:173–193. doi: 10.1007/s00439-009-0702-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.