

Abstract
The lymphatic vasculature is essential for the recirculation of extracellular fluid, fat absorption, and immune function and as a route of tumor metastasis. The dissection of molecular mechanisms underlying lymphangiogenesis has been accelerated by the identification of tissue-specific lymphatic endothelial markers and the study of congenital lymphedema syndromes. We report the results of genetic analyses of a kindred inheriting a unique autosomal-recessive lymphedema-choanal atresia syndrome. These studies establish linkage of the trait to chromosome 1q32-q41 and identify a loss-of-function mutation in PTPN14, which encodes a nonreceptor tyrosine phosphatase. The causal role of PTPN14 deficiency was confirmed by the generation of a murine Ptpn14 gene trap model that manifested lymphatic hyperplasia with lymphedema. Biochemical studies revealed a potential interaction between PTPN14 and the vascular endothelial growth factor receptor 3 (VEGFR3), a receptor tyrosine kinase essential for lymphangiogenesis. These results suggest a unique and conserved role for PTPN14 in the regulation of lymphatic development in mammals and a nonconserved role in choanal development in humans.
Main Text
The lymphatic system is composed of a network of vessels that serves as a unidirectional transport system for fluid, cells, and macromolecules from the interstitial spaces back into the central circulation. Perturbations in the development, maintenance, or function in the lymphatic vascular network can lead to lymphedema, a sustained accumulation of interstitial fluid with secondary pathology as a consequence of increased hydrodynamic pressure and decreased perfusion in the affected areas.1 The progression of events that initiates and organizes lymphangiogenesis remains elusive, in part due to a paucity of lymphatic endothelial cell-specific markers until relatively recently. In the past decade, the identification of major lymphangiogenic growth factors and lymphatic endothelial markers has contributed significantly to the understanding of the molecular mechanisms of lymphangiogenesis.2 A number of genes whose functions are crucial for progression through the different stages of lymphangiogenesis have been identified through animal models or human studies of congenital lymphedema (recently reviewed by Tammela and Alitalo3).
Mutation of VEGFR3 (MIM 136352) in humans causes Milroy disease4–6 (MIM 153100), isolated lymphedema at birth secondary to absent or hypoplastic subcutaneous lymphatic vessels.7,8 Heterozygosity for inactivating mutation of Vegfr3 also results in lymphatic vessel hypoplasia, chylous ascites, and lymphedema in the spontaneously occurring Chy mouse.9 The essential role of VEGFR3 signaling in lymphangiogenesis has been underscored by the embryonic lethal phenotype of mice lacking the gene encoding the Vegfr3 ligand, Vegfc. In null embryos, lymphatic development was arrested but lymphatic specification and blood vessel development were unaffected.9 In contrast, overexpression of Vegfc in transgenic mouse keratinocytes resulted in hyperplasia of cutaneous lymphatic vessels.10,11
Apart from disease associated with deficiency of VEGFR3, several syndromic forms of lymphedema have been characterized at the genetic level in humans. These include mutation of the transcription factors FOXC2 (MIM 602402), in lymphedema-distichiasis syndrome12 (LD [MIM 153400]); SOX18 (MIM 601618), in hypotrichosis-lymphedema-telangiectasia syndrome13 (MIM 607823); CCBE1 (MIM 612753), in Hennekam lymphangiectasia-lymphedema syndrome14 (MIM 235510); and NEMO (MIM 300248), in osteopetrosis lymphedema, anhidrotic ectodermal dysplasia, and immunodeficiency syndrome15 (MIM 300301). The molecular bases of other rare syndromes in which lymphedema is a major feature, such as cholestasis-lymphedema syndrome (MIM 214900), have not yet been determined. In 1982, a multigenerational consanguineous Middle Eastern kindred was described with autosomal-recessive inheritance of bilateral posterior choanal atresia (MIM 608911), high arched palate, and other developmental abnormalities (hypoplastic nipples, pericardial effusion, and pectus excavatum) in one of the individuals.16 This pedigree was reported again in 1991 after five out of seven individuals affected with choanal atresia developed hard, nonpitting, lower-extremity lymphedema with onset between 4 and 5 yrs of age.17 We enrolled members of this pedigree (Figure 1) in a linkage study approved by the institutional review board of the Mount Sinai School of Medicine. All individuals provided informed consent, and the study was performed in accordance with both institutional and national ethical guidelines. We collected peripheral blood for extraction of genomic DNA and generation of Epstein-Barr virus (EBV)-transformed lymphoblastoid cell lines. Initially, six affected pedigree members were genotyped with short tandem repeat polymorphic markers, and when homozygosity was observed in at least four subjects, their parents were genotyped for marker informativeness. Upon identifying a marker that was homozygous in all affected individuals and completely informative in their parents, all available members of the pedigree were genotyped with flanking markers and the data were analyzed with the homozygosity mapping program MapmakerHomoz, as described previously.18 Significant linkage was achieved with a peak multipoint LOD score of 7.01 to an interval on chromosome 1q32-q41 (Figure S1a, available online). Haplotypes across this interval were constructed manually and validated with the Genehunter 2 program19 for individual nuclear families. Haplotype analysis defined a 13.2 cM critical region (Figure 1) that spanned approximately 8.6 Mb and contained 52 predicted genes (Figure S1b). Positional candidate genes were screened by direct sequencing of amplicons produced from either genomic DNA or reverse-transcribed cDNA. PROX1 (MIM 601546), a candidate gene of particular interest given its function as a master regulator of lymphangiogenesis,20 was excluded after extensive sequence analysis of exonic and flanking intronic regions with the use of primers designed from public genomic sequence with the Primer3 application (available upon request).21
Figure 1.
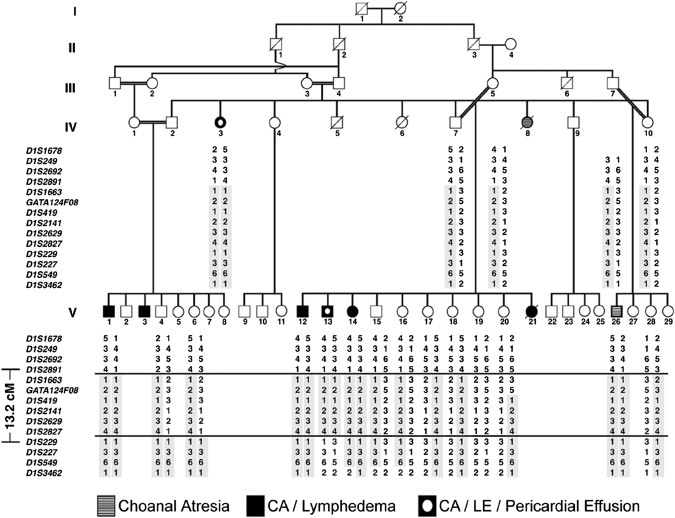
Haplotype Analysis of Choanal Atresia-Lymphedema Pedigree
A consanguineous pedigree is shown in which affected individuals manifest choanal atresia, lymphedema, and pericardial effusion. Individual V-26 was an infant at the time of evaluation and may have been too young to manifest lymphedema. Haplotype analysis of the critical region for choanal atresia-lymphedema syndrome on chromosome 1q32-q41 is shown. Markers inherited in homozygosity in affected individuals are boxed in gray. The telomeric boundary of the critical region is at marker D1S2891 and the centromeric boundary is at marker D1S229.
Because none of the remaining positional candidates were known to play a role in lymphangiogenesis, genes with a high probability of regulating intracellular signaling were prioritized for sequence analysis. Protein tyrosine phosphatase (PTP) mutations have been associated with lymphedema in the case of PTPN11 (MIM 176876) and Noonan syndrome (MIM 163950). PTPN11 encodes SHP-2, a PTP with an established role in relaying signals from several receptor tyrosine kinases through the RAS-MAPK signaling pathway.22 SHP-2 has been shown to interact with VEGFR3, and RAS proteins have recently been demonstrated to regulate VEGFR3 expression modulating lymphatic endothelium, suggesting a potential mechanism for lymphedema in RAS signaling disorders.23 PTPN14 (MIM 603155), which encodes a nonreceptor PTP known alternatively as Pez, PTPD2, or PTP36 in the mouse,24–26 was thus an attractive candidate.
We screened PTPN14 by generating overlapping RT-PCR amplicons from lymphoblastoid cell-derived cDNA (HQ116786) (Figure S1c). Primer pairs used for the PTPN14 mutational analysis and other experiments are provided in Table S1. As shown for an amplicon spanning exons 3–8 (Figure 2A, left), the mutant transcript was truncated relative to the expected product size. Sequence analysis of the truncated transcript revealed absence of exon 7 (Figure 2B, top). Exon 7 failed to amplify from genomic DNA (HQ116785) templates obtained from affected individuals, suggesting a genomic deletion. This hypothesis was confirmed by amplification and sequence analysis of a long-range PCR product that spanned exons 6–8 (Figure 2A, right). A 2016 bp deletion (Figure 2B, bottom) inherited in homozygosity in all affected individuals, clearly demonstrating that the mutation segregated with the disease. This deletion was not present in 222 control chromosomes of Arab (n = 62) or mixed European (n = 160) origin. The sample size was adequate for detection of rare polymorphisms with over 80% power within the general European population. There were no genomic structural elements, such as flanking repetitive sequences, that suggested an obvious mechanism for the deletion. Analysis of the transcripts and expressed sequence tags mapping to this locus did not reveal evidence for alternative coding transcripts in which the exon was excluded, nor were any alternative splicing isoforms observed during RT-PCR amplification of the transcript from wild-type (WT) or mutant lymphoblastoid cells.
Figure 2.
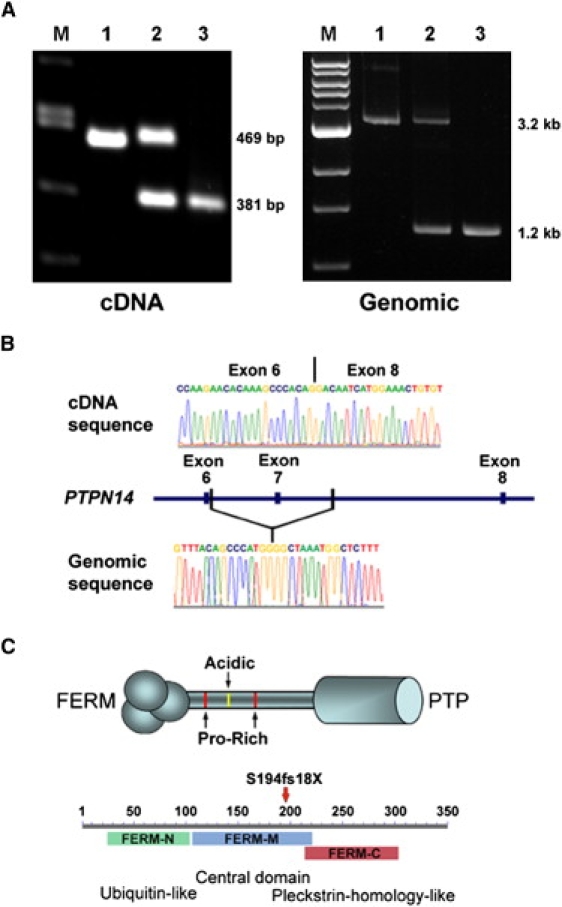
cDNA and Genomic Mutation Analysis of PTPN14
(A) Amplification products of PTPN14 cDNA (left) or genomic DNA (right) are shown. Lymphoblastoid cell total RNA was used to generate cDNA (Superscript II reverse transcriptase; Roche) as template for generating RT-PCR products spanning PTPN14 exons 3–8 from WT (left lane 1), obligate heterozygous (left lane 2), and homozygous affected individuals (left lane 3). The expected sizes of the full-length and truncated amplicon lacking exon 7 are shown at right. A 3.2 kb amplification product from genomic DNA (right lane 1) contained a ∼2 kb deletion evident in amplicons derived from heterozygous (right lane 2) and homozygous (right lane 3) individual DNA samples.
(B) The absence of exon 7 was confirmed in mutant cDNA transcripts (top), and the boundaries of the genomic deletion (bottom) were defined by sequence analysis.
(C) Structural features of PTPN14 include an N-terminal FERM domain, a central poorly conserved linker region with proline-rich SH3-like motifs and an acidic polyglutamate domain, and a C-terminal phosphatase domain (top). The location of the frameshift introduced by loss of exon 7 within the tripartite FERM domain is indicated by the red arrow (bottom).
PTPN14 contains two conserved structural elements: an amino terminal FERM domain (band 4.1-ezrin-radixin-moesin family of adhesion molecules) and a carboxy terminal PTP domain.25,26 The subcellular localization of the protein is dependent on serum concentration and cell density27 and is regulated through serine/threonine phosphorylation.28 The mutant PTPN14 transcript was predicted to encode a frameshifted sequence after residue 193 and a premature termination codon after residue 211 (S194fs212X), interrupting the FERM domain after the second of three subdomains (Figure 2C). Efforts to express a construct corresponding to the mutant protein tagged with the FLAG epitope resulted in only a low level of expression restricted to the insoluble fraction (not shown), suggesting that the protein was likely to be unstable and catalytically inactive.
Despite extensive efforts, we were not able to identify any additional individuals with the choanal atresia/lymphedema syndrome, precluding formal genetic proof that the PTPN14 deletion caused the disease phenotype. To overcome this limitation, we capitalized on the available high-throughput murine gene targeting resources to generate a mouse model of PTPN14 deficiency (Figure 3A). Two embryonic stem cell lines (XE198 and RRR484) in which exon-trapping vectors were integrated within the Ptpn14 gene were obtained from the International Gene Trap Consortium.29 The trap vectors were located in introns 13 and 5, respectively, and were predicted to interrupt the protein after residues 354 and 170. The latter mutant interrupted the FERM domain, providing a good model of the human mutation. Feeder-independent embryonic stem cells were maintained in a pluripotential state in culture medium containing leukocyte inhibitory factor (500 U/ml) and grown on gelatinized culture flasks as recommended. These clones were sequence validated and used to generate chimeric founders. Germline transmission was observed from three chimeric animals generated from RRR484 but not from XE198 chimeras. Genotyping was performed by screening tail-cut DNA for the presence of the β-geo fusion gene and for the loss of the WT amplicon flanking the gene trap insertion site. The insertion site in intron 5 was mapped precisely by sequencing of chimeric amplicons, including the ends of the trap vector and the flanking mouse genomic DNA. Mice homozygous for the trap allele grew slower than littermate WT controls, but the difference in weight became nonsignificant after six months of age (Figure S2a). Histologic analysis of postnatal cranial sections revealed no evidence of choanal atresia (Figure S2b), and morphologic evaluation of adult animal facial structures did not reveal any overtly dysmorphic features (Figure S2c). Lymphedema was not observed in any animals at birth, but after 5 mo, approximately 14% of the mutant animals displayed forelimb and/or hindlimb swelling or periorbital edema (Figure 3B).
Figure 3.
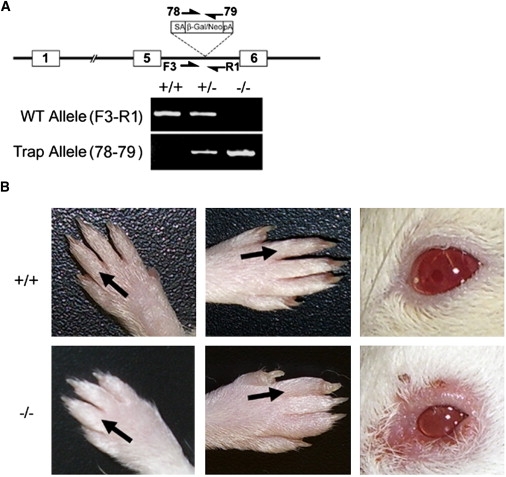
A Mouse Gene Trap Model of Ptpn14 Deficiency
(A) Diagram of the insertion of a gene trap viral vector into the genomic interval between exons 5 and 6 of Ptpn14. Primer pairs F3-R1 and 78-79 were used to screen for the WT and trap alleles, respectively. The results of PCR assays using tail-cut genomic DNA from WT (+/+), heterozygous (−/+), or homozygous (−/−) trap allele mice are shown below.
(B) Lymphedema in the upper and lower extremities and periorbital region in adult mice homozygous for the Ptpn14 trap allele. Visible swelling of the dorsal forelimbs and/or hindlimbs and/or periorbital area was evident in a subset of animals. The distal extremities and periorbital region of a WT animal are shown (top) for comparison.
Given the genetic evidence that PTPN14 plays a role in lymphatic function, we then investigated the effect of PTPN14 deficiency on lymphatic vascular patterning. We performed whole-mount immunohistochemistry experiments to visualize the lymphatic capillaries in ears from symptomatic Ptpn14trap/trap and WT mice. These studies revealed that the lymphatic capillaries were hyperplastic in symptomatic mutant animals (Figure 4A), a finding in striking contrast to the lymphatic hypoplasia resulting from loss of VEGFR3 function.9 Of note, the Foxc2-deficient mouse model of lymphedema-distichiasis syndrome30 also manifests lymphatic hyperplasia. Work in this model has revealed that Foxc2 and Vegfr3 act cooperatively in lymphatic patterning, with Vegfr3 potentially acting as an upstream regulator of Foxc2.31
Figure 4.
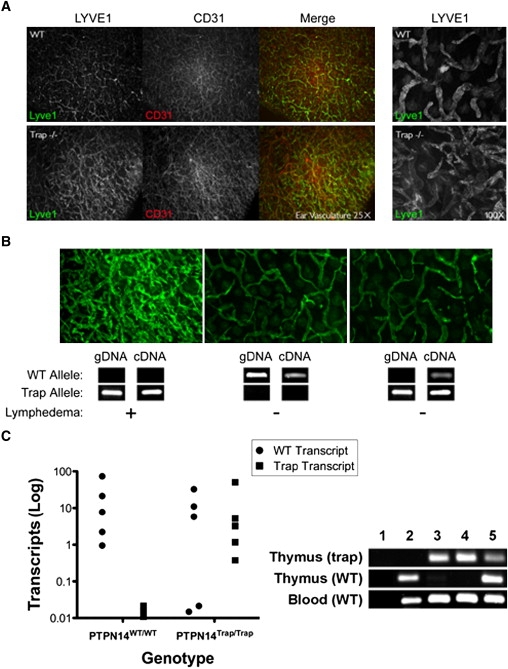
Lymphatic Hyperplasia in Symptomatic Ptpn14 Mutant Mice
(A) Immunohistochemical staining of ear skin sections from WT (top) and mutant (bottom) mice. After treatment with Nair for hair removal, ear leaflets were separated and the central cartilage removed. The epidermal layer was removed by treatment with 0.5 M ammonium thiocyanate, and dermal skin was fixed with 100% acetone and 80% methanol before antibody incubations. Lymphatic vessels were stained with rabbit anti-LYVE1 (Upstate) followed by anti-rabbit Alex Fluor 488 (Molecular Probes), and blood vessels were visualized with panendothelial rat anti-CD31 (BD Biosciences) followed by anti-rat Alexa Fluor 594 (Molecular Probes). Images were captured with a Zeiss Axiophot2 fluorescence microscope after mounting (Dako Cytomation). Immunostaining of the lymphatic endothelial marker LYVE1 (green) and the vascular endothelial predominant marker CD31 (red) are shown at low magnification (25×). A higher-magnification view (100×) of LYVE1 staining is shown at right. The sections from mutant animals have an increased density of LYVE1-positive lymphatic capillaries compared to WT ear sections.
(B) Ear sections from symptomatic mutant (left), WT (center), and nonsymptomatic mutant (right) mice are shown with confirmatory genotyping (gDNA) and expression analysis (cDNA) for Ptpn14 WT and trap alleles shown below. Genomic DNA was obtained from tail cuts, and cDNA was obtained from peripheral blood. The Ptpn14 trap transcript was amplified with primers specific for exon 5 and the β-geo open reading frame. WT transcripts arising by skipping of the trap exon were detected with the use of primers flanking the exon 5–6 splice junction. WT transcripts were readily detected with cDNA derived from asymptomatic mice but not with that from symptomatic mice.
(C) Real-time quantitative PCR with thymocyte-derived cDNA from WT and asymptomatic trapped animals for the calculation of generated WT and trapped transcripts. 20 ng of cDNA and 0.2 μM of each primer (Table S1) with SYBR Green Master (Rox) (Roche) were amplified in an ABI PRISM 7900HT detection system for 40 cycles in triplicate. Transcript abundance was calculated with the formula 2500 × 1.93̂((mean Gapdh Ct)−(mean transcript Ct)), with Ct as the threshold cycle. The right panel shows differential splicing of WT transcripts in asymptomatic trapped animals in thymocyte- versus leukocyte-derived cDNA. WT and trap products from qPCR in the left panel were analyzed by gel electrophoresis. Lanes: 1, negative control; 2, WT; 3–5, homozygous Ptpn14trap. Trap amplicons derived from the thymus were present in all mutant samples but not in the WT sample (top panel). WT amplicons were present in only one (lane 5) of the three mutant samples derived from the thymus (middle panel) but in all samples derived from blood (bottom panel).
Unlike the mouse model, the lymphedema phenotype in the human disease was completely penetrant by late childhood, raising the possibility that skipping of the gene trap could produce WT transcript and ameliorate the phenotype. Analysis of mRNA splicing with the use of WT and mutant allele-specific oligonucleotides confirmed the presence of WT transcripts in mice homozygous for the trap allele but without a lymphatic phenotype, consistent with leakiness of the trap (Figure 4B). This phenomenon has previously been described for other trap alleles.32,33 We assessed the transcript abundance of the correctly spliced and gene-trapped isoforms in a set of phenotypically normal trap homozygote and WT mice using thymocyte-derived RNA (Figure 4C). Interestingly, whereas the abundance of a reference gene transcript (Gapdh) was relatively uniform across all samples (not shown), the expression of the WT transcript was highly variable. The trap isoform also showed variable levels of expression but was present in all mutant samples. Of note, the WT isoform was not observed in two trap homozygotes. When peripheral blood leukocytes were used as the RNA source, expression of the WT isoform was detected, confirming that alternative splicing of the gene trap exon was not uniform in different tissues (Figure 4C, right panel). Because the trap allele is hypomorphic rather than a true null, a role for PTPN14 in choanal development in mice cannot be definitively excluded.
The expression pattern of PTPN14 is broad,26 including breast, kidney, skeletal muscle, lung, placenta, heart, and pancreas, and is not consistent with the observed lymphatic and choanal atresia phenotype, so we then sought to confirm the physiologic relevance of PTPN14 to lymphangiogenesis. The robust expression of PTPN14 in human umbilical vein endothelial cells (HUVECs) has been proposed to suggest a potential role in endothelial cell function.27 We carried out RT-PCR and immunoblot analyses in lymphatic endothelial cells isolated from human foreskin (generously provided by M. Skobe) and established that PTPN14 was transcribed and translated in these cells (Figure 5A), consistent with a potential role for the gene in lymphatic endothelial development and/or function.
Figure 5.
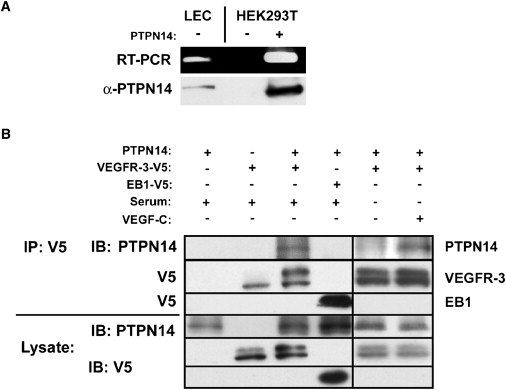
PTPN14 Can Modulate VEGFR3 Receptor Activation in a Specific Fashion
(A) cDNA and total protein lysates were prepared from human foreskin lymphatic endothelial cells (LECs) for RT-PCR and immunoblot analyses. PTPN14 polyclonal rabbit primary antisera (a generous gift from Y. Khew-Goodall) were raised against a peptide corresponding to PTPN14 residues 683–696 as described previously, and subsequent aliquots were generated against this same epitope in rabbits. PTPN14 transcription (top) and protein expression (bottom) were confirmed in LECs, absent in HEK293 cells (−), and present in HEK293 cells expressing Flag-tagged PTPN14 (+).
(B) HEK293 cells were cotransfected with combinations of V5-tagged VEGFR3, V5-tagged EB1, and FLAG-tagged PTPN14 as indicated at right. Confluent HEK293 cell monolayers were washed with PBS on ice and then scraped with lysis buffer (20 mM Tris pH 7.6, 150 mM NaCl, 50 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 1% [v/v] Triton X-100, 0.5% [w/v] sodium deoxycholate, 0.1% [w/v] SDS) and protease inhibitors (Complete Protease Inhibitor Tablets, Roche). Lysates were immunoprecipitated with mouse anti-V5 antibody (Invitrogen), and coimmunoprecipitation of FLAG-tagged PTPN14 (top row) was detected by immunoblotting with anti-PTPN14 antibody. Blots were stripped and reprobed with anti-V5 antibody to confirm the immunoprecipitation of V5-tagged VEGFR3 (second row) and V5-tagged EB1 (third row). PTPN14 coimmunoprecipitated with V5-tagged VEGFR3, but not with a control protein, V5-tagged EB1, under basal conditions in serum-replete medium (lanes 3 and 4). Human recombinant vascular endothelial growth factor-C (VEGFC; R&D Systems) stimulation of transfected cells resulted in a more robust interaction relative to serum-starved cells (lanes 5 and 6). Experiments were repeated three to five times, and representative data are shown.
The mechanism by which the protein could mediate such an effect is unclear, because PTPN14 has a dynamic subcellular localization pattern, with targeting to intercellular junctions in confluent, quiescent HUVECs and nuclear translocation in proliferating cells.27 Given the critical importance of VEGFR3 in lymphangiogenesis, we tested the hypothesis that the phosphatase and the VEGFR3 receptor might form a physical complex in cells expressing both proteins. We coexpressed FLAG-tagged PTPN14 (generously provided by Y. Khew-Goodall27) and a full-length human VEGFR3 cloned into the pcDNA6-V5-HisA expression vector. Coimmunoprecipitation of FLAG-tagged PTPN14 was observed after immunoprecipitation of V5-tagged VEGFR3, but not with the negative control, V5-tagged EB1 (Figure 5B). The abundance of coimmunoprecipitated PTPN14 was enhanced by stimulation of transfected cells with VEGFC. These results were consistent with the hypothesis that VEGFC-mediated signaling may enhance the recruitment of PTPN14 to a protein complex that includes VEGFR3. The potential of these proteins to interact in vivo is also supported by independent work demonstrating that the expression patterns of Ptpn14 and Vegfr3 are broad and exhibit substantial overlap.25,34 The notion that PTPN14 might function in a multiprotein complex is further supported by the established role of its closest relative, PTPN21 (MIM 603271), which regulates proliferative signaling pathways through a scaffolding function.35,36 Two structural motifs in PTPN14, the FERM domain37 and a proline-rich potential SH3 motif,25 could function as adaptors in signal transduction.
The mechanism by which PTPN14 functions in lymphatic development is still uncertain. The accumulation of the protein at adherens junctions and its ability to dephosphorylate β-catenin implicate PTPN14 in promoting cell adhesion,38 conceivably by maintaining the integrity of lymphatic capillaries. In this model, loss of the protein leads to lymphatic dysfunction, capillary leakage, and compensatory hypertrophy. However, in light of the observed cytostatic effect of overexpression in 3T3 cells39 we have not ruled out the possibility that PTPN14 may function as a growth suppressor in lymphatic endothelial cells, with hyperplasia as a consequence of loss of function.
At present, we do not have evidence that PTPN14 plays a role in other syndromic or nonsyndromic forms of lymphedema. We have not identified any other kindred with recessively inherited choanal atresia and childhood-onset lymphedema as primary phenotypic features, despite extensive literature searching. Additionally, screening of a collection of familial lymphedema pedigrees that did not manifest choanal atresia with the markers used in this pedigree did not identify any potentially linked families (D. Finegold, R. Ferrell, personal communication). Gorham disease (MIM 123880), which involves the overgrowth of lymphatic vessels into bones,40 is a disorder with potential linkage to PTPN14 and/or VEGFR3 on the basis of its pathophysiology. However, although pericardial effusion is an overlapping phenotype between Gorham disease and choanal atresia-lymphedema syndrome, the typical musculoskeletal pain or progressive degenerative bone deformities were not present in the two clinical reports describing the phenotype in the current pedigree.
The role of PTPs in human disease has been a subject of increasing interest, particularly with respect to cancer development.41 PTEN (MIM 601728) is a well-characterized cause of multiple distinct hamartoma tumor syndromes due to germline inactivating or deletion mutations.42 More recently, germline gain-of-function (GOF) mutations in PTPN11 have been shown to cause Noonan and Leopard (MIM 151100) syndromes,43,44 with sporadic GOF mutations detected in a subset of leukemias.45 With regard to non-Mendelian disease, a functional SNP in PTPN22 (MIM 600716) encoding a GOF variant (R620Y) has been associated with a variety of autoimmune conditions.46 To date, the only proposed role for PTPN14 in human disease has been as a potential tumor suppressor, on the basis of the presence of sporadic mutations in colorectal cancer cells.47 The phenotype of the family under study does not include early-onset cancer. Rather, our findings identify PTPN14 as a unique regulator of lymphatic development in mammals and choanal development in humans. Future studies will be necessary to elucidate the molecular mechanisms by which the protein exerts its in vivo biological effects.
Acknowledgments
The authors would like to thank Monica Zak for her excellent assistance in obtaining the full family pedigree and to thank all of the family members who participated in these studies. The authors declare that there are no conflicts of interest.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
International Gene Trap Consortium, http://www.genetrap.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim/
Primer3, http://frodo.wi.mit.edu/primer3/
UCSC Human Genome Bioinformatics (browser build hg18, March 2006), http://www.genome.ucsc.edu
Accession Numbers
The GenBank accession numbers for the genomic and cDNA sequences reported in this paper are HQ116785 and HQ116786.
References
- 1.Cueni L.N., Detmar M. The lymphatic system in health and disease. Lymphat. Res. Biol. 2008;6:109–122. doi: 10.1089/lrb.2008.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karpanen T., Alitalo K. Molecular biology and pathology of lymphangiogenesis. Annu. Rev. Pathol. 2008;3:367–397. doi: 10.1146/annurev.pathmechdis.3.121806.151515. [DOI] [PubMed] [Google Scholar]
- 3.Tammela T., Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 4.Ferrell R.E., Levinson K.L., Esman J.H., Kimak M.A., Lawrence E.C., Barmada M.M., Finegold D.N. Hereditary lymphedema: evidence for linkage and genetic heterogeneity. Hum. Mol. Genet. 1998;7:2073–2078. doi: 10.1093/hmg/7.13.2073. [DOI] [PubMed] [Google Scholar]
- 5.Irrthum A., Karkkainen M.J., Devriendt K., Alitalo K., Vikkula M. Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am. J. Hum. Genet. 2000;67:295–301. doi: 10.1086/303019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karkkainen M.J., Ferrell R.E., Lawrence E.C., Kimak M.A., Levinson K.L., McTigue M.A., Alitalo K., Finegold D.N. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat. Genet. 2000;25:153–159. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
- 7.Bollinger A., Isenring G., Franzeck U.K., Brunner U. Aplasia of superficial lymphatic capillaries in hereditary and connatal lymphedema (Milroy's disease) Lymphology. 1983;16:27–30. [PubMed] [Google Scholar]
- 8.Brice G., Child A.H., Evans A., Bell R., Mansour S., Burnand K., Sarfarazi M., Jeffery S., Mortimer P. Milroy disease and the VEGFR-3 mutation phenotype. J. Med. Genet. 2005;42:98–102. doi: 10.1136/jmg.2004.024802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karkkainen M.J., Saaristo A., Jussila L., Karila K.A., Lawrence E.C., Pajusola K., Bueler H., Eichmann A., Kauppinen R., Kettunen M.I. A model for gene therapy of human hereditary lymphedema. Proc. Natl. Acad. Sci. USA. 2001;98:12677–12682. doi: 10.1073/pnas.221449198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeltsch M., Kaipainen A., Joukov V., Meng X., Lakso M., Rauvala H., Swartz M., Fukumura D., Jain R.K., Alitalo K. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science. 1997;276:1423–1425. doi: 10.1126/science.276.5317.1423. [DOI] [PubMed] [Google Scholar]
- 11.Veikkola T., Jussila L., Makinen T., Karpanen T., Jeltsch M., Petrova T.V., Kubo H., Thurston G., McDonald D.M., Achen M.G. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. 2001;20:1223–1231. doi: 10.1093/emboj/20.6.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang J., Dagenais S.L., Erickson R.P., Arlt M.F., Glynn M.W., Gorski J.L., Seaver L.H., Glover T.W. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am. J. Hum. Genet. 2000;67:1382–1388. doi: 10.1086/316915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irrthum A., Devriendt K., Chitayat D., Matthijs G., Glade C., Steijlen P.M., Fryns J.P., Van Steensel M.A., Vikkula M. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am. J. Hum. Genet. 2003;72:1470–1478. doi: 10.1086/375614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alders M., Hogan B.M., Gjini E., Salehi F., Al-Gazali L., Hennekam E.A., Holmberg E.E., Mannens M.M., Mulder M.F., Offerhaus G.J. Mutations in CCBE1 cause generalized lymph vessel dysplasia in humans. Nat. Genet. 2009;41:1272–1274. doi: 10.1038/ng.484. [DOI] [PubMed] [Google Scholar]
- 15.Döffinger R., Smahi A., Bessia C., Geissmann F., Feinberg J., Durandy A., Bodemer C., Kenwrick S., Dupuis-Girod S., Blanche S. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat. Genet. 2001;27:277–285. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 16.Qazi Q.H., Kanchanapoomi R., Beller E., Collins R. Inheritance of posterior choanal atresia. Am. J. Med. Genet. 1982;13:413–416. doi: 10.1002/ajmg.1320130409. [DOI] [PubMed] [Google Scholar]
- 17.Har-El G., Borderon M.L., Weiss M.H. Choanal atresia and lymphedema. Ann. Otol. Rhinol. Laryngol. 1991;100:661–664. doi: 10.1177/000348949110000812. [DOI] [PubMed] [Google Scholar]
- 18.Gelb B.D., Edelson J.G., Desnick R.J. Linkage of pycnodysostosis to chromosome 1q21 by homozygosity mapping. Nat. Genet. 1995;10:235–237. doi: 10.1038/ng0695-235. [DOI] [PubMed] [Google Scholar]
- 19.Kruglyak L., Daly M.J., Reeve-Daly M.P., Lander E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 20.Wigle J.T., Harvey N., Detmar M., Lagutina I., Grosveld G., Gunn M.D., Jackson D.G., Oliver G. An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J. 2002;21:1505–1513. doi: 10.1093/emboj/21.7.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 22.Gelb B.D., Tartaglia M. Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Hum. Mol. Genet. 2006;15(Spec No 2, Spec No 2):R220–R226. doi: 10.1093/hmg/ddl197. [DOI] [PubMed] [Google Scholar]
- 23.Ichise T., Yoshida N., Ichise H. H-, N- and Kras cooperatively regulate lymphatic vessel growth by modulating VEGFR3 expression in lymphatic endothelial cells in mice. Development. 2010;137:1003–1013. doi: 10.1242/dev.043489. [DOI] [PubMed] [Google Scholar]
- 24.Møller N.P., Møller K.B., Lammers R., Kharitonenkov A., Sures I., Ullrich A. Src kinase associates with a member of a distinct subfamily of protein-tyrosine phosphatases containing an ezrin-like domain. Proc. Natl. Acad. Sci. USA. 1994;91:7477–7481. doi: 10.1073/pnas.91.16.7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawada M., Ogata M., Fujino Y., Hamaoka T. cDNA cloning of a novel protein tyrosine phosphatase with homology to cytoskeletal protein 4.1 and its expression in T-lineage cells. Biochem. Biophys. Res. Commun. 1994;203:479–484. doi: 10.1006/bbrc.1994.2207. [DOI] [PubMed] [Google Scholar]
- 26.Smith A.L., Mitchell P.J., Shipley J., Gusterson B.A., Rogers M.V., Crompton M.R. Pez: a novel human cDNA encoding protein tyrosine phosphatase- and ezrin-like domains. Biochem. Biophys. Res. Commun. 1995;209:959–965. doi: 10.1006/bbrc.1995.1591. [DOI] [PubMed] [Google Scholar]
- 27.Wadham C., Gamble J.R., Vadas M.A., Khew-Goodall Y. Translocation of protein tyrosine phosphatase Pez/PTPD2/PTP36 to the nucleus is associated with induction of cell proliferation. J. Cell Sci. 2000;113:3117–3123. doi: 10.1242/jcs.113.17.3117. [DOI] [PubMed] [Google Scholar]
- 28.Ogata M., Takada T., Mori Y., Uchida Y., Miki T., Okuyama A., Kosugi A., Sawada M., Oh-hora M., Hamaoka T. Regulation of phosphorylation level and distribution of PTP36, a putative protein tyrosine phosphatase, by cell-substrate adhesion. J. Biol. Chem. 1999;274:20717–20724. doi: 10.1074/jbc.274.29.20717. [DOI] [PubMed] [Google Scholar]
- 29.Stryke D., Kawamoto M., Huang C.C., Johns S.J., King L.A., Harper C.A., Meng E.C., Lee R.E., Yee A., L'Italien L. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31:278–281. doi: 10.1093/nar/gkg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kriederman B.M., Myloyde T.L., Witte M.H., Dagenais S.L., Witte C.L., Rennels M., Bernas M.J., Lynch M.T., Erickson R.P., Caulder M.S. FOXC2 haploinsufficient mice are a model for human autosomal dominant lymphedema-distichiasis syndrome. Hum. Mol. Genet. 2003;12:1179–1185. doi: 10.1093/hmg/ddg123. [DOI] [PubMed] [Google Scholar]
- 31.Petrova T.V., Karpanen T., Norrmén C., Mellor R., Tamakoshi T., Finegold D., Ferrell R., Kerjaschki D., Mortimer P., Ylä-Herttuala S. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat. Med. 2004;10:974–981. doi: 10.1038/nm1094. [DOI] [PubMed] [Google Scholar]
- 32.Voss A.K., Thomas T., Gruss P. Compensation for a gene trap mutation in the murine microtubule-associated protein 4 locus by alternative polyadenylation and alternative splicing. Dev. Dyn. 1998;212:258–266. doi: 10.1002/(SICI)1097-0177(199806)212:2<258::AID-AJA10>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 33.Voss A.K., Thomas T., Gruss P. Efficiency assessment of the gene trap approach. Dev. Dyn. 1998;212:171–180. doi: 10.1002/(SICI)1097-0177(199806)212:2<171::AID-AJA3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 34.Kukk E., Lymboussaki A., Taira S., Kaipainen A., Jeltsch M., Joukov V., Alitalo K. VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development. 1996;122:3829–3837. doi: 10.1242/dev.122.12.3829. [DOI] [PubMed] [Google Scholar]
- 35.Carlucci A., Gedressi C., Lignitto L., Nezi L., Villa-Moruzzi E., Avvedimento E.V., Gottesman M., Garbi C., Feliciello A. Protein-tyrosine phosphatase PTPD1 regulates focal adhesion kinase autophosphorylation and cell migration. J. Biol. Chem. 2008;283:10919–10929. doi: 10.1074/jbc.M707248200. [DOI] [PubMed] [Google Scholar]
- 36.Cardone L., Carlucci A., Affaitati A., Livigni A., DeCristofaro T., Garbi C., Varrone S., Ullrich A., Gottesman M.E., Avvedimento E.V., Feliciello A. Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novel positive regulator of src signaling. Mol. Cell. Biol. 2004;24:4613–4626. doi: 10.1128/MCB.24.11.4613-4626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Funakoshi-Tago M., Pelletier S., Moritake H., Parganas E., Ihle J.N. Jak2 FERM domain interaction with the erythropoietin receptor regulates Jak2 kinase activity. Mol. Cell. Biol. 2008;28:1792–1801. doi: 10.1128/MCB.01447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wadham C., Gamble J.R., Vadas M.A., Khew-Goodall Y. The protein tyrosine phosphatase Pez is a major phosphatase of adherens junctions and dephosphorylates beta-catenin. Mol. Biol. Cell. 2003;14:2520–2529. doi: 10.1091/mbc.E02-09-0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ogata M., Takada T., Mori Y., Oh-hora M., Uchida Y., Kosugi A., Miyake K., Hamaoka T. Effects of overexpression of PTP36, a putative protein tyrosine phosphatase, on cell adhesion, cell growth, and cytoskeletons in HeLa cells. J. Biol. Chem. 1999;274:12905–12909. doi: 10.1074/jbc.274.18.12905. [DOI] [PubMed] [Google Scholar]
- 40.Radhakrishnan K., Rockson S.G. Gorham's disease: an osseous disease of lymphangiogenesis? Ann. N Y Acad. Sci. 2008;1131:203–205. doi: 10.1196/annals.1413.022. [DOI] [PubMed] [Google Scholar]
- 41.Tonks N.K. Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 42.Blumenthal G.M., Dennis P.A. PTEN hamartoma tumor syndromes. Eur. J. Hum. Genet. 2008;16:1289–1300. doi: 10.1038/ejhg.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oishi K., Zhang H., Gault W.J., Wang C.J., Tan C.C., Kim I.K., Ying H., Rahman T., Pica N., Tartaglia M. Phosphatase-defective LEOPARD syndrome mutations in PTPN11 gene have gain-of-function effects during Drosophila development. Hum. Mol. Genet. 2009;18:193–201. doi: 10.1093/hmg/ddn336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tartaglia M., Mehler E.L., Goldberg R., Zampino G., Brunner H.G., Kremer H., van der Burgt I., Crosby A.H., Ion A., Jeffery S. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 45.Tartaglia M., Niemeyer C.M., Fragale A., Song X., Buechner J., Jung A., Hählen K., Hasle H., Licht J.D., Gelb B.D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003;34:148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 46.Vang T., Congia M., Macis M.D., Musumeci L., Orrú V., Zavattari P., Nika K., Tautz L., Taskén K., Cucca F. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat. Genet. 2005;37:1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z., Shen D., Parsons D.W., Bardelli A., Sager J., Szabo S., Ptak J., Silliman N., Peters B.A., van der Heijden M.S. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.