

Abstract
Gene-gene interactions have long been recognized to be fundamentally important for understanding genetic causes of complex disease traits. At present, identifying gene-gene interactions from genome-wide case-control studies is computationally and methodologically challenging. In this paper, we introduce a simple but powerful method, named “BOolean Operation-based Screening and Testing” (BOOST). For the discovery of unknown gene-gene interactions that underlie complex diseases, BOOST allows examination of all pairwise interactions in genome-wide case-control studies in a remarkably fast manner. We have carried out interaction analyses on seven data sets from the Wellcome Trust Case Control Consortium (WTCCC). Each analysis took less than 60 hr to completely evaluate all pairs of roughly 360,000 SNPs on a standard 3.0 GHz desktop with 4G memory running the Windows XP system. The interaction patterns identified from the type 1 diabetes data set display significant difference from those identified from the rheumatoid arthritis data set, although both data sets share a very similar hit region in the WTCCC report. BOOST has also identified some disease-associated interactions between genes in the major histocompatibility complex region in the type 1 diabetes data set. We believe that our method can serve as a computationally and statistically useful tool in the coming era of large-scale interaction mapping in genome-wide case-control studies.
Introduction
Genome-wide case-control studies use high-throughput genotyping technologies to assay hundreds of thousands of SNPs and relate them to clinical conditions or measurable traits. To understand underlying causes of complex disease traits, it is often necessary to consider joint genetic effects (epistasis) across the whole genome. The concept of epistasis1 was introduced around 100 years ago. It is generally defined as interactions among different genes. Recently, Phillips2 highlighted the essential role of gene-gene interactions in the structure and evolution of genetic systems. Three terminologies are used to describe gene-gene interactions:
-
•
Functional epistasis is a functional description that addresses the molecular interactions.
-
•
Compositional epistasis, originally defined by Bateson,1 is referred to as the blocking of one allelic effect by another allele at a different locus.
-
•
Statistical epistasis, attributed to Fisher,3 is defined as the statistical deviation from the additive effects of two loci on the phenotype.
The existence of epistasis has been widely accepted as an important contributor to genetic variation in complex diseases such as asthma, cancer, diabetes, hypertension, and obesity.4 As a matter of fact, many researchers believe that it is critical to model complex interactions in order to elucidate the joint genetic effects that may cause complex diseases. They have demonstrated the presence of gene-gene interactions in complex diseases such as breast cancer5 and coronary heart disease.6 The problem of detecting gene-gene interactions in genome-wide case-control studies has attracted extensive research interest. The difficulty in this problem is the heavy computational burden. For example, in order to detect pairwise interactions from 500,000 SNPs genotyped in thousands of samples, we need 1.25 × 1011 statistical tests in total. A recent review4 presented a detailed analysis on many popular methods that detect epistasis on the basis of the statistical definition, including MDR,5 PLINK,7 Tuning ReliefF,8 Random Jungle,9 BEAM,10 and three proposed search strategies.11
Among them, BEAM and MDR were reported to have difficulties in handling 500,000 SNPs genotyped in thousands of samples.4 Both methods need a prescreening process to reduce the number of SNPs in order to analyze the large data sets. Marchini et al.11 demonstrated that it is feasible to test association allowing for interactions in a genome-wide scale. Random Jungle can handle genome-wide data efficiently. However, both Marchini's method and Random Jungle aim at testing associations allowing for interactions, which is easier than testing interactions (we have detailed explanations of a test of association allowing for interactions and a test of interactions in the Discussion). PLINK was recommended as the most computationally feasible method that is able to detect gene-gene interactions in genome-wide data.4 PLINK finished a pairwise interaction examination of 89,294 SNPs selected from the WTCCC Crohn disease data set in 14 days. To accelerate the analysis process in genome-wide association studies (GWAS), the parallel computation was recommended.4,12
Here, we propose a fast method, named “BOolean Operation-based Screening and Testing” (BOOST), for the analysis of all pairwise interactions in genome-wide SNP data. In our method, we design a Boolean representation of genotype data, which promotes not only space efficiency but also CPU efficiency because it involves only Boolean values and allows for the use of fast logic (bitwise) operations to obtain contingency tables. On the basis of this data representation, we propose a two-stage (screening and testing) search method. In the screening stage, we use a noniterative method to approximate the likelihood ratio statistic in evaluating all pairs of SNPs and select those passing a specified threshold. Most nonsignificant interactions will be filtered out, and the survival of significant interactions is guaranteed. In the testing stage, we employ the classical likelihood ratio test to measure the interaction effects of selected SNP pairs. Experiments on WTCCC data sets show that our method is faster than current methods. This efficiency helps to identify interesting interaction patterns from the type 1 diabetes data set and the rheumatoid arthritis data set.
Material and Methods
Notation
Suppose we have L SNPs and n samples. We use Xl to denote the l-th SNP, , and Y to denote the class label (1 for case and 2 for control). SNPs are biallelic genetic markers in genome-wide case-control studies. In general, we use capital letters (e.g., A, B, …) to denote major alleles and use lowercase letters (e.g., a, b, …) to denote minor alleles. For each SNP, there are three genotypes: the homozygous reference genotype (AA), the heterozygous genotype (Aa), and the homozygous variant genotype (aa). The popular way of coding the genotype data is to use {1, 2, 3} to represent {AA, Aa, aa}, respectively.
Definition of Interaction via Logistic Regression Models
Interactions are often defined via logistic regression models.13 The logistic regression model with only main effects, i.e., the main effect model, has the following form:
| (Equation 1) |
The logistic regression model with both main effect terms and interaction terms, i.e., the full model, has the following form:
| (Equation 2) |
Please note that the superscript Xp of in both equations is merely a label and does not represent the exponent. The term represents the coefficient of Xp at category i. This representation extends to and as well. There are five coefficients in Equation 1 and nine coefficients in Equation 2. This is because one category of both Xp and Xq is used as the reference. This notation is adopted by Agresti14 to make the representations of logistic regression models and log-linear models (introduced later) more compact.
Let LM and LF be the log-likelihoods of the main effect model and the full model, respectively. According to the likelihood ratio test, interaction effects are defined as the difference of the log-likelihoods of these two models evaluated at their maximum likelihood estimations (MLEs), i.e., . Hence, interaction effects can be interpreted as the departure from linear models naturally.4
However, it is computationally unaffordable to directly use this measure to evaluate all pairs of SNPs in a genome-wide case-control study because there are hundreds of billions of pairs to be tested. Therefore, faster test procedures without the loss of statistical powers are needed in GWAS. Noticing the equivalence between a logistic regression model and its corresponding log-linear model,14 here we propose to test two-locus interactions on the basis of log-linear models. The advantage of so doing is that the test statistic can be quickly approximated without iteration.
Log-Linear Models for Contingency Tables
To test the interaction effect between two SNPs (Xp, Xq) and disease status Y by using log-linear models, a contingency table of these three variables will be used (see Table 1). The size of the contingency table is I × J × K, where I = 3, J = 3 and K = 2. In Table 1, nijk is used to denote the observed count in the cell (i, j, k). It is considered as a realization of a random variable Nijk assumed as Poisson distributed. We use πijk to denote the probability that an observation falls in the cell (i, j, k). A natural constraint of πijk is
| (Equation 3) |
Table 1.
The Genotype Counts in Cases and Controls
| Y = 1 | Xq = 1 | Xq = 2 | Xq = 3 | Y = 2 | Xq = 1 | Xq = 2 | Xq = 3 |
|---|---|---|---|---|---|---|---|
| Xp = 1 | n111 | n121 | n131 | Xp = 1 | n112 | n122 | n132 |
| Xp = 2 | n211 | n221 | n231 | Xp = 2 | n212 | n222 | n232 |
| Xp = 3 | n311 | n321 | n331 | Xp = 3 | n312 | n322 | n332 |
Cases are denoted with Y = 1 and controls with Y = 2.
We use the dot convention to indicate summation over a subscript; e.g., is the marginal probability of Xp = i, and is the number of observations with Xp = i. The notation extends to two dimensions as well. For example, is the marginal probability of Xp = i and Xq = j, and is the corresponding count. Clearly, we have .
Log-linear models treat Nijk as independent Poisson random variables with their means as follows:
| (Equation 4) |
The likelihood function is
| (Equation 5) |
Correspondingly, the log-likelihood function is
| (Equation 6) |
In the space of log-linear models, the homogeneous association model is the equivalent form of the logistic regression model with only main effects (defined in Equation 1), and the saturated model matches the full logistic regression model (defined in Equation 2). Table 2 summarizes the equivalence between log-linear models and logistic models for a three-way contingency table. The details are provided in the Appendix. In the following text, we explain how these two models are used to test interactions.
Table 2.
Equivalence between Log-Linear Models and Logistic Models for a Three-Way Table with Binary Response Variable Y
| Log-Linear Model | Logistic Model | MLE of μijk | ||
|---|---|---|---|---|
| Block independence model (MB): | β0 | |||
| Partial independence model (MP): | ||||
| Homogeneous association model (MH): | iterative estimation | |||
| Saturated model (MS): | nijk |
The models MB and MP are used in the discussion of the difference between the test of interactions and the test of associations. The details of these two models are provided in the Appendix.
Measuring Interaction via Log-Linear Models
On the basis of the equivalence between the log-linear model and its corresponding logistic regression model, we construct our test statistic using the homogeneous association model MH and the saturated model MS. Let LH and LS be the log-likelihood of MH and MS, respectively. According to Equation 6 and the MLE of μijk in MS (see Table 2 and the Appendix), the maximum log-likelihood of MS is
| (Equation 7) |
The log-likelihood of MH is maximized at its MLE :
| (Equation 8) |
In other words,
| (Equation 9) |
Notice that always exists and is unique because of the concavity of LH. To measure interaction effects based on the likelihood ratio test, we have
| (Equation 10) |
Because Equation 4 implies that
| (Equation 11) |
Equation 10 can be further reduced as
| (Equation 12) |
where is the Kullback-Leibler divergence of and .
The new measure provides us another interpretation of interactions. Equation 12 shows that the difference of the two log-likelihoods is proportional to the Kullback-Leibler divergence of the joint distribution obtained under the saturated model MS, and the distribution obtained under the homogeneous association model MH. The distribution is constructed via lower-order distributions (see the Appendix). From the perspective of log-linear models, interaction effects can be understood as the information contained in the joint distribution but not in its lower-order factorization, which is known as “synergy” in physics.15 If no interaction effects exist, the joint distribution can be well characterized by its lower-order factorization.
Boolean Operation-Based Screening and Testing
Boolean Representation of Genotype Data
The data set containing L SNPs and n samples is usually stored in an matrix. Each cell in this matrix takes a value from {1, 2, 3}, the elements of which represent the homozygous reference genotype, the heterozygous genotype, and the homozygous variant genotype, respectively. In our method, we introduce a Boolean representation of genotype data (the details are provided in the Appendix). This Boolean representation enables us to collect contingency tables in a fast manner.
Screening and Testing
Directly using to test interactions in GWAS still has some difficulties, because no closed-form solution exists for the homogenous association model MH. Iterative methods are needed in model fitting to compute . This will be computationally intensive when we face hundreds of billions of SNP pairs.
To solve this issue, we propose to approximate the homogenous association model MH with the Kirkwood superposition approximation (KSA):15
| (Equation 13) |
where is a normalization term. The benefit of using KSA is two-fold:
First, is an upper bound of ; i.e.,
| (Equation 14) |
where is the log-likelihood evaluated at the MLE of the KSA model (see the proof in the Appendix).
Noticing that the calculation of is straightforward and no iteration is involved, the approximated measure can be obtained easily on the basis of the contingency table collected by the Boolean operation. Therefore, the KSA model can be applied to evaluate hundreds of billions of SNP pairs. Because we are interested only in interactions with large values, we can first filter out those SNP pairs with by using a threshold τ, and we can then conduct statistical tests on the remaining SNP pairs.
Second, the bound in Equation 14 is tight. When the joint distribution is (Equation 13), the equality holds; i.e., . This bound is very close to the statistic of the likelihood ratio test. To illustrate the tightness of the bound, we use the simulation method proposed by Li et al.16 to generate a data set containing 2000 SNPs and 1000 samples based on HapMap data. Figure 1A shows the linkage disequilibrium (LD) pattern of the simulated data, which is very similar to the real data. Using this data, we calculate based on the KSA and based on log-linear models for all pairs of 2000 SNPs. Figure 1B shows the comparison of these two models. It can be seen that consistently overestimates . For the region [25, + ∞], is almost identical to .
Figure 1.
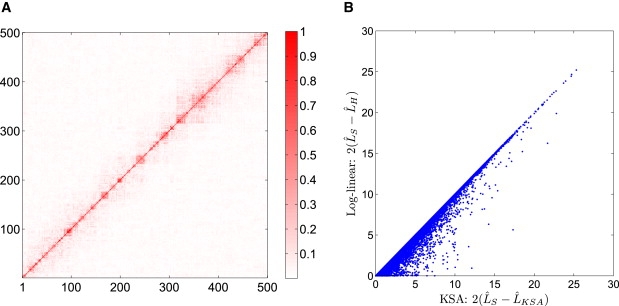
KSA Performance in Simulation
(A) The LD (measured by r2) pattern of simulated data from the Hapmap data. To show the block structure clearly, we show only the LD of the first 500 SNPs here. The LD block structure of all 2000 SNPs is very similar.
(B) Comparison of the values and based on KSA and log-linear models. KSA overestimation is illustrated here. For the region [25, + ∞), is almost identical to .
In summary, most nonsignificant interactions can be filtered out because of the tightness of the bound (Equation 14) and the survival of significant interactions is guaranteed. On the basis of this upper bound, we propose our method, BOOST:
Stage 1: Screening
We evaluate all pairwise interactions by using the KSA in the screening stage. For each pair, the calculation of is based on the contingency table collected by using Boolean operations. Because , an interaction obtained by the KSA without passing a specified threshold τ, i.e., , would not be considered in stage 2. The threshold τ corresponds to the significant threshold (with the Bonferroni correction) specified by users. Because the Bonferroni correction tends to be conservative, a smaller threshold can be used to put more SNP pairs into the testing stage. We set τ = 30 in our experiments to test the computational capacity of our method. The threshold τ = 30 corresponds to the unadjusted p = 4.89 × 10−6, which is a very weak significance level for a genome-wide study.
Stage 2: Testing
For each pair with , we test the interaction effect using the likelihood ratio statistic . We fit the log-linear models MH and MS and calculate this test statistic using Equation 12. After that, we conduct the χ2 test with four degrees of freedom (df = 4) to determine whether the interaction effect is significant. The p value is adjusted by the Bonferroni correction, with the number of tests , where L is the total number of SNPs before screening.
To approximate MH, we may also choose some other log-linear models, such as the block independence model MB or the partial independence model MP (see Table 2). However, such approximations will lead to very loose bounds, leaving millions of SNP pairs to be examined in the testing stage. Using the KSA, we have empirically observed that 300,000∼600,000 SNP pairs are examined in the testing stage when the WTCCC data are analyzed. When the partial independence model is used, the number of SNP pairs is up to 108∼109.
Results
Experiments on Simulation Data
The performance of our approach is evaluated through comparative studies with existing works. Our goal is to discover epistatic interactions from genome-wide data. Among many methods recently proposed, we mainly compare BOOST with PLINK7 with respect to the power of gene-gene interaction identification. The reasons for choosing PLINK for comparison are as follows:
-
•
A recent review4 tested many available methods and recommended PLINK as a powerful tool for testing interactions on a genome-wide scale.
-
•
Both PLINK and BOOST use an exhaustive search strategy. The comparison of their performance is fair.
We conduct the following simulation studies to compare BOOST with PLINK (tested with the “-fast-epistasis” option and without the “-case-only” option):
-
•
Case 1: Disease loci with main effects.
-
•
Case 2: Disease loci without main effects.
-
•
Case 3: Genetic heterogeneity.
-
•
Case 4: Null simulation for testing type I errors.
Case 1: Disease Loci with Main Effects
We consider four epistasis models whose odds tables are given in Table S7, available online. Model 1 is a multiplicative model.11 Model 2 is an epistasis model17 that has been used to describe handedness18 and the color of swine.19 Model 3 is a classical epistasis model.20,21 Model 4 is the well known XOR (exclusive OR) model.
Let p(D|Gi) denote the probability of an individual being affected given its genotype combination Gi (i.e., the penetrance of Gi), and let denote the probability of an individual not being affected given its genotype Gi. On the basis of the definition of the odds of a disease,
| (Equation 15) |
the penetrance p(D|Gi) of the genotype Gi can be calculated by using
| (Equation 16) |
The disease prevalence p(D) and genetic heritability h2 are given as
| (Equation 17) |
| (Equation 18) |
In our simulation, the prevalence p(D) and the heritability h2 are controlled by the parameters α and θ (see Table S6). We first specify the disease prevalence p(D) and the genetic heritability h2, and we then numerically solve the parameters (α and θ) on the basis of the above equations. For example, we set p(D) = 0.1 and h2 = 0.03 in model 1. Then we obtain α = 0.09989 and θ = 3.4481 for minor allele frequency (MAF) = 0.1.
In the simulation, we set h2 = 0.03 for model 1 and h2 = 0.02 for models 2, 3, and 4. We generate genotype data on the basis of the Hardy-Weinberg principle. We set the MAFs of disease-associated SNPs to be 0.1, 0.2, and 0.4. We generate the MAFs of unassociated SNPs uniformly from [0.05, 0.5]. We simulate 100 data sets under each setting for each disease model. Each data set contains 1000 SNPs. To take sample size into consideration, we simulate both 800 samples and 1600 samples with the balanced design.
Figure 2 presents the comparison results with the significance thresholds selected as 0.1, 0.2, and 0.3 after the Bonferroni correction. For model 1 with MAF = 0.2, 0.4 and model 2 with MAF = 0.1, the statistical power of PLINK is higher. This is because these model settings are well captured by the allele interaction test. For all other settings, BOOST outperforms PLINK.
Figure 2.
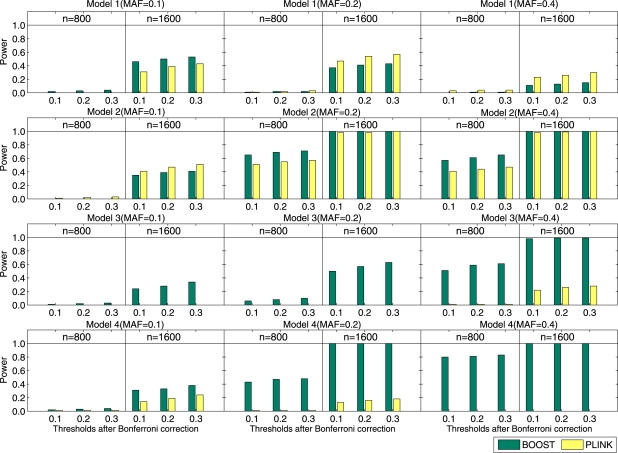
The Performance Comparison between BOOST and PLINK on Four Disease Models
Under each parameter setting, 100 data sets are generated. Both 800 samples and 1600 samples with balanced design are simulated. The power is calculated as the proportion of the 100 data sets in which the interactions of the disease-associated SNPs are detected. The absence of bars indicates no power.
Case 2: Disease Loci without Main Effects
Disease models displaying no main effects22 have been carefully discussed, and a wide spectrum of these models23 has been provided. In this experiment, we use all of these 70 pure epistatic models without main effect to compare performance. For convenience, these models are listed in Tables S8–S14. The heritability h2 controls the phenotypic variation of these 70 models, which ranges from 0.01 to 0.4. The MAF ranges from 0.2 to 0.4. For each model, the statistical power is evaluated under different sample sizes, including n = 400, n = 800, and n = 1, 600 (half controls and half cases). For each setting, 100 data sets are generated. Each data set contains 1000 SNPs.
Please check Figures S4–S7 to see the comparison results for the 70 models. For some models, such as model epi1–5, BOOST and PLINK perform equally well. For most of these models, BOOST is superior to PLINK because the interaction patterns cannot be well characterized by allele interactions.
Case 3: Genetic Heterogeneity
Genetic heterogeneity refers to the phenomenon that a disease is affected by different subsets of genes. It plays a substantial role in complex human diseases.24 Here, we set up a simulation study to show the performance of BOOST and PLINK when genetic heterogeneity is present. We choose some epistatic models used in case 2 to generate the data. The heritability h2 of these models ranges from 0.01 to 0.4. Different sample sizes, including n = 400, n = 800 and n = 1600, are simulated for each model. The details of simulation are provided in the Appendix.
The performance of both BOOST and PLINK is given in Figure S8. Genetic heterogeneity affects the performance of both BOOST and PLINK. In general, their performance degrades as heritability h2 decreases. The sample size plays an important role when genetic heterogeneity is present. When the sample size increases from 400 to 1600, the power of both BOOST and PLINK increases a lot.
Case 4: Null Simulation for Testing Type I Errors
To compare BOOST and PLINK in terms of type I errors, we conduct null simulation in two scenarios:
-
•
Scenario 1: Without LD. We generate 1000 null data sets. Each data set contains 1000 SNPs and 1000 samples. All of the SNPs are generated independently, with MAFs uniformly distributed in [0.05, 0.5]. The result is shown in Figure 3A. It can be seen that the type I error of BOOST agrees with the nominal error rate and the type I error of PLINK is a little bit less than the nominal error rate.
-
•
Scenario 2: With LD. The simulation program “genomeSIMLA”25 is used to simulate the SNP data on the basis of the marker information on the Affymetrix 500K chip from human chromosome 1. LD exists among SNPs. We generate 100 null data sets, each of which contains 38,836 SNPs and 1000 samples. The result is shown in Figure 3B. Because of the LD pattern, the error rates of both methods are lower than the nominal error rate, confirming that the Bonferroni correction is conservative. Surprisingly, unlike the situation in scenario 1, the error rate of BOOST is less than that of PLINK. The reason is that some cells of a contingency table may be empty when LD exists. This leads to the true degree of freedom dftrue ≤ 4. Because we calculate p values by using the χ2 distribution with df = 4, BOOST has a lower type I error rate than PLINK. This simulation study also implies that it is possible to increase the power of BOOST by using a more accurate degree of freedom in statistical tests.
Figure 3.
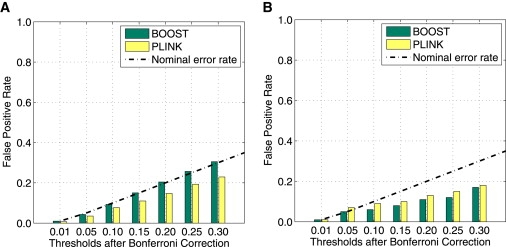
Comparison of the Type I Error Rates in Null Simulation
(a) Null simulation with no LD.
(b) Null simulation with LD.
Experiments on WTCCC data
We have applied BOOST to analyze data (14,000 cases in total and 3000 shared controls) from the WTCCC on seven common human diseases: bipolar disorder (BD), coronary artery disease (CAD), Crohn disease (CD), hypertension (HT), rheumatoid arthritis (RA), type 1 diabetes (T1D), and type 2 diabetes (T2D). The procedure of quality control is presented in the Appendix. The results under different constraints are reported in Table 3. For T1D, we discovered many gene-gene interactions in the MHC region (see detailed descriptions in the following section). For the other six diseases, however, we did not find nontrivial interactions (except one SNP pair in CD).
Table 3.
The Number of Interactions Identified from the WTCCC Data Sets of Seven Diseases under Different Constraints
| BD | CAD | CD | HT | RA | T1D | T2D | |
|---|---|---|---|---|---|---|---|
| C1 | 10 | 16 | 8 | 7 | 350 | 4499 | 18 |
| C1 & C2 | 0 | 0 | 1 | 0 | 0 | 789 | 0 |
| C1 & C2 & C3 | 0 | 0 | 1 | 0 | 0 | 91 | 0 |
Abbreviations are as follows: BD, bipolar disorder; CAD, coronary artery disease; CD, Crohn disease; HT, hypertension; RA, rheumatoid arthritis; T1D, type 1 diabetes; T2D, type 2 diabetes. C1 is the significance threshold constraint: the significance threshold is 0.05 for the Bonferroni-corrected interaction p value. C2 is the distance constraint: the physical distance between two interacting SNPs is at least 1Mb. This constraint is used to avoid interactions that might be attributed to the LD effects.4C3 is the main effect constraint: The single-locus p value should not be less than 10−6. This constraint is used to see whether there exist strong interactions without significant main effects, because those SNPs with p ≥ 10−6 are usually filtered out in the typical single-locus scan.
T1D and RA
The MHC region in chromosome 6 has long been investigated as the most variable region in the human genome with respect to infection, inflammation, autoimmunity, and transplant medicine.26 The recent study conducted by the WTCCC27 has shown that both T1D and RA are strongly associated with the MHC region via single-locus association mapping. The top-left panel of Figure 4 shows that the single-locus association map does not reveal much difference between T1D and RA. In our study, BOOST reports 4499 interactions in the T1D data set (see Table 3), in which 4489 interactions (99.8%) are in the MHC region. Clayton's analysis28 on the T1D data set found that with the exception of strong interactions within the MHC region, interactions are small and have a modest effect on prediction. Our results have verified Clayton's finding from another perspective. As a comparison, BOOST reports 350 interactions in the RA data set, in which 280 interactions (80.0%) are in the MHC region. Our genome-wide interaction map provides evidence that the MHC region is associated with these two diseases in different ways. The bottom panel of Figure 4 gives detailed interaction maps in the MHC region for T1D and RA data. We further calculate composite LD using the method by Zaykin et al.29 The LD map of MHC region is provided in the top-right panel of Figure 4. These interaction maps, different from the LD map, reveal a distinct pattern difference between T1D and RA. Specifically, there are three subregions in the MHC region: namely, the MHC class I region (29.8Mb–31.6Mb), the MHC class III region (31.6Mb–32.3Mb), and the MHC class II region (32.3Mb–33.4Mb). A closer inspection of the T1D interaction map indicates that strong interaction effects widely exist between genes within and across three classes, whereas most significant interactions in RA involve only loci closely placed in the MHC class II region. The contrast of the interaction patterns between T1D and RA may explain their different etiologies, which are not revealed by single-locus association mapping.
Figure 4.
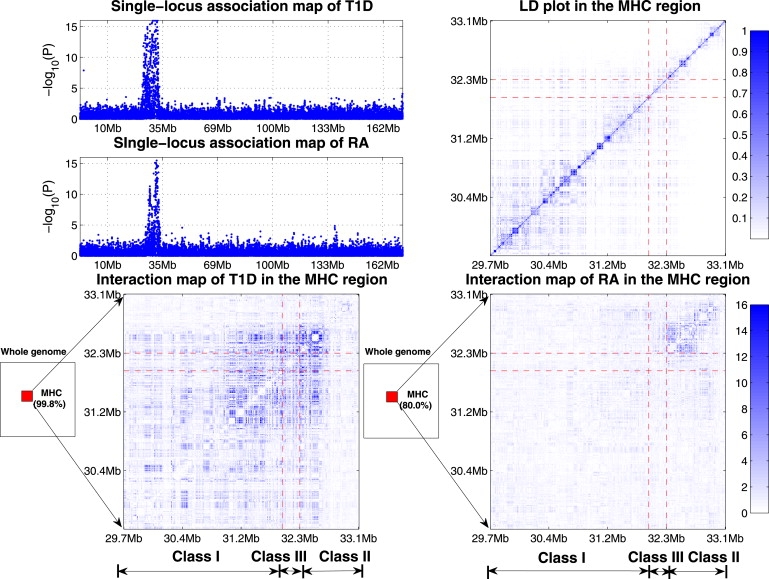
Comparison between the Single-Locus Association Mapping and the Interaction Mapping for T1D and RA
Top-left panel: Single-locus association mapping of T1D and RA. These two share a very similar hit region in chromosome 6.
Top-right panel: The LD map of the MHC region in control samples.
Bottom panel: Genome-wide interaction mapping of T1D and RA. 99.8% of T1D interactions and 80.0% of RA interactions are in the MHC region. Strong interaction effects widely exist between genes in and across the MHC class I, II, and III in T1D, whereas most significant interactions of RA involve only loci closely placed in the MHC class II region (The p values are truncated at p = 1.0 × 10−16).
Interactions without Significant Main Effects Detected in T1D
The mathematical property of interactions without significant main effects has been discussed in detail.22 The existence of these interactions has been shown from the experiment results based on relatively small numbers of SNPs.5,6 Here, we provide the result identified in the genome-wide scale. The MHC region is a highly polymorphic region with a high gene density. Although previous reports27,30 using the single-locus scan have identified strong associations between MHC genes (such as HLA-DQB1 and HLA-DRB1) and T1D, it is still unclear which and how many loci within the MHC region determine T1D susceptibility. Interactions without significant main effects can provide additional information to help pinpoint disease-associated loci, because SNPs involved in those interactions are usually filtered out in the single-locus scan.
Among the selected 789 interacting pairs in T1D, 91 pairs have nonsignificant loci under the single-locus scan (all of them are listed in Table S6). A careful inspection of these 91 interactions has identified two interesting interaction patterns between the MHC class I and class II. One interaction pattern involves the 31350k–31390k region (see Figure 5) and the 32810k–32860k region (see Figure 6) in chromosome 6 (please check more results in the Appendix). The interactions between two regions in these two figures are listed in Table 4. All SNPs in these interactions display weak main effects, whereas their joint effects are statistically significant. The potential pathways involving HLA_B, HLA_DQA2, and PSMB8 are shown in Figure 7. HLA_B, HLA_DQA2, and PSMB8 potentially interact in the antigen-processing and -presentation pathway.31–34 HLA_B and HLA_DQA2 potentially interact in the type 1 diabetes mellitus pathway.30,35,36 As Nejentsev et al.30 argued that both the MHC class I and II genes should be considered to better understand type 1 diabetes susceptibility, our results provide further evidence that the interaction effects between these two classes may contribute to the etiology of type 1 diabetes.
Figure 5.
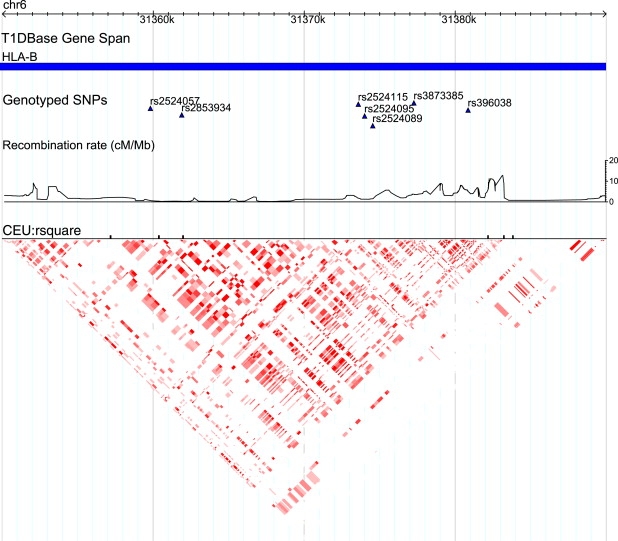
The 31350k–31390k Region of Chromosome 6
HLA-B in the MHC class I is located in this region. The recombination rate and LD plot from HapMap show that a block structure exists from 31360k to 31380k. This region is mapped through the SNPs rs2524057, rs2853934, rs2524115, rs396038, rs3873385, rs2524095, and rs2524089. The SNPs rs2524095 and rs2524089 are involved in the interactions with the 32930k–32960k region shown in Figure S2.
Figure 6.
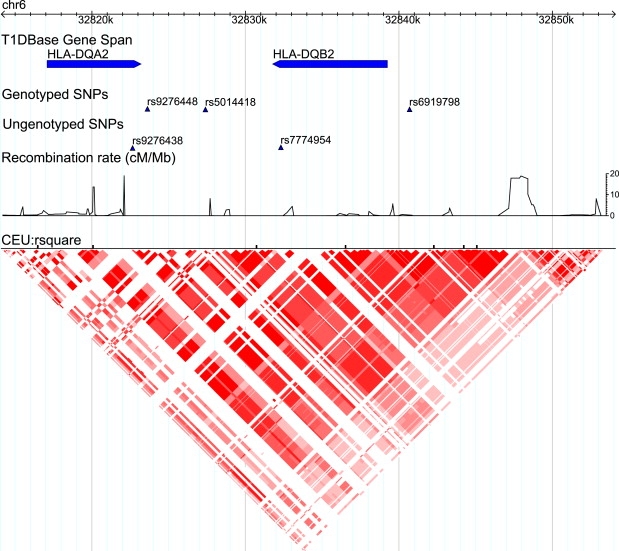
The 32810k–32860k Region of Chromosome 6
HLA-DQA2 and HLA-DQB2 in the MHC class II reside in this region. The recombination rate and LD plot from HapMap show that a block structure exists from 32820k to 32847k. This region is mapped through the genotyped SNPs rs9276448, rs5014418, and rs6919798. The ungenotyped SNPs rs9276438 and rs7774954 reside in HLA-DQA2 and HLA-DQB2, respectively. They are in strong LD with those genotyped SNPs.
Table 4.
|
SNP 1 |
SNP 2 |
Interaction |
||
|---|---|---|---|---|
| SNP | Single-Locus p Value | SNP | Single-Locus p Value | BOOST p Value |
| rs2524057 | 4.807 × 10−1 | rs9276448 | 8.878 × 10−3 | 5.362 × 10−14 |
| rs2524057 | 4.807 × 10−1 | rs5014418 | 1.116 × 10−2 | 2.738 × 10−13 |
| rs2853934 | 8.336 × 10−2 | rs9276448 | 8.878 × 10−3 | 2.507 × 10−13 |
| rs2524115 | 1.215 × 10−1 | rs9276448 | 8.878 × 10−3 | 6.456 × 10−13 |
| rs3873385 | 3.368 × 10−1 | rs9276448 | 8.878 × 10−3 | 3.186 × 10−14 |
| rs3873385 | 3.368 × 10−1 | rs5014418 | 1.116 × 10−2 | 3.841 × 10−14 |
| rs3873385 | 3.368 × 10−1 | rs6919798 | 6.077 × 10−2 | 4.257 × 10−13 |
| rs396038 | 9.939 × 10−2 | rs9276448 | 8.878 × 10−3 | 5.894 × 10−13 |
The SNPs in the SNP 1 column reside in HLA-B, and the SNPs in the SNP 2 column are located at the block across HLA-DQA2 and HLA-DQB2. They show strong interactions without displaying significant main effects.
Figure 7.
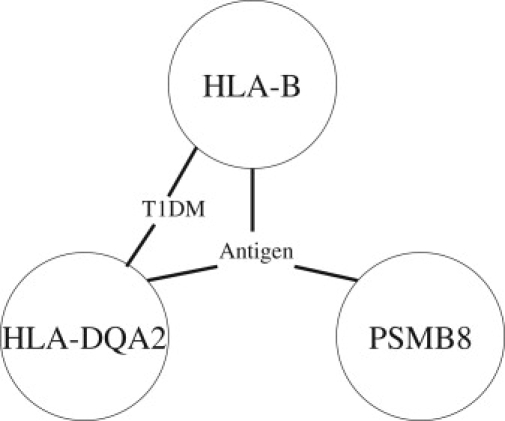
Potential Pathways Involving HLA_B, HLA_DQA2, and PSMB8
T1DM represents the type 1 diabetes mellitus pathway. Antigen represents the antigen processing and presentation pathway.
Discussion
Relationship between Our Method and Other Two-Stage Methods
The analysis of GWAS data is a challenging computational problem. To speed up this process, many methods4,5,11 have been coupled with some prescreening algorithms to reduce the number of SNPs. Most of the currently available screening algorithms are based on single-locus tests and can be finished very quickly. However, for some SNPs with weak main effects but significant interactions, these screening algorithms will filter them out. Our screening method does not have this issue. It uses a fast approximation to evaluate all SNP pairs with the guarantee that significant interactions will not be filtered out no matter whether individual SNPs display main effects or not.
Relationship between Our Method and PLINK
Both BOOST and PLINK use the exhaustive search to find epistatic interactions in GWAS. The key difference between BOOST and PLINK is the way that they test interaction effects:
-
•
PLINK tests interactions based on alleles.7 Three genotype categories are collapsed into two allele categories. Correspondingly, 3 × 3 contingency tables are collapsed into 2 × 2 tables. The difference of the odds ratios from the two 2 × 2 tables (one for cases and the other for controls) is used to construct a χ2 test with df = 1.
-
•
BOOST tests interactions based on genotypes, using the χ2 test with df = 4.
In general, if the underlying interaction could be well characterized by an allele interaction, then the statistical power of PLINK would be higher than that of BOOST. However, the type of underlying interaction is generally unknown and may vary widely.22 BOOST is more flexible because it covers a larger model space than PLINK. BOOST can be modified to test the allelic model by collapsing 3 × 3 contingency tables to 2 × 2 contingency tables (in the same way that PLINK does). The two-stage strategy in BOOST can then be applied to these 2 × 2 contingency tables. The statistical power of the modified BOOST will be roughly the same as PLINK because they both are based on the same allelic model. The ignorable difference is due to the difference between the Wald test and the likelihood ratio test. In the released software of BOOST, the allelic test has also been implemented. Regarding the running time, the BOOST allelic test is similar to the BOOST genotype test.
Relationship between Our Method and INTERSNP
Recently, INTERSNP37 has implemented the interaction test in GWAS using log-linear models. Regarding the interaction test, both INTERSNP and our work are developed on the basis of the standardized definition using logistic regression models.13 INTERSNP has directly used an iterative method to fit the log-linear model MH. It is still very time consuming to test interactions in GWAS. Therefore, INTERSNP suggests the use of some prior knowledge to reduce the number of SNPs, including the single-locus test, genetics criteria, and pathway information. Genetics criteria and pathway information provide biological constraints that are very useful. But using the single-locus test in the filtering, which has been discussed in the earlier section, will filter out those SNPs with weak main effects but significant interactions. Moreover, how to choose the threshold in filtering is also critical. On the contrary, we propose to use the noniterative approximation to directly examine all SNPs pairs. We show the computational performance of BOOST and INTERSNP in the following section.
Computation Time
From a practical point of view, a key issue of detecting gene-gene interactions in genome-wide case-control studies is the computational efficiency. Cordell4 reported that PLINK took about 14 days to test pairwise interactions of the selected 89,294 SNPs on a single node of a computer cluster. Random Jungle can analyze the large data sets quickly. However, Random Jungle aims at detecting association allowing for interactions rather than detecting interactions (see detailed explanations in the next subsection). Besides, Random Jungle has difficulty in finding interacting SNP pairs displaying weak main effects because trees built in Random Jungle rely on the main effects of SNPs. BEAM took about 8 days to handle 47,727 SNPs using 5 × 107 Markov chain Monte Carlo iterations. Currently, BEAM has difficulties in handling 500,000 to 1,000,000 SNPs genotyped in 5000 or more samples. Cordell4 recommended PLINK as a powerful method of testing interactions in GWAS.
We tested the running time of PLINK on our desktop computer. In addition, we also tested INTERSNP on the same data sets because INTERSNP also uses log-linear models to test interactions. The results are shown in Table 5. BOOST is roughly 63 times faster than PLINK and 95 times faster than INTERSNP. It can finish the analysis of all pairs of roughly 360,000 SNPs within 60 hr (around 2.5 days) on a standard desktop (3.0 GHz CPU with 4G memory running the Windows XP Professional x64 edition system). Parallel computing12 can be used to further improve the computation time for BOOST, PLINK, and INTERSNP. The WTCCC phase 2 study will analyze over 60,000 samples of various diseases using either the Affymetrix v6.0 chip or the Illumina 660K chip. The shared control samples will increase from 3000 to 6000. Such an increase in the number of SNPs and the sample size is more demanding on the computation efficiency. We anticipate that BOOST is still applicable for analyzing the new data sets.
Table 5.
Time Comparison of BOOST, PLINK, and INTERSNP
| Data Size | BOOST | PLINK | INTERSNP |
|---|---|---|---|
| n = 5000, L = 1000 | < 2s | 106s | 160s |
| n = 5000, L = 5000 | 42s | 2703s | 4277s |
| n = 5000, L = 10,000 | 170s | 10,915s | 15,805s |
PLINK is tested with the “–fast-epistasis” option and without the “–case-only” option. All timings are carried out on a 3.0 GHz CPU with 4G memory running the Windows XP Professional system.
Test of interactions versus Test of Associations
To test association between a specific SNP Xp and the phenotype Y, a typical method is to test the difference between the deviance of the null model (Equation 19) and the deviance of the alternative model (Equation 20) with df = 2:
| (Equation 19) |
| (Equation 20) |
This is known as a “test of single-SNP association.”
In the above test, SNP Xp is allowed to interact with other SNPs. As a matter of fact, if the disease is influenced by SNP Xp itself and its interaction effect with another SNP Xq, the statistical power of detecting SNP Xp will be increased when allowing for interactions. This is known as a “test of two-locus associations allowing for interactions”4. Typically, this is accomplished by testing the difference between the log-likelihood of the null model (Equation 19) and that of the alternative model (Equation 21) with df = 8:
| (Equation 21) |
Marchini et al.11 highlighted the importance of testing associations allowing for interactions in a genome-wide scale and successfully demonstrated its feasibility. They reported that performing all pairwise tests of associations allowing for interactions with df = 8 at 300,000 loci with 1000 cases and 1000 controls can be finished in 33 hr on a 10-node cluster. According to the equivalence between log-linear models and logistic models, it is clear that the feasibility of this exhaustive search method relies on the closed-form solution of the block independence model MB and the closed-form solution of the saturated model MS (see the Appendix for the details of MB and MS).
The differences of these tests are:
-
•
The test of single-SNP association is to compare MP with MB (see Table 2 for descriptions of MP and MB).
-
•
The test of associations allowing for interactions is to compare MS with MB.
-
•
The test of interaction is to compare MS with MH.
As we mentioned above, no closed-form solution exists for the test of interactions. In this sense, the test of interactions is more difficult than the test of associations allowing for interactions.
On Statistical Epistasis
It is extensively debated to what extent statistical epistasis implies biological or functional epistasis.4 The statistical epistasis is exploited in the literature, perhaps because of the following reasons:
-
•
The definition of statistical epistasis yields an appropriate measure for describing biological phenomena that one locus's effect on the phenotype depends on another locus.2 This facilitates mathematical analysis of epistasis.
-
•
On the basis of the statistical definition, gene-gene interactions can be connected to Kullback-Leibler divergence used in the information theory (see Equation 12) and high-order mutual information in physics.15 This definition may bridge the gap between the biological understanding and the physical interpretation.
-
•
Compositional epistasis, conceived by Bateson, is closer to the biological understanding of gene-gene interactions than statistical epistasis.2 Compositional epistasis has recently been shown to be empirically testable via a statistical approach.38 In some cases, compositional and statistical epistatis are equivalent to each other.38 Therefore, statistical epistasis can still provide useful information for biological understanding.
Currently, PLINK, INTERSNP, and BOOST are designed to test statistical epistasis. We realize that detecting statistical epistasis in a genome-wide scale is easier than finding compositional epistasis because the test of compositional epistasis for each SNP pair requires enumerating all possible genetic interaction models.2 The detection of compositional epistasis will be investigated in our future work.
Conclusion
The large number of SNPs genotyped in genome-wide case-control studies poses a great computational challenge in the identification of gene-gene interactions. During the last few years, there have been fast-growing interests in developing and applying computational and statistical approaches to finding gene-gene interactions. In this paper, we present a method named “BOOST” to address this problem. Not only is BOOST computationally efficient, it has also shown good statistical power for a wide spectrum of epistasis models. We have successfully applied our method to analyze seven data sets from the WTCCC. Our experimental results demonstrate that interaction mapping is both computationally and statistically feasible for hundreds of thousands of SNPs genotyped in thousands of samples.
In this work, we focus mainly on the genome-wide case-control studies; i.e., the disease phenotype can be represented as a binary variable. In the current stage, our method cannot be applied to GWAS involving continuous phenotypes unless those continuous phenotypes can be discretized. There are two ways to handle covariates in our models. If the covariate is discrete or can be discretized, our method can be directly extended to handle it. If not, logistic regression can be used in the postprocessing step to adjust the covariate. In the postprocessing step, the computational burden of logistic regression is affordable because the number of selected interactions is limited.
There are some limitations of BOOST with respect to statistical power. BOOST uses a fixed degree of freedom (df = 4) to conduct the genotype test. When the contingency table is too sparse due to the low minor allele frequency, the degree of freedom of the statistical test should be reduced. To improve the performance of BOOST, we can first use BOOST to report interactions with a loose threshold and then use the penalized logistic regression39 with the adaptive degree of freedom to adjust these interactions. There are several other issues that we have not addressed, such as population substructures and imputation of the missed genotypes. We will investigate them in our future work.
Acknowledgments
We thank the editor and the anonymous reviewers for their constructive suggestions and comments. This work was partially supported with grant GRF621707 from the Hong Kong Research Grant Council, grants RPC06/07.EG09, RPC07/08.EG25, and RPC10EG04 from the Hong Kong University of Science and Technology, and a grant from Sir Michael and Lady Kadoorie Funded Research Into Cancer Genetics.
Appendix
Log-Linear models
Here, we briefly describe four log-linear models, including the homogeneous association model MH, the saturated model MS, the block independence model MB, and the partial independence model MP. These four models are used in the main text. Please see details in Agresti.14
Homogeneous Association Model MH
The homogeneous association model MH factorizes the joint distribution πijk using the joint distributions of all pairs. The hypothesis is
| (Equation 22) |
where ψij, ϕik and ωjk are some lower-order distributions. The name “homogeneous association” comes from the fact that the association between any two of three variables is the same at all levels of the third variable.14
The homogeneous association model MH is defined as
| (Equation 23) |
Unfortunately, no closed-form expression exists for the MLE of μijk (denoted as ) in Equation 23. Iterative approaches, such as the Newton-Raphson method, are needed in order to estimate the parameters.
Saturated Model MS
The saturated model MS defines the joint distribution with all factors. The saturated log-linear model is
| (Equation 24) |
The MLE of μijk in Equation 24 is
| (Equation 25) |
Block Independence Model MB
When the joint distribution cannot be completely factorized, it may be factorized into blocks. The hypothesis is
| (Equation 26) |
The corresponding log-linear model is
| (Equation 27) |
Under this structure, the MLE of μijk is
| (Equation 28) |
Partial Independence Model MP
The joint distribution may be factorized when some variables are given. For example, given Y, the hypothesis is
| (Equation 29) |
The corresponding log-linear model is
| (Equation 30) |
Then the MLE of μijk is
| (Equation 31) |
Connection between Log-Linear Models and Logistic Models
For convenience, we use the homogeneous association model MH as an example to describe the equivalence between a log-linear model and its corresponding logistic model. Its logit is
| (Equation 32) |
The first term is a constant that does not depend on i or j. The second term depends only on the category i of Xp. The third term depends only on the category j of Xq. Therefore, this logit has the following form:
| (Equation 33) |
Clearly, this is equivalent to the logistic model with only main effect terms defined in Equation 1. Using the similar inference mentioned above, it is straightforward to find the connection between the saturated model MS and the full logistic regression model defined in Equation 2.
Proof of
To show this, we need only to show . By Equation 4 and Equation 13, we have
| (Equation 34) |
Taking the logarithm on both sides of Equation 34 yields
| (Equation 35) |
where
| (Equation 36) |
This shows that the KSA model can be written in the form of Equation 23. For any model with this structure, we have shown that the log-likelihood LH evaluated at its MLE achieves its maximum in Equation 9. Therefore, we have
| (Equation 37) |
Boolean Representation and Operation of Genotype Data
For a data set containing L SNPs genotyped from n samples, an matrix W is usually used to store the data, where each row represents genotype data for one specific SNP and each column represents one sample. A toy example including three SNPs genotyped from 16 samples is illustrated below, where the first eight columns in W (denoted as Ui) represent control samples and the others represent case samples (denoted as Di).
To evaluate the interaction effect between SNP p and SNP q, we need two rows (Xp,Xq) in W to collect the contingency table. It is very time consuming to collect contingency tables for all SNP pairs in a genome-wide case-control study, because hundreds of billions of SNPs pairs exist for typical genotyping chips.
In our method, we introduce a Boolean representation of genotype data. Instead of using one row for each SNP, the new representation uses three rows, with each row for one specific genotype. Each row consists of two-bit strings, one for control samples and the other for case samples. Each bit in the string represents one sample, and its value (0 or 1) indicates whether the sample has the corresponding genotype. For the above toy example, the corresponding Boolean representation is as follows:
Both W and Wbit contain the same amount of information. To demonstrate this equivalence, we underline some matched items between W and Wbit. For example, the five 2′s in the first row of W are represented as five 1's in the second row of Wbit. Although the dimension of Wbit is three times as large as that of W, its space usage in the computer is smaller because each byte can store 8 bits. For a data set with 4000 samples and 500,000 SNPs (about the same size as the WTCCC data set), the new data representation needs around 700M bytes, whereas the general data representation requires 1900M bytes. More importantly, using Wbit is more CPU efficient than using W in collecting the contingency table (Table 1). This is because we can directly carry out the fast logic (bitwise) operation with Wbit. For example, to collect n121 in Table 1 (n121 represents the number of cases with Xq = 1 and Xq = 2), we just need to conduct the logical AND operation on the case bit strings of row Xp = 1 and Xq = 2, then count the number of 1's in the result. The 64-bit registers can perform 64-bit AND operation in one instruction, and the counting of “1” bits in a bit string (also called hamming weight) can be accomplished with an efficient algorithm (see http://en.wikipedia.org/wiki/Hamming_weight).
Genetic Heterogeneity Simulation
The simulation models are chosen on the basis of the performance of BOOST and PLINK in case 2. For each setting of h2 and MAF, there are five models. We choose the one under which BOOST and PLINK perform best (i.e., have the highest statistical power). For example, both BOOST and PLINK have the best performance on model epi33 among models epi31–epi35 (with the same setting of h2 = 0.05 and MAF = 0.2). Therefore, for this setting of h2 and MAF, we select model epi33. The reason for so doing is to make sure that both BOOST and PLINK have reasonably good performance when genetic heterogeneity is absent. Then we can observe how genetic heterogeneity degrades their performance. All selected models are given in Table S5. In the simulation, 100 data sets are generated under each model setting. In each data set, 1000 SNPs are simulated. Different sample sizes (n = 400, 800, and 1600) are simulated. To simulate genetic heterogeneity, 50% case samples are generated at loci X1 and X2 and another 50% case samples are generated at loci X3 and X4. The distribution of case samples is based on a specific disease model given in Table S6. Each data set has two pairs of associated SNPs. Therefore, there are 200 pairs of SNPs for each parameter setting. We set the counter T to be zero initially. If one pair of these 200 pairs is detected (on the basis of the Bonferroni correction), then T = T + 1. After testing 100 data sets, the power is calculated as T/200.
Quality Control
We first check the quality of control samples:
-
•
Those genotype data with a Chiamo score27 < 0.95 are considered as missing data. SNPs with more than 10% missing data are removed.
-
•
Those SNPs with a minor allele frequency < 0.05 are removed.
-
•
We also perform the Hardy-Weinberg Equilibrium (HWE) test for each SNP. Those SNPs with a p value ≤ 0.001 are removed.
Next, we check the quality of case samples. The strategy is similar to that for control samples except that the HWE test is not performed. The number of remaining SNPs is given in Table S1.
More Results of T1D Data Analysis
We have identified 91 interactions in which all loci are nonsignificant in the single-locus scan. These 91 interactions show two interesting interaction patterns between MHC class I and class II. We have shown one pattern in the main article. We have also identified another interaction pattern in chromosome 6 in the 31350k–31390k region (shown in Figure S1) and the 32930k–32960k region (shown in Figure S2). The six interactions between these two regions are listed in Table S2. It can be observed again that all SNPs in this table display weak main effects whereas their joint effects are statistically significant. We further report the odds ratios for those interactions in Table S3 and Table S4. For the first interaction group given in Table S3, the genotype combinations Aa/Bb, Aa/bb, aa/Bb, and aa/bb, where the uppercase and lowercase letters represent the major alleles and minor alleles, respectively, have significantly higher disease risks than others. The interaction effect of these genotypes can generally approximate the multiplicative model (see the left panel of Figure S3). For the second interaction group given in Table S4, the genotype combination aa/bb has a significantly higher disease risk than others. The interaction effect of this genotype is considered as a joint recessive effect (see the right panel of Figure S3).
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
BOOST software, http://bioinformatics.ust.hk/BOOST.html
References
- 1.Bateson W., Mendel G. Cambridge University Press; Cambridge: 1909. Mendel's Principles of Heredity. [Google Scholar]
- 2.Phillips P.C. Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 2008;9:855–867. doi: 10.1038/nrg2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher R.A. The correlations between relatives on the supposition of mendelian inheritance. Philosophical Transactions of the Royal Society of Edinburgh. 1918;52:399–433. [Google Scholar]
- 4.Cordell H.J. Detecting gene-gene interactions that underlie human diseases. Nat. Rev. Genet. 2009;10:392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ritchie M.D., Hahn L.W., Roodi N., Bailey L.R., Dupont W.D., Parl F.F., Moore J.H. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am. J. Hum. Genet. 2001;69:138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson M.R., Kardia S.L., Ferrell R.E., Sing C.F. A combinatorial partitioning method to identify multilocus genotypic partitions that predict quantitative trait variation. Genome Res. 2001;11:458–470. doi: 10.1101/gr.172901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moore J., White B. Tuning reliefF for genomewide genetic analysis. Lect. Notes Comput. Sci. 2007;4447:166–175. [Google Scholar]
- 9.Schwarz D., Kónig I., Ziegler A. On safari to random jungle: A fast implementation of random forests for high dimensional data. Bioinformatics. 2010;26:1752–1758. doi: 10.1093/bioinformatics/btq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y., Liu J.S. Bayesian inference of epistatic interactions in case-control studies. Nat. Genet. 2007;39:1167–1173. doi: 10.1038/ng2110. [DOI] [PubMed] [Google Scholar]
- 11.Marchini J., Donnelly P., Cardon L.R. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat. Genet. 2005;37:413–417. doi: 10.1038/ng1537. [DOI] [PubMed] [Google Scholar]
- 12.Ma L., Runesha H., Dvorkin D., Garbe J., Da Y. Parallel and serial computing tools for testing single-locus and epistatic SNP effects of quantitative traits in genome-wide association studies. BMC Bioinformatics. 2009;9:315. doi: 10.1186/1471-2105-9-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordell H.J. Epistasis: what it means, what it doesn't mean, and statistical methods to detect it in humans. Hum. Mol. Genet. 2002;11:2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- 14.Agresti A. Second Edition. Wiley and Sons; 2002. Categorical Data Analysis. Wiley Series in Probability and Statistics. [Google Scholar]
- 15.Matsuda H. Physical nature of higher-order mutual information: Intrinsic correlations and frustration. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 2000;6:3096–3102. doi: 10.1103/physreve.62.3096. [DOI] [PubMed] [Google Scholar]
- 16.Li J., Chen Y. Generating samples for association studies based on HapMap data. BMC Bioinformatics. 2008;9:44. doi: 10.1186/1471-2105-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neuman R.J., Rice J.P. Two-locus models of disease. Genet. Epidemiol. 1992;9:347–365. doi: 10.1002/gepi.1370090506. [DOI] [PubMed] [Google Scholar]
- 18.Levy J., Nagylaki T. A model for the genetics of handedness. Genetics. 1992;72:117–128. doi: 10.1093/genetics/72.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lerner I. W.H. Freeman; San Francisco: 1968. Heredity, Evolution, and Society. [Google Scholar]
- 20.Li W., Reich J. A complete enumeration and classification of two-locus disease models. Hum. Hered. 2000;50:334–349. doi: 10.1159/000022939. [DOI] [PubMed] [Google Scholar]
- 21.Frankel W.N., Schork N.J. Who's afraid of epistasis? Nat. Genet. 1996;14:371–373. doi: 10.1038/ng1296-371. [DOI] [PubMed] [Google Scholar]
- 22.Culverhouse R., Suarez B.K., Lin J., Reich T. A perspective on epistasis: limits of models displaying no main effect. Am. J. Hum. Genet. 2002;70:461–471. doi: 10.1086/338759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velez D.R., White B.C., Motsinger A.A., Bush W.S., Ritchie M.D., Williams S.M., Moore J.H. A balanced accuracy function for epistasis modeling in imbalanced datasets using multifactor dimensionality reduction. Genet. Epidemiol. 2007;31:306–315. doi: 10.1002/gepi.20211. [DOI] [PubMed] [Google Scholar]
- 24.McClellan J., King M.C. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 25.Dudek S., Motsinger A., Velez D., Williams S., Ritchie M. Data simulation software for whole-genome association and other studies in human genetics. Pacific Symposium on Biocomputing. 2006:499–510. [PubMed] [Google Scholar]
- 26.Lechler R., Warrens A. Academic Press; 2000. HLA in health and disease. [Google Scholar]
- 27.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clayton D.G. Prediction and interaction in complex disease genetics: experience in type 1 diabetes. PLoS Genet. 2009;5:e1000540. doi: 10.1371/journal.pgen.1000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaykin D.V., Meng Z., Ehm M.G. Contrasting linkage-disequilibrium patterns between cases and controls as a novel association-mapping method. Am. J. Hum. Genet. 2006;78:737–746. doi: 10.1086/503710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nejentsev S., Howson J.M., Walker N.M., Szeszko J., Field S.F., Stevens H.E., Reynolds P., Hardy M., King E., Masters J., Wellcome Trust Case Control Consortium Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature. 2007;450:887–892. doi: 10.1038/nature06406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown M.G., Driscoll J., Monaco J.J. Structural and serological similarity of MHC-linked LMP and proteasome (multicatalytic proteinase) complexes. Nature. 1991;353:355–357. doi: 10.1038/353355a0. [DOI] [PubMed] [Google Scholar]
- 32.Ortiz-Navarrete V., Seelig A., Gernold M., Frentzel S., Kloetzel P.M., Hämmerling G.J. Subunit of the ‘20S’ proteasome (multicatalytic proteinase) encoded by the major histocompatibility complex. Nature. 1991;353:662–664. doi: 10.1038/353662a0. [DOI] [PubMed] [Google Scholar]
- 33.Villadangos J.A. Presentation of antigens by MHC class II molecules: getting the most out of them. Mol. Immunol. 2001;38:329–346. doi: 10.1016/s0161-5890(01)00069-4. [DOI] [PubMed] [Google Scholar]
- 34.Rocha N., Neefjes J. MHC class II molecules on the move for successful antigen presentation. EMBO J. 2008;27:1–5. doi: 10.1038/sj.emboj.7601945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howson J.M., Walker N.M., Clayton D., Todd J.A., Type 1 Diabetes Genetics Consortium Confirmation of HLA class II independent type 1 diabetes associations in the major histocompatibility complex including HLA-B and HLA-A. Diabetes Obes. Metab. 2009;Suppl 1:31–45. doi: 10.1111/j.1463-1326.2008.01001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Husain Z., Kelly M.A., Eisenbarth G.S., Pugliese A., Awdeh Z.L., Larsen C.E., Alper C.A. The MHC type 1 diabetes susceptibility gene is centromeric to HLA-DQB1. J. Autoimmun. 2008;30:266–272. doi: 10.1016/j.jaut.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Herold C., Steffens M., Brockschmidt F.F., Baur M.P., Becker T. INTERSNP: genome-wide interaction analysis guided by a priori information. Bioinformatics. 2009;25:3275–3281. doi: 10.1093/bioinformatics/btp596. [DOI] [PubMed] [Google Scholar]
- 38.VanderWeele T.J. Epistatic interactions. Statistical Application in Genetics and Molecular Biology. 2010;9 doi: 10.2202/1544-6115.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park M.Y., Hastie T. Penalized logistic regression for detecting gene interactions. Biostatistics. 2008;9:30–50. doi: 10.1093/biostatistics/kxm010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.