

Abstract
We characterized an autosomal-recessive syndrome of focal epilepsy, dysarthria, and mild to moderate intellectual disability in a consanguineous Arab-Israeli family associated with subtle cortical thickening. We used multipoint linkage analysis to map the causative mutation to a 3.2 Mb interval within 16p13.3 with a LOD score of 3.86. The linked interval contained 160 genes, many of which were considered to be plausible candidates to harbor the disease-causing mutation. To interrogate the interval in an efficient and unbiased manner, we used targeted sequence enrichment and massively parallel sequencing. By prioritizing unique variants that affected protein translation, a pathogenic mutation was identified in TBC1D24 (p.F251L), a gene of unknown function. It is a member of a large gene family encoding TBC domain proteins with predicted function as Rab GTPase activators. We show that TBC1D24 is expressed early in mouse brain and that TBC1D24 protein is a potent modulator of primary axonal arborization and specification in neuronal cells, consistent with the phenotypic abnormality described.
Main Text
The epilepsies and intellectual disabilities (ID) are clinically heterogeneous groups of disorders each affecting about 3% of the population at some time in life. Intellectual disability presents in association with seizures in around 21% of cases.1 The degree of comorbidity between epilepsy and ID strongly suggests an overlap of underlying genetic determinants. Traditionally, mutation identification in epilepsy and ID has been driven by positional and functional gene candidate approaches. Although mutations in ion channel subunits have emerged as the most significant cause of idiopathic epilepsies,2 no pattern for enrichment of mutations in genes of a particular function has emerged for ID or epilepsy associated with ID. For such situations, genome-wide copy number profiling,3 systematic resequencing,4 and, most recently, targeted sequence enrichment and next-generation sequencing5–7 are proving to be the most effective approaches for discovering the disease-associated variation.
We examined a large Arab family from northern Israel with epilepsy and ID (Figure 1A). Focal seizures with prominent eye blinking and facial and limb jerking began at around 2 months of age and persisted throughout life. The patients described an aura with the sensation of the tongue being anaesthetized. Convulsive seizures also occurred but were generally controlled by antiepileptic medication. Early motor and speech development was mildly delayed in some children, and in adult life there was borderline to moderate ID associated with mild dysarthria and ataxia. MRI showed features that we interpreted as abnormal cortical thickening, most obvious in the anteromesial frontal areas (Figure 1B).
Figure 1.
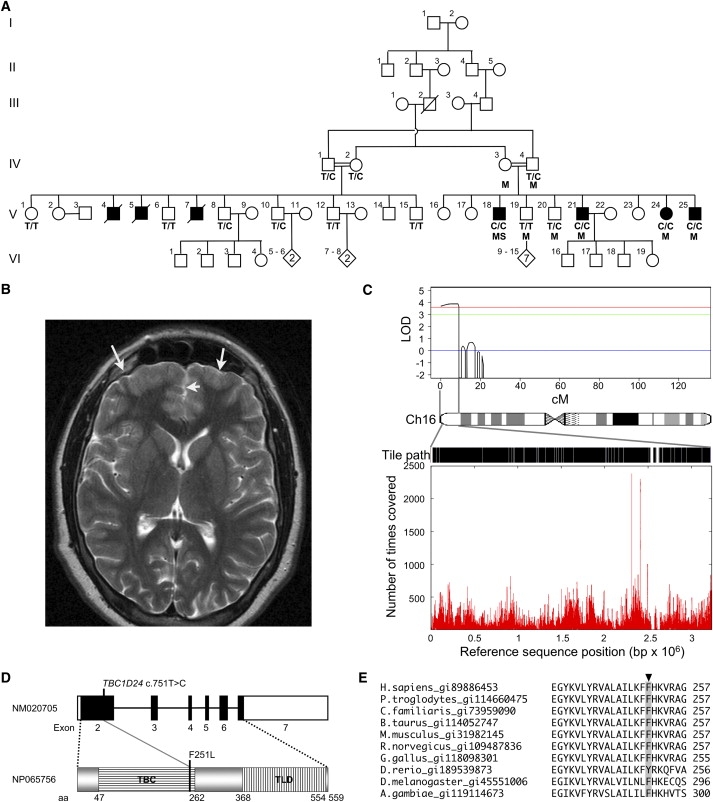
Mapping and Identification of a Mutation in TBC1D24
(A) Pedigree showing evidence of likely autosomal-recessive inheritance and consanguinity. Individuals used in homozygosity mapping are marked with M, the individual sequenced by next-generation sequencing is marked with S, and C/C, C/T, and T/T indicate TBC1D24 c.751 alleles. Solid squares indicate affected individuals.
(B) Axial MRI image of V-25 that we interpreted as showing thickened cortex in the frontal poles with loss of gray-white matter definition, consistent with a developmental malformation (arrows).
(C) Work flow for the identification of the mutation in TBC1D24: linkage homozygous-by-descent analysis (top) identified a single peak on chromosome 16p13.3 covering 9cM (or 3.2 Mb); the entire sequence, excluding repeats, was tiled as indicated by the black sections on the tile path; sequence coverage (bottom), determined by the number of times a base was covered by a sequence read, shows variation over the 3.2 Mb interval.
(D) A schematic representation of the exonic structure of TBC1D24 and the domain structure of the TBC1D24 protein. The position of the c.751C>T, p.F251L mutation within the TBC domain (translated from exon 2) is shown. Exons are indicated as boxes, with the translated region shown in black. The TBC and TLD domains are shown.
(E) A portion of a CLUSTALW multiple protein alignment of TBC1D24 orthologs shows that p.F251 (highlighted with gray background) is conserved in mammals, chicken, fruit fly, mosquito, and a highly similar tyrosine (Y) substitution in zebra fish. Orthologs of TBC1D24 were identified by tblastn search, and the Homologene database and alignments were performed with the EBI CLUSTALW server.19
The pedigree structure with multiple consanguineous unions suggested autosomal-recessive inheritance. For the purpose of linkage mapping, eight samples were genotyped (as indicated in Figure 1A) with the 250K Nsp Affymetrix chip. The study was approved by the Tel Aviv Sourasky Medical Center ethics committee. Informed consent was obtained from participating subjects. The genotype calls were produced with CRLMM,8 which is implemented in the R statistical programming language OLIGO package. A subset of 7406 SNPs with high heterozygosity (mean heterozygosity = 0.48) was created for the linkage mapping with the program Linkdatagen,9 setting the bin size to 0.5 cM. Genotyping errors were identified with Merlin.10,11 Multipoint linkage analysis was carried out with Merlin by using a rare autosomal-recessive model with the following parameters for the disease allele a and for normal allele A: Pr(a) = 0.0001, Pr(disease|aa) = 0.9999, Pr(disease|AA or Aa) = 0.0001, reflecting a fully penetrant model. That model identified a single candidate region encompassing 160 genes on chromosome 16, between rs1088638 and rs9922740, LOD = 3.86 (Figure 1C).
A custom 385K tiling microarray (Roche/Nimblegen) was constructed with genomic DNA sequence corresponding to the entire linked interval, excluding regions masked by the Sequence Search and Alignment by Hashing Algorithm12 (2.3 out of 3.2 Mb). This was used as a probe for enrichment of the linkage interval from genomic DNA from individuals V-18 and V-21 (Figure 1A; Nimblegen).5 Genomic DNA fragments were retrieved from individual V-18 with an estimated mean 180-fold enrichment. These DNA fragments were concatenated by ligation and sheared by sonication to be converted to an Illumina GAII-compatible library6 (GeneWorks). A single lane from the Illumina GAII returned 8.8 × 106 65 bp reads (GeneWorks). Using default parameters of the Phred/Phrap/Consed program (version 19.0),13 we found that 2.7 × 106 (31%) of the reads assembled to a repeat masked segment of the hg18 reference sequence corresponding to the linkage interval. Coverage to a depth of at least one sequence read incorporated 95.51% of bases from open reading frames (ORFs) of RefSeq genes included in the March 2006 (hg18) build of the UCSC Genome Browser. A minimum of 10-fold coverage was achieved for 76.92% of bases in all ORFs in the interval. Over the entire 3.2 Mb linkage interval, 89.93% of bases were covered with at least one sequence read (see Figure S1 available online). Coverage was biased against sequences with high GC content and repeats, as defined by Repeat Masker (p < 0.05 by χ2 analysis; Table S1).
We identified 1029 single nucleotide variants, at a minimum of 10-fold coverage, in which the variant base was represented in at least 85% of reads. Of these variants, 102 were not represented in dbSNP130. These novel variants were categorized as ORF, splice site, 5′UTR, 3′UTR, intronic, or intergenic based on their genomic context in relation to known genes (Table S2). Variants were prioritized for analysis according to the order listed above, focusing on mutations in ORF or splice sites that were predicted to affect translated sequences. This filtering removed all but six variants, and only three of these—one in TBC1D24 (c.751T>C, p.F251L [NM_020705]), another in the alternatively spliced C16orf13 (c.406C>T, p.L136P [NM_032366.3]), and a third in PRSS41 (c.856A>G, p.S286G [NM_001135086.1])—were predicted to result in amino acid changes (Table 1). Subsequent segregation analysis on an extended pedigree to that originally used for mapping led us to exclude the c.406C>T change in C16orf13, which was not present in the obligate carrier male IV-1 (Figure 1A). PRSS41 was shown to not be expressed in the brain and to therefore be unlikely to contribute to the phenotype (Figure S2). As a consequence, only a single missense change, TBC1D24 (c.751T>C, p.F251L) remained (Figure 1D). This change was not present in 210 control chromosomes from a matched Arab population.
Table 1.
Summary of Unique Variants in Open Reading Frames
| bp on Chr16a | Gene | RefSeq | DNA Variant | Predicted Protein Variant |
|---|---|---|---|---|
| 625,295 | C16orf13 | NM_032366.3 and NM_001040160.1 | c.406T>C and c.243T>C | p.(L136P) and p.(S82P) |
| 1,664,017 | CRAMP1L | NM_020825.3 | c.3780C>T | p.(G1260G) |
| 1,756,740 | MAPK8IP3 | NM_015133.3 | c.2952G>A | p.(S984S) |
| 1,809,941 | HAGH | NM_005326.4 | c.390C>T | p.(I130I) |
| 2,486,901 | TBC1D24 | NM_020705.1 | c.751T>C | p.(F251L) |
| 2,795,033 | PRSS41 | NM_001135086.1 | c.856A>G | p.(S286G) |
Base pair (bp) position is based on the UCSC Genome Browser hg18 (March 2006) reference sequence.
The following data support the proposition that the TBC1D24 (c.751T>C, p.F251L) variant is pathogenic. First, the phenylalanine (F) at position p.251 is conserved in mammals and, to a lesser extent, in other vertebrate and invertebrate species (Figure 1E). Second, we showed that TBC1D24 is expressed in mouse embryonic stem cell-derived neurons, cultured embryonic day 18.5 (E18.5) mouse hippocampal neurons, and the developing mouse brain (Figures S3A, S3B, and S3C, respectively). Collectively, these expression profiles correlate high expression of Tbc1d24 with terminal differentiation of neurons, which is consistent with a gene influencing cortical development, early-onset seizures, and ID. Third, to determine the currently unknown role of the TBC1D24 protein, we overexpressed the wild-type and mutant TBC1D24 proteins in mouse E18.5 primary hippocampal neurons. Cell morphology in neurons overexpressing the TBC1D24 protein with the p.F251L change did not differ noticeably from those transfected only with green fluorescent protein (GFP). However, the overexpression of the wild-type TBC1D24 protein significantly increased the length of primary axons, as defined by Tau1 staining and the number of neurite termini (p < 0.05 by Student's two-tailed t test; Figures 2A and 2B, respectively), at both 5 and 7 days posttransfection, demonstrating increased arborization (Figure 2D). Furthermore, we observed ectopic axon specification in cells overexpressing the wild-type protein (Figure 2C). Taken together, these cell culture data suggest that TBC1D24 protein is likely to have an important role in normal human brain development. Moreover, the absence of these effects in cells overexpressing the p.F251L mutant suggests that this missense change results in a loss of TBC1D24 protein function (Figure 2). These three lines of evidence demonstrate that the TBC1D24 c.751T>C, p.F251L change represents the most likely disease-causing mutation in this family.
Figure 2.
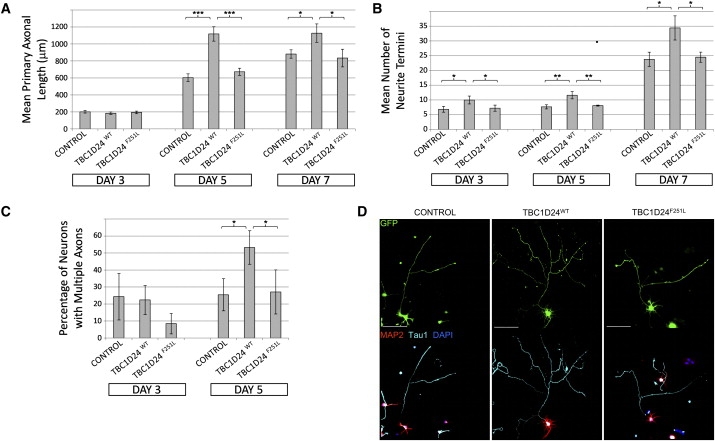
TBC1D24 Is a Potent Modulator of Neuronal Cell Morphology and Primary Axon Specification
For all experiments: ED18.5 hippocampal neurons were isolated via established methods20 and then transfected with the Amaxa Mouse Neuron Nucleofector kit (Lonza). Cells were transfected with 1 μg of the supplied pmaxGFP vector alone (control) or in addition to 2 μg of either a commercially available expression vector RC210346 (NM_020705; OriGene Technologies) for the human TBC1D24 ORF (TBC1D24WT) or the same RC210346 vector with the c.751T>C mutation introduced by site-directed mutagenesis (TBC1D24F251L). Following transfection, neurons were plated onto poly-D-lysine (Sigma)-coated coverslips at a density of 1.25 × 105 cells per well in Neurobasal media containing 2% (vol/vol) B27 (Invitrogen) and 10% fetal calf serum (FCS, Thermoscientific). Once cells were attached (∼4 hr), the FCS was removed and the cells were cultured for a further 3, 5, or 7 days, with half of the media changed every 4 days. Cells were fixed with 4% paraformaldehyde for 15 min at room temperature and then permeabilized, and nonspecific antigens were blocked with a solution of phosphate-buffered saline containing 1% Tween 20 (PBST) and 10% normal horse serum (Sigma). Primary antibodies were incubated overnight at 4°C at the following dilutions: chicken anti-MAP2 (1:2000, Chemicon, Millipore Bioscience Research Products) to identify dendrites or mouse anti-Tau1 (1:2000, Chemicon) to identify axons. Either donkey anti-mouse-Alexa647 (Invitrogen) or donkey anti-chicken-Cy3 (Chemicon) secondary antibodies were used at a dilution of 1:800. Nuclei were stained with DAPI as per the manufacturer's protocol (Invitrogen). Coverslips were mounted with Slowfade antifade mounting media (Invitrogen). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005 by Student's two-tailed t test, assuming equal variances.
(A) Overexpression of TBC1D24WT but not TBC1D24F251L promotes axonal growth in cultured hippocampal neurons. Graph depicts average length of primary axons ± standard deviation (SD) from triplicate experiments measured with the “measure cumulative distances” plugin for the ImageJ software package (National Institutes of Health), with at least 20 measurements made for each treatment.
(B) Overexpression of TBC1D24WT but not TBC1D24F251L promotes arborization of cultured hippocampal neurons. Graph depicts the average number of neurite termini per cell ± SD from triplicate experiments, with at least 10 neurons counted for each treatment.
(C) Overexpression of TBC1D24WT but not TBC1D24F251L promotes ectopic axonal specification in hippocampal neurons after 5 days' culture. Graph shows average percentage of transfected cells with multiple axons ± SD from triplicate experiments, with at least 10 neurons counted for each treatment.
(D) Example images of transfected neurons at 5 days of differentiation, showing either native GFP fluorescence or indirect immunofluorescent staining with Tau1, MAP2, and DAPI, as described. Scale bars represent 100 μM.
The TBC domain protein family members that have been characterized are Rab GTPase activators (GAPs) that catalyze GTP hydrolysis, switching their cognate GTPase from the active GTP-bound form to the inactive GDP-bound form. In TBC1D24, the N-terminal TBC subdomain lacks the crucial arginine and glutamine residues required for GAP activity;14 thus, TBC1D24 may have an alternate function in the cell, as has been described for TBC1D3.15 The Rab GTPases are known to control neuronal cell morphology and migration because their mutations have previously been demonstrated to be deleterious to normal brain development, such as in OPHN1.16 The TBC1D24 protein has two main domains, the TBC domain (residues 47–262) and the TLD domain (of unknown function; residues 368–554). TBC1D24 may produce up to three protein isoforms as a result of alternative splicing, and the p.F251L change is predicted to affect all of these.
Targeted sequence enrichment and next-generation sequencing are revolutionizing novel disease gene discovery. In this case, the technology has allowed us to take a direct, unbiased and highly effective approach from phenotype to mutation. The refined linkage interval also harbors two ion channel genes: CACNA1H (known to have susceptibility alleles for epilepsy [MIM 607904])17 and CLCN7 (MIM 602727), which would have been classed as plausible candidates over uncharacterized TBC1D24 via a traditional, positional candidate approach. In humans, 44 TBC domain-containing genes have been identified.18 Unmasking a defect in TBC1D24, causing epilepsy and ID, has implicated a member of this large, conserved family of genes in human disease.
Acknowledgments
We are grateful for the cooperation of the family involved in this study, as well as to Bev Johns for technical assistance and Rob King, Andre Rickers, and Graeme Woolford from GeneWorks for help with the resequencing. M.B. and J.G. were supported by the National Health and Medical Research Council (NH&MRC) with a Career Development Award and a Principal Research Fellowship, respectively. This project was supported by NH&MRC program grant 400121 and also by International Science Linkages, established under the Australian Government's innovation statement, “Backing Australia's Ability.”
Contributor Information
Samuel F. Berkovic, Email: samuelfb@unimelb.edu.au.
Jozef Gecz, Email: jozef.gecz@adelaide.edu.au.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
CLUSTALW at EBI, http://www.ebi.ac.uk/Tools/clustalw2/index.html
Homologene, http://www.ncbi.nlm.nih.gov/homologene/
Linkdatagen, http://bioinf.wehi.edu.au/software/linkdatagen/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
Primer Bank, http://pga.mgh.harvard.edu/primerbank/
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Airaksinen E.M., Matilainen R., Mononen T., Mustonen K., Partanen J., Jokela V., Halonen P. A population-based study on epilepsy in mentally retarded children. Epilepsia. 2000;41:1214–1220. doi: 10.1111/j.1528-1157.2000.tb00328.x. [DOI] [PubMed] [Google Scholar]
- 2.Helbig I., Scheffer I.E., Mulley J.C., Berkovic S.F. Navigating the channels and beyond: Unravelling the genetics of the epilepsies. Lancet Neurol. 2008;7:231–245. doi: 10.1016/S1474-4422(08)70039-5. [DOI] [PubMed] [Google Scholar]
- 3.Mefford H.C., Eichler E.E. Duplication hotspots, rare genomic disorders, and common disease. Curr. Opin. Genet. Dev. 2009;19:196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O'Meara S., Latimer C., Dicks E., Menzies A. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albert T.J., Molla M.N., Muzny D.M., Nazareth L., Wheeler D., Song X., Richmond T.A., Middle C.M., Rodesch M.J., Packard C.J. Direct selection of human genomic loci by microarray hybridization. Nat. Methods. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 6.Hodges E., Xuan Z., Balija V., Kramer M., Molla M.N., Smith S.W., Middle C.M., Rodesch M.J., Albert T.J., Hannon G.J., McCombie W.R. Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 2007;39:1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 7.Gnirke A., Melnikov A., Maguire J., Rogov P., LeProust E.M., Brockman W., Fennell T., Giannoukos G., Fisher S., Russ C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carvalho B., Bengtsson H., Speed T.P., Irizarry R.A. Exploration, normalization, and genotype calls of high-density oligonucleotide SNP array data. Biostatistics. 2007;8:485–499. doi: 10.1093/biostatistics/kxl042. [DOI] [PubMed] [Google Scholar]
- 9.Bahlo M., Bromhead C.J. Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics. 2009;25:1961–1962. doi: 10.1093/bioinformatics/btp313. [DOI] [PubMed] [Google Scholar]
- 10.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 11.Abecasis G.R., Wigginton J.E. Handling marker-marker linkage disequilibrium: Pedigree analysis with clustered markers. Am. J. Hum. Genet. 2005;77:754–767. doi: 10.1086/497345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ning Z., Cox A.J., Mullikin J.C. SSAHA: A fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon D. Viewing and editing assembled sequences using Consed. Curr. Protoc. Bioinformatics. 2003;Chapter 11 doi: 10.1002/0471250953.bi1102s02. Unit 11.2. [DOI] [PubMed] [Google Scholar]
- 14.Pan X., Eathiraj S., Munson M., Lambright D.G. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature. 2006;442:303–306. doi: 10.1038/nature04847. [DOI] [PubMed] [Google Scholar]
- 15.Frittoli E., Palamidessi A., Pizzigoni A., Lanzetti L., Garrè M., Troglio F., Troilo A., Fukuda M., Di Fiore P.P., Scita G., Confalonieri S. The primate-specific protein TBC1D3 is required for optimal macropinocytosis in a novel ARF6-dependent pathway. Mol. Biol. Cell. 2008;19:1304–1316. doi: 10.1091/mbc.E07-06-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Billuart P., Bienvenu T., Ronce N., des Portes V., Vinet M.C., Zemni R., Roest Crollius H., Carrié A., Fauchereau F., Cherry M. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature. 1998;392:923–926. doi: 10.1038/31940. [DOI] [PubMed] [Google Scholar]
- 17.Heron S.E., Khosravani H., Varela D., Bladen C., Williams T.C., Newman M.R., Scheffer I.E., Berkovic S.F., Mulley J.C., Zamponi G.W. Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann. Neurol. 2007;62:560–568. doi: 10.1002/ana.21169. [DOI] [PubMed] [Google Scholar]
- 18.Gao X., Jin C., Xue Y., Yao X. Computational analyses of TBC protein family in eukaryotes. Protein Pept. Lett. 2008;15:505–509. doi: 10.2174/092986608784567483. [DOI] [PubMed] [Google Scholar]
- 19.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaech S., Banker G. Culturing hippocampal neurons. Nat. Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.