

Abstract
Occult macular dystrophy (OMD) is an inherited macular dystrophy characterized by progressive loss of macular function but normal ophthalmoscopic appearance. Typical OMD is characterized by a central cone dysfunction leading to a loss of vision despite normal ophthalmoscopic appearance, normal fluorescein angiography, and normal full-field electroretinogram (ERGs), but the amplitudes of the focal macular ERGs and multifocal ERGs are significantly reduced at the central retina. Linkage analysis of two OMD families was performed by the SNP High Throughput Linkage analysis system (SNP HiTLink), localizing the disease locus to chromosome 8p22-p23. Among the 128 genes in the linkage region, 22 genes were expressed in the retina, and four candidate genes were selected. No mutations were found in the first three candidate genes, methionine sulfoxide reductase A (MSRA), GATA binding 4 (GATA4), and pericentriolar material 1 (PCM1). However, amino acid substitution of p.Arg45Trp in retinitis pigmentosa 1-like 1 (RP1L1) was found in three OMD families and p.Trp960Arg in a remaining OMD family. These two mutations were detected in all affected individuals but in none of the 876 controls. Immunohistochemistry of RP1L1 in the retina section of cynomolgus monkey revealed expression in the rod and cone photoreceptor, supporting a role of RP1L1 in the photoreceptors that, when disrupted by mutation, leads to OMD. Identification of RP1L1 mutations as causative for OMD has potentially broader implications for understanding the differential cone photoreceptor functions in the fovea and the peripheral retina.
Main Text
Occult macular dystrophy (OMD) is an autosomal-dominant form of inherited macular dystrophy characterized by progressive decrease of visual acuity due to macular dysfunction, which was first reported by Y.M. et al. in 1989.1–3 The disorder was called “occult” because of the fact that the macular dysfunction of this disease is hidden by a normal fundus appearance. Typical OMD, as described by Y.M. et al., is characterized by central cone dysfunction and in some cases rod dysfunction, leading to a loss of vision despite normal ophthalmoscopic appearance, normal fluorescein angiography, and normal full-field electroretinograms (ERGs). However, the amplitudes of the focal macular ERGs and multifocal ERGs are significantly reduced, indicating dysfunction of the central retina.1,2,4 OMD is known for its broad range of age at disease onset, from 6 to 81 yrs. Brockhurst et al. have reported age at onset of four out of eight OMD patients at over 65 yrs5 and similar findings have also been observed in earlier cases.1,2 The patient III-3 in family 1 did not notice any visual disturbance in her right eye even at the age of 81 yrs.
The four families shown in Figure 1 demonstrate dominant inheritance of the OMD phenotype. None of the patients had ocular diseases other than OMD, except senile cataract or diabetic retinopathy. Control family members were confirmed to be normal via a complete ophthalmic examination including focal macular ERGs or multifocal ERGs. For this study, the ethics review committees of the National Hospital Organization Tokyo Medical Center, the Niigata University Graduate School of Medical and Dental Sciences, and the Nagoya University Medical School approved the study, and written informed consent was obtained from both affected and unaffected subjects.
Figure 1.
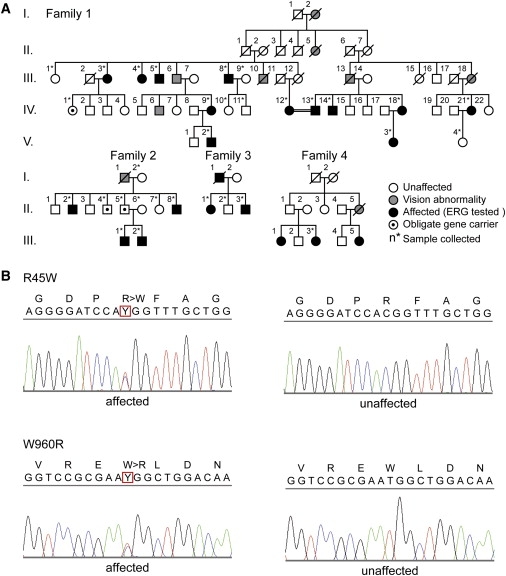
Autosomal OMD Families and DNA Sequencing of RP1L1
(A) The four families shown demonstrate dominant inheritance of the OMD phenotype. In all presented families, none of the patients had ocular diseases other than OMD, except senile cataract and diabetic retinopathy. Control family members were confirmed to be normal via a complete ophthalmic examination including focal macular ERGs or multifocal ERGs.
(B) DNA sequencing of both p.Arg45Trp and p.Trp960Arg mutations found in four independent families.
Linkage analysis of OMD families 1 and 2 was performed. Eighteen individuals from family 1 and eleven individuals from family 2 were genotyped by Affymetrix's Genome-Wide Human SNP array 6.0 in accordance with the manufacturer's instructions (Affymetrix, Santa Clara, CA). DNA samples from family 2 were subjected to whole-genome amplification with the use of REPLI-g (QIAGEN, Tokyo, Japan) prior to SNP genotyping. With SNP HiTLink6 used as a pipeline, SNPs with a Hardy-Weinberg p value > f 0.001, a call rate of 1, and a maximum confidence score > 0.02 were used for the analysis. SNPs with the minor allele frequency of 0 in controls were eliminated from the analysis. Parametric multipoint linkage analysis (autosomal-dominant model with a setting of liability classes; age-dependent penetrance of 0.19, 0.55, and 0.91 for 0–20, 21–40, and > 41 yrs old, respectively, and disease frequency of 0.000001) was performed with Allegro version 2,7 intermarker distance from 80 kb to 120 kb with the use of SNP HiTLink. Because of the limitation of computational capacity, family 1 was divided into two branches (branch 1-1: descendants of II-1; branch 1-2: descendants of II-7) for multipoint linkage analysis. Haplotypes were reconstructed by Allegro.
The parametric linkage study of family 1 using SNP microarrays and SNP HiTLink mapped the disease locus to an approximately 10 Mb region of chromosome 8p22-p23 with a maximum LOD score of 3.77 (Figure 2). Parametric linkage analysis of affected individuals only produced similar results (Figure 3 and Figure S2 available online). A common haplotype between rs365309 and rs2632841 was shared by all of the affected individuals (Table 1). With the additional linkage study of family 2, the cumulative parametric multipoint LOD score rose to over 4 (Figure S1). A total of 128 known genes were found within the approximately 10 Mb linkage-associated region, containing 22 retina-expressed genes as candidates for mutational analyses. No mutations were found in the first three candidate genes, methionine sulfoxide reductase A (MSRA), GATA binding 4 (GATA4), and pericentriolar material 1 (PCM1). However, a c.362C>T (p.Arg45Trp) substitution in retinitis pigmentosa 1-like 1 (RP1L1 [MIM 608581]) was found in all affected individuals in family 1. We further extended the mutational analysis of RP1L1 to three other families with autosomal OMD, and we identified the p.Arg45Trp alteration in families 2 and 4 and a c.3107T>C (p.Trp960Arg) mutation in family 3 (Table 2). Additionally, known and unknown natural variants were found in RP1L1, as shown in Table S2. Unknown SNPs were submitted to the dbSNP database.
Figure 2.
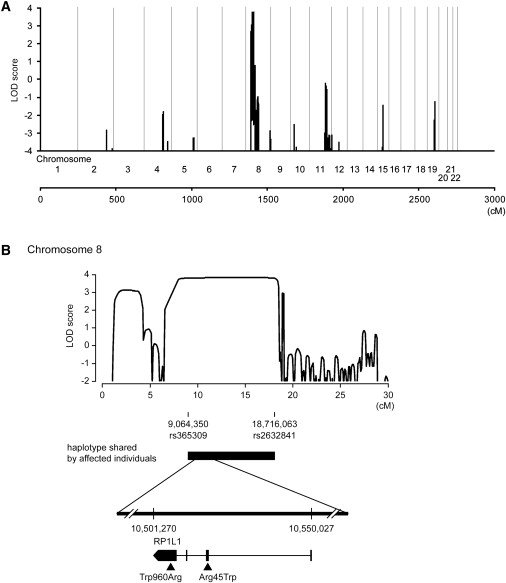
Linkage Analysis and Haplotype Analysis of Family 1
(A) Parametric multipoint linkage analysis of family 1. Horizontal axis indicates cumulative position (cM) from the short arm of chromosome 1. As a result of computational capacity, family 1 was divided into two branches for calculation of LOD scores. No other chromosomes except chromosome 8 yielded a positive LOD score.
(B) Parametric multipoint linkage analysis of family 1 and mutations in RP1L1. A maximum LOD score of 3.77 was obtained at 8p32.1-8p22. A haplotype bounded by rs365309 (physical position: 9,064,350 in the hg18 assembly of the UCSC Genome Browser) and rs2632841 (18,716,063) was shared by all affected individuals. Horizontal axis indicates the position (cM) on the short arm of chromosome 8. Vertical axis indicates the parametric multipoint LOD score. Mutations (p.Arg45Trp and p.Trp960Arg) are demonstrated.
Figure 3.
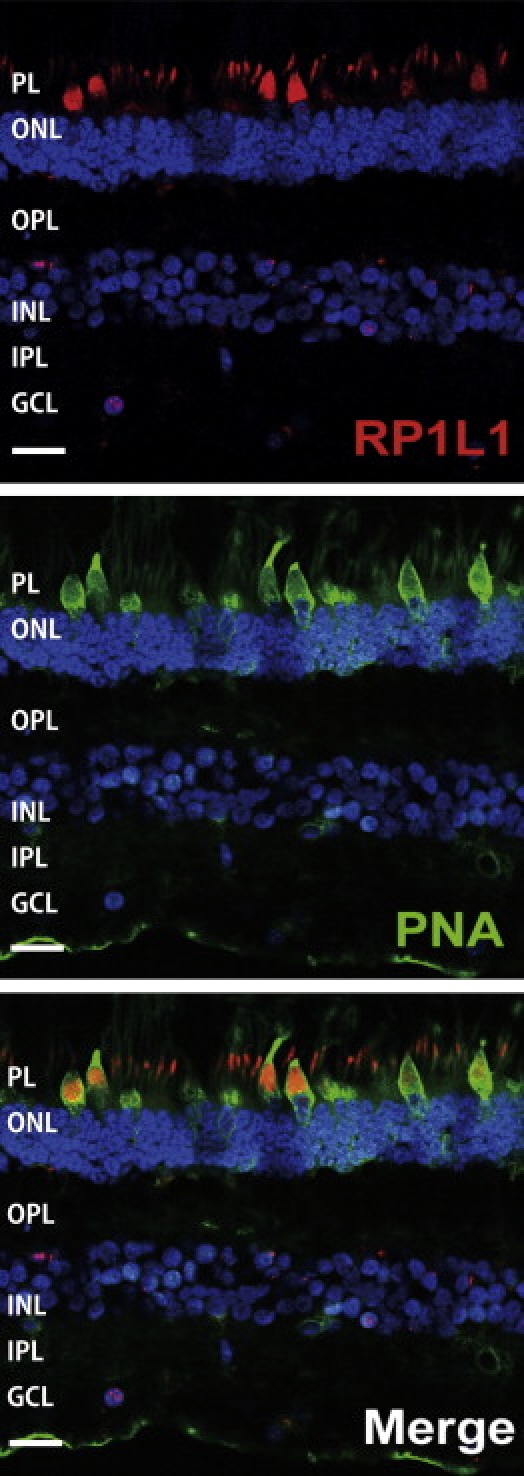
Immunohistochemistry of RP1L1 in the Cynomolgus Monkey Retina
Localization of RP1L1 in the rod and cone photoreceptors in the Cynomolgus monkey (Macaca fascicularis). Retina labeled with anti-human RP1L1 (red, top); same section labeled with retinal cone specific marker, peanut agglutinin lectin (PNA, green, middle); merged image (bottom). Yellow signal present in cone photoreceptor resulted from combination of the red signal of RP1L1 and the green signal of PNA. Cell nuclei were stained with DAPI (blue). PL, photoreceptor layer; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer, Scale bars represent 20 μm.
Table 1.
Disease-Linked Haplotypes in Families 1 and 2
|
Family 1 |
Family 2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|
|
Branch 1-1 |
Branch 1-2 |
||||||||
| Probe Set ID | dbSNP rs ID | Position | III-8 | IV-9 | IV-13 | IV-18 | IV-21 | II-2 | III-2 |
| SNP_A-8338925 | rs365309 | 9,064,350 | (A) | (A) | A | A | B | A | A |
| SNP_A-8281994 | rs1530483 | 9,065,671 | B | B | B | B | B | B | B |
| SNP_A-8360926 | rs10086673 | 10,342,727 | B | B | (B) | B | B | (A) | A |
| SNP_A-2082488 | rs9329223 | 10,369,164 | A | A | A | A | A | (B) | B |
| SNP_A-2013182 | rs6601491 | 10,453,427 | B | B | B | B | B | (A) | A |
| RP1L1 p.Arg45Trp | c.133C>T | 10,517,989 | T | T | T | T | T | T | T |
| SNP_A-8345504 | rs10097570 | 10,586,268 | A | A | A | A | (A) | (B) | B |
| SNP_A-1790165 | rs10111051 | 10,590,882 | A | A | A | A | (A) | (B) | B |
| SNP_A-8587750 | rs2163379 | 10,769,460 | A | A | A | (A) | A | (B) | B |
| SNP_A-8500791 | rs7460507 | 11,006,485 | B | B | B | B | B | A | A |
| SNP_A-8525908 | rs9772321 | 12,536,010 | A | A | A | A | A | A | A |
| SNP_A-8283296 | rs1021087 | 13,500,502 | A | (A) | (A) | A | A | B | B |
| SNP_A-8441723 | rs6987209 | 14,501,302 | B | B | B | B | B | B | B |
| SNP_A-2044287 | rs7818067 | 15,580,087 | A | A | A | A | A | A | A |
| SNP_A-4273924 | rs6992112 | 16,689,526 | A | A | A | A | A | A | (A) |
| SNP_A-8447659 | rs471041 | 17,707,836 | B | B | B | B | B | B | B |
| SNP_A-8399664 | rs2638658 | 18,713,620 | A | (A) | (A) | A | A | (B) | B |
| SNP_A-4233785 | rs2632841 | 18,716,063 | B | B | (B) | B | A | B | B |
Disease-linked haplotypes of the two patients (IV-9 and III-8) who are descendants from II-1 (branch 1-1 of family 1), the three patients (IV-13, IV-18, and IV-21) who are descendants from II-7 (branch 1-2 of family 1), and the two patients (II-2 and III-2) from family 2 are shown. Haplotypes are unequivocally determined, except those with brackets that are inferred to minimize the number of recombination events. Disease-linked haplotypes of the two branches of family 1 are the same, confirming that all affected individuals in family 1 share the same haplotype. Recombination events in the family was observed at rs365309 (telomeric boundary) and at rs2632841 (centromereic boundary). When disease haplotypes are compared between families 1 and 2, who share the p.Arg45Trp mutation in RP1L1 in common, disease-linked haplotypes flanking the RP1L1 locus are different between these families, suggesting that the p.Arg45Trp mutation originated independently.
Table 2.
Summary of RP1L1 Mutations in Families with OMD
| ID in Pedigree | Clinical Stage | Sex | Age at Diagnosis | Age at Onset in Estimation | Mutation | Best Corrected Visual Acuity (Right / Left) |
|---|---|---|---|---|---|---|
| 1 III3 | affected | F | 81 | 50 | c.362C>T | 1.2 / 0.1 |
| 1 III4 | affected | F | 71 | 25 | c.362C>T | 0.4 / 0.5 |
| 1 III5 | affected | M | 74 | 30 | c.362C>T | 0.2 / 0.3 |
| 1 III8 | affected | M | 82 | 20 | c.362C>T | 0.2 / 0.2 |
| 1 IV1 | unaffected | F | 60 | - | c.362C>T | 1.2 / 1.2 |
| 1 IV9 | affected | F | 49 | unknown | c.362C>T | 1.2 / 1.2 |
| 1 IV12 | affected | F | 69 | 50 | c.362C>T | 0.1 / 0.07 |
| 1 IV13 | affected | M | 70 | 20 | c.362C>T | 0.1 / 0.1 |
| 1 IV14 | affected | M | 66 | 30 | c.362C>T | 0.2 / 0.3 |
| 1 IV18 | affected | F | 58 | 12 | c.362C>T | 0.1 / 0.1 |
| 1 IV21 | affected | F | 58 | 47 | c.362C>T | 0.1 / 0.4 |
| 1 V2 | affected | M | 20 | 13 | c.362C>T | 0.3 / 0.3 |
| 1 V3 | affected | F | 19 | 6 | c.362C>T | 0.2 / 0.15 |
| 2 II2 | affected | M | 69 | unknown | c.362C>T | 0.2 / 0.2 |
| 2 II4 | unaffected | M | 58 | - | c.362C>T | 1.0 / 1.0 |
| 2 II5 | unaffected | M | 55 | - | c.362C>T | 1.0 / 1.0 |
| 2 II8 | affected | M | 52 | unknown | c.362C>T | 0.2 / 0.3 |
| 2 III1 | affected | M | 23 | 23 | c.362C>T | 0.2 / 0.3 |
| 2 III2 | affected | M | 20 | 20 | c.362C>T | 0.3 /0.3 |
| 3 II1 | affected | F | 29 | 12 | c.3107T>C | 0.2 / 0.2 |
| 3 II3 | affected | M | 19 | 13 | c.3107T>C | 0.2 / 0.3 |
| 4 III3 | affected | F | 52 | 30 | c.362C>T | 0.15 / 0.15 |
Summary of individuals from autosomal OMD families 1–4, in whom p.Arg45Trp or p.Trp960Arg mutations of RP1L1 were found. Three unaffected individuals at the age of 55–60 were found with the mutation. These individuals suggest a reduced penetrance of the mutation or a possible onset at a later age.
In these four families, all of the affected individuals carried one of the two mutations identified in this study, c.362C>T or c.3107T>C. We identified three apparently unaffected individuals carrying the p.Arg45Trp mutation, which suggest a reduced penetrance of the mutation or possibility a later onset of the disease for these individuals. Both mutations were absent in 1752 Japanese control chromosomes.
Immunohistochemistry of RP1L1 in the macula section of primate Cynomolgus monkeys (Macaca fascicularis) was performed. The eyes from a 6-yr-old normal male cynomolgus monkey were obtained from Tsukuba Primate Research Center, National Institute of Biomedical Innovation, Japan. All experimental procedures were approved by the Animal Welfare and Animal Care Committee of the National Institute of Biomedical Innovation, in compliance with guidelines of the Association for Research in Vision and Ophthalmology. Cynomolgus eyes were removed and immediately fixed overnight with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. After washing in PBS, eyes were cryoprotected in the gradient sucrose dissolved in PBS and embedded into optimal cutting temperature (OCT) compound (Tissue Tek, Miles, IL, USA). Frozen retinal sections cut at 8 μm thickness with cryostat were incubated at 4°C with a 1:500 dilution of human RP1L1 polyclonal antibody raised against the N terminus of human RP1L1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Immunofluorescence was visualized with Alexa 568 goat anti-rabbit IgG (Invitrogen, Carlsbad, CA, USA), Alexa 488 PNA (Invitrogen) for detection of cone photoreceptor, and DAPI (Invitrogen) for nuclear staining. Fluorescence images were analyzed with a confocal laser microscope (Radiance 2000, Bio-Rad Laboratories, Hercules, CA, USA).
To our surprise, the immunohistochemistry of RP1L1 in the macula section of Cynomolgus monkeys revealed expression in retinal rod and cone photoreceptors by human RP1L1 antibody (Figure 3). This expression pattern is significantly different from the previous study of mouse RP1L1, in which RP1L1 was localized exclusively in axoneme of rods.8 Furthermore, the human amino acid sequence is only 39% identical to that of the mouse, due to a lack of both polymorphic 16 amino acid repeats or a lack of the highly repetitive Glu-rich region, making mouse RP1L1 protein considerably shorter than the human protein, which may lead to different functional roles in the primate retina. Recent investigation of photoreceptor structure in OMD patients using advanced optical coherence tomography suggests that the predominant defect involves the cone photoreceptor.9,10 Our optical coherence tomography observations also show loss of the cone outer segment tip and irregularity of the inner segment/outer segment junction in the center of the macula of all examined case individuals in family 1 (data not shown). Y.M. et al. have observed that not only cone but also rod sensitivity in the macula was abnormal in some of the older patients.1,2 It is likely that the initial event may be macular cone specific but may later extend to rod abnormality. Further investigation of RP1L1 function is required in order to answer these clinical observations.
RP1L1 was originally cloned as a gene derived from common ancestor as retinitis pigmentosa 1 (RP1 [MIM 180100]) on the same chromosome 8.11,12 RP1L1 shares 35% amino acid identity with RP1, a gene responsible for 5%–10% of autosomal-dominant retinitis pigmentosa (RP [MIM 268000]) worldwide.13–15 When RP1L1 was first identified, a number of attempts were made to identify mutations in RP1L1 in various RP patients, with no success. The present study demonstrates that RP1L1 mutation is responsible for OMD, but not for RP. Patients with RP carrying the most common RP1 alteration, p.Arg677X, exhibit night and peripheral vision disturbance beginning in the third decade of life. RP1 is found exclusively in the retina and is localized to both rods and cones. Rod-cone functional comparison in RP patients has indicated that rod sensitivity loss is at least 2 log units greater than cone sensitivity loss.13 Thus phenotypic characteristics of RP caused by RP1 mutations and those of OMD caused by RP1L1 mutations perfectly agree with the different localizations of RP1 and RP1L1 in retina.
The outer segments of rod and cone photoreceptors are highly specialized cilia containing hundreds of disc membranes stacked in an orderly array along the photoreceptor axoneme. Previous studies have shown that RP1 is part of the axoneme and is required for this correct orientation and higher-order stacking of outer segment discs.16 This is achieved by the interaction of RP1 with the microtubule in the connecting cilia.17 RP1 contains microtubule-binding domains (amino acids 28–228) of neuronal microtubule-associated protein (MAP) doublecortin (DCX), which is required to maintain axoneme length and stability.18 The RP1L1 p.Arg45Trp alteration resides in one of the two DCX domains (amino acids 33–113 and 147–228), which is required for interaction with RP1 to assemble and stabilize axonemal microtubules.8 In primates, both RP1L1 and RP1 proteins may cooperatively function in the rod and cone photoreceptors to perform this task. The mutation in RP1L1 is likely to dominantly affect the cooperative function with RP1 in rod and cone photoreceptors, given that in a previous publication, the RP1L1 heterozygous knockout mice were reported to be normal whereas homozygous knockout mice were reported to develop subtle retinal degeneration. Our findings in OMD may shed light for further investigation of patients with cone dystrophy.
In conclusion, we identified RP1L1 mutations that cause autosomal-dominant OMD, and furthermore, our findings revealed that RP1L1 plays essential roles in the cone functions in human and that disruption of RP1L1 function leads to OMD.
Acknowledgments
This research was supported in part by grants to Takeshi Iwata and Kazushige Tsunoda by the Ministry of Health, Labour, and Welfare of Japan. This work was also supported in part to Shoji Tsuji by KAKENHI (Grant-in-Aid for Scientific Research) on Priority Areas, Applied Genomics, the Global COE Program, and Scientific Research (A) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
SNP HiTLink software, http://www.dynacom.co.jp/u-tokyo.ac.jp/snphitlink/
Accession Numbers
The dbSNP accession numbers for the SNPs reported in this paper are ss252841181 and ss252841182.
References
- 1.Miyake Y., Ichikawa K., Shiose Y., Kawase Y. Hereditary macular dystrophy without visible fundus abnormality. Am. J. Ophthalmol. 1989;108:292–299. doi: 10.1016/0002-9394(89)90120-7. [DOI] [PubMed] [Google Scholar]
- 2.Miyake Y., Horiguchi M., Tomita N., Kondo M., Tanikawa A., Takahashi H., Suzuki S., Terasaki H. Occult macular dystrophy. Am. J. Ophthalmol. 1996;122:644–653. doi: 10.1016/s0002-9394(14)70482-9. [DOI] [PubMed] [Google Scholar]
- 3.Wildberger H., Niemeyer G., Junghardt A. Multifocal electroretinogram (mfERG) in a family with occult macular dystrophy (OMD) Klin. Monatsbl. Augenheilkd. 2003;220:111–115. doi: 10.1055/s-2003-38161. [DOI] [PubMed] [Google Scholar]
- 4.Piao C.H., Kondo M., Tanikawa A., Terasaki H., Miyake Y. Multifocal electroretinogram in occult macular dystrophy. Invest. Ophthalmol. Vis. Sci. 2000;41:513–517. [PubMed] [Google Scholar]
- 5.Brockhurst R.J., Sandberg M.A. Optical coherence tomography findings in occult macular dystrophy. Am. J. Ophthalmol. 2007;143:516–518. doi: 10.1016/j.ajo.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 6.Fukuda Y., Nakahara Y., Date H., Takahashi Y., Goto J., Miyashita A., Kuwano R., Adachi H., Nakamura E., Tsuji S. SNP HiTLink: a high-throughput linkage analysis system employing dense SNP data. BMC Bioinfomatics. 2009;10:121. doi: 10.1186/1471-2105-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gudbjartsson D.F., Thorvaldsson T., Kong A., Gunnarsson G., Ingolfsdottir A. Allegro version 2. Nat. Genet. 2005;37:1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 8.Yamashita T., Liu J., Gao J., LeNoue S., Wang C., Kaminoh J., Bowne S.J., Sullivan L.S., Daiger S.P., Zhang K. Essential and synergistic roles of RP1 and RP1L1 in rod photoreceptor axoneme and retinitis pigmentosa. J. Neurosci. 2009;29:9748–9760. doi: 10.1523/JNEUROSCI.5854-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park S.J., Woo S.J., Park K.H., Hwang J.M., Chung H. Morphologic photoreceptor abnormality in occult macular dystrophy on spectral-domain optical coherence tomography. Invest. Ophthalmol. Vis. Sci. 2010;51:3673–3679. doi: 10.1167/iovs.09-4169. [DOI] [PubMed] [Google Scholar]
- 10.Sisk R.A., Berrocal A.M., Lam B.L. Loss of foveal cone photoreceptor outer segments in occult macular dystrophy. Ophthalmic Surg Lasers Imaging. 2010;41:1–3. doi: 10.3928/15428877-20100215-49. [DOI] [PubMed] [Google Scholar]
- 11.Conte I., Lestingi M., den Hollander A., Alfano G., Ziviello C., Pugliese M., Circolo D., Caccioppoli C., Ciccodicola A., Banfi S. Identification and characterization of the retinitis pigmentosa 1-like1 gene (RP1L1): a novel candidate for retinal degenerations. Eur. J. Hum. Genet. 2003;11:155–162. doi: 10.1038/sj.ejhg.5200942. [DOI] [PubMed] [Google Scholar]
- 12.Bowne S.J., Daiger S.P., Malone K.A., Heckenlively J.R., Kennan A., Humphries P., Hughbanks-Wheaton D., Birch D.G., Liu Q., Pierce E.A. Characterization of RP1L1, a highly polymorphic paralog of the retinitis pigmentosa 1 (RP1) gene. Mol. Vis. 2003;9:129–137. [PMC free article] [PubMed] [Google Scholar]
- 13.Pierce E.A., Quinn T., Meehan T., McGee T.L., Berson E.L., Dryja T.P. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat. Genet. 1999;22:248–254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- 14.Sullivan L.S., Heckenlively J.R., Bowne S.J., Zuo J., Hide W.A., Gal A., Denton M., Inglehearn C.F., Blanton S.H., Daiger S.P. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat. Genet. 1999;22:255–259. doi: 10.1038/10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobson S.G., Cideciyan A.V., Iannaccone A., Weleber R.G., Fishman G.A., Maguire A.M., Affatigato L.M., Bennett J., Pierce E.A., Danciger M. Disease expression of RP1 mutations causing autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2000;41:1898–1908. [PubMed] [Google Scholar]
- 16.Liu Q., Lyubarsky A., Skalet J.H., Pugh E.N., Jr., Pierce E.A. RP1 is required for the correct stacking of outer segment discs. Invest. Ophthalmol. Vis. Sci. 2003;44:4171–4183. doi: 10.1167/iovs.03-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Q., Zuo J., Pierce E.A. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J. Neurosci. 2004;24:6427–6436. doi: 10.1523/JNEUROSCI.1335-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gleeson J.G., Allen K.M., Fox J.W., Lamperti E.D., Berkovic S., Scheffer I., Cooper E.C., Dobyns W.B., Minnerath S.R., Ross M.E., Walsh C.A. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.