

Abstract
Retinitis pigmentosa (RP) is a heterogeneous group of inherited retinal degenerations caused by mutations in at least 45 genes. Using homozygosity mapping, we identified a ∼4 Mb homozygous region on chromosome 2p15 in patients with autosomal-recessive RP (arRP). This region partially overlaps with RP28, a previously identified arRP locus. Sequence analysis of 12 candidate genes revealed three null mutations in FAM161A in 20 families. RT-PCR analysis in 21 human tissues revealed high levels of FAM161A expression in the retina and lower levels in the brain and testis. In the human retina, we identified two alternatively spliced transcripts with an intact open reading frame, the major one lacking a highly conserved exon. During mouse embryonic development, low levels of Fam161a transcripts were detected throughout the optic cup. After birth, Fam161a expression was elevated and confined to the photoreceptor layer. FAM161A encodes a protein of unknown function that is moderately conserved in mammals. Clinical manifestations of patients with FAM161A mutations varied but were largely within the spectrum associated with arRP. On funduscopy, pallor of the optic discs and attenuation of blood vessels were common, but bone-spicule-like pigmentation was often mild or lacking. Most patients had nonrecordable electroretinographic responses and constriction of visual fields upon diagnosis. Our data suggest a pivotal role for FAM161A in photoreceptors and reveal that FAM161A loss-of-function mutations are a major cause of arRP, accounting for ∼12% of arRP families in our cohort of patients from Israel and the Palestinian territories.
Main Text
Retinitis pigmentosa (RP [MIM 268000]) is the most common inherited retinal degeneration, with an estimated worldwide prevalence of 1:4000.1–3 The disease is highly heterogeneous and can be inherited as autosomal recessive (50%–60%), autosomal dominant (30%–40%), or X-linked (5%–15%).4 At present, 32 genetic loci have been implicated in nonsyndromic autosomal-recessive RP (arRP) (RetNet), most of which account for a few percent of RP cases each. In five of the 32 previously reported arRP loci (RP22, RP28, RP29, RP32, RP54), the causative gene is as yet unknown. Two unrelated Indian arRP families were linked to the RP28 locus on chromosome 2p14-p15.5,6 Recently, one of the genes in the linked interval, MDH1 (MIM ∗154200), was considered as a good candidate for RP because of its role in the Krebs cycle, but it was found to be negative for mutations via ultra high-throughput sequencing analysis in these two RP28-linked families.7
Homozygosity mapping using high-density SNP microarrays in consanguineous as well as nonconsanguineous families is currently the most efficient tool for identifying novel arRP genes.8–11 The Israeli and Palestinian populations that we serve are an efficient resource for the identification of disease-causing genes with the use of this approach because of the relatively high level of consanguinity, large number of siblings per family, and presence of subpopulations isolated over hundreds of years (The Israeli National Genetic Database).12–14
In the present study, we used homozygosity mapping to identify the cause of disease in families from Israel and the Palestinian territories with arRP. Patients were of various origins, including North African Jews, Ashkenazi Jews, Syrian Jews, and Arab Muslims. The tenets of the Declaration of Helsinki were followed, and informed consent was obtained from all patients who participated in this study, prior to donation of a blood sample. DNA was extracted with the FlexiGene DNA Kit (QIAGEN) from the index patient as well as from other affected and unaffected family members. Whole-genome SNP analysis was performed with the use of Affymetrix 250K microarrays. The criterion for hybridization quality was set to p < 0.05, and a region of homozygosity was determined by a minimal number of 100 consecutive homozygous markers. A relatively large number of patients with arRP, mainly from consanguineous families, had large homozygous regions on chromosome 2p. In 14 patients who belong to nine unrelated families of North African Jewish origin, we identified the most common haplotype (haplotype A; Figure 1A). In addition, four patients from two Syrian Jewish families shared a less common haplotype, haplotype B (Figure 1A). The two haplotypes share a homozygous region of ∼4 Mb (60.67–64.51 Mb) that overlaps with the RP28 locus reported in the two families of Indian origin.5,6 The shared homozygous region contains 22 annotated genes (Figure 1B), none of which is expressed exclusively in the retina. Using the Primer3 software,15 we designed primers flanking all coding exons and exon-intron boundaries of 12 genes (B3GNT2 [MIM ∗605581], CCT4 [MIM ∗605142], COMMD1 [MIM ∗607238], C2ORF86, EHBP1 [MIM ∗609922], FAM161A, MDH1, OTX1 [MIM ∗600036], PELI1, TMEM17, UGP2 [MIM ∗191760], VSP54) located within the linked region that is overlapping with RP28 and performed sequence analysis in two patients representing the two haplotypes. The analysis revealed two homozygous null mutations in the FAM161A gene (Figures 1C and 1D, Figure S1, Table 1, Table S1): c.1355_6delCA (p.Thr452SerfsX3) in patient MOL0766 V:2 (haplotype A) and c.1567C>T (p.Arg523X) in patient MOL0276 II:2 (haplotype B). Sequence analysis of FAM161A in 12 additional patients with a homozygous region on chromosome 2p revealed one patient, MOL0777 II:2 (from a consanguineous Arab Muslim family; Figure S1), with a large homozygous haplotype (haplotype C; Figure 1A) covering > 50 Mb, who was homozygous for a null mutation in exon 5, c.1786C>T (p.Arg596X) (Figures 1C and 1D, Table 1). The three mutations that we identified are expected to produce an abnormal transcript that is likely to be recognized by the nonsense-mediated mRNA decay system, resulting in the absence of FAM161A protein. Less likely, the mutations might produce an abnormal protein lacking a considerable portion of the normal protein. In any case, these mutations cause loss of function in a haplosufficiency mechanism.
Figure 1.
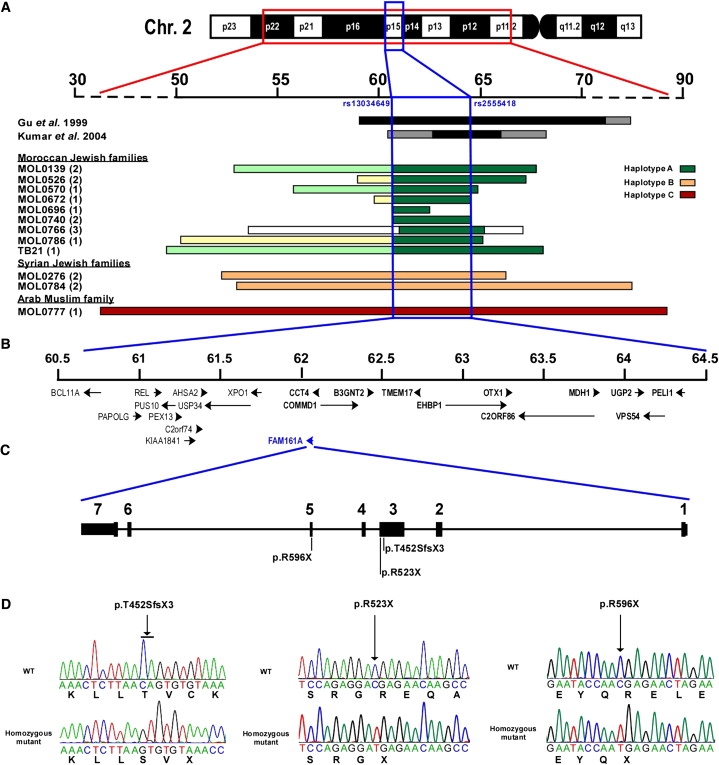
Autozygosity Mapping Results, Chromosomal Region, Gene Structure, and Mutations Identified in FAM161A
(A) The chromosomal region harboring the homozygous haplotypes at chromosome 2 (upper panel). Rulers are based on the February 2009 USCS Genome Browser build (hg19). The homozygous regions in the two previously reported RP28 Indian families are depicted as black (homozygous regions) or gray (cosegregating heterozygous regions) bars. The lower set of horizontal bars represents the homozygous region in our set of patients, as identified by whole-genome SNP analysis, depicted for each of the studied families. The family origin, family number, and number of analyzed patients per family (in parentheses) are shown on the left. Each of the three core haplotypes (A, B, C) is color-coded, with flanking homozygous regions marked by various colors indicating a deviation from the common haplotype. The region flanked by SNP markers rs13034649 and ra2555418 covering ∼4 Mb between 60.67 and 64.51 Mb was considered the shared homozygous region. A smaller homozygous region that was identified in family MOL0696 was not used to define the shared homozygous region because of its relatively small size (∼1.8 Mb; 60.67–62.44 Mb).
(B) The genes located within the shared homozygous region are ordered on the basis of their genomic location, with an arrow indicating gene orientation. Twelve of the 22 genes were screened for mutations and marked in bold. The FAM161A gene is highlighted in blue.
(C) Intron-exon structure of FAM161A and location of the null mutations. All seven exons are coding exons. Exon 2 contains an in-frame ATG codon that might be used for initiation of translation. Exon 4 is alternatively spliced (see Figure 3 for more details).
(D) FAM161A mutations identified in patients with arRP. The chromatograms of a wild-type and a homozygous mutant individual are depicted for each of the three mutations. The nucleotide change is shown below the chromatogram, and the amino acid is shown below the second base of each codon. An arrow indicates the mutation location.
Table 1.
RP Families with FAM161A Mutations
| Family | No. of Affected Individuals | Level of Consanguinitya | Origin | Size and Range (Mb) of Homozygous Region | Mutation in FAM161A (Effect on Protein) |
|---|---|---|---|---|---|
| MOL0053 | 4 | none | Libyan Jew | na | p. Arg523X p. Arg523X |
| MOL0100 | 2 | none | Moroccan Jew | na | p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0139 | 2 | 2:1 | Moroccan Jew | 14.90 (52.91–67.81) |
p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0195 | 3 | none | Ashkenazi Jew (M)b, Tunisian Jew (P)b | na | p.Thr452SerfsX3 (M)b p. Arg523X (P)b |
| MOL0276 | 3 | 3:3 | Syrian Jew | 14.01 (52.32–66.33) |
p. Arg523X p. Arg523X |
| MOL0284 | 1 | none | Ashkenazi Jew | na | p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0286 | 1 | none | Moroccan Jew | na | p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0303 | 1 | none | Bulgarian Jew | na | p.Thr452SerfsX3 p. Arg523X |
| MOL0352 | 1 | none | Libyan Jew | na | p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0446 | 1 | none | Libyan and Moroccan Jew | na | p.Thr452SerfsX3 p. Arg523X |
| MOL0526 | 2 | none | Moroccan Jew | 8.00 (59.24–67.24) |
p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0570 | 1 | none | Moroccan Jew | 11.61 (55.88–67.49) |
p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0672 | 1 | none | Moroccan Jew | na | p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0696 | 2 | none | Moroccan Jew | 1.77 (60.67–62.44) |
p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0740 | 2 | none | Moroccan Jew | 6.80 (60.72–67.52) |
p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0766 | 4 | 2:3 | Moroccan Jew | 13.53 (53.71–67.24) |
p.Thr452SerfsX3 p.Thr452SerfsX3 |
| MOL0777 | 2 | 2:2 | Arab Muslim | 52.82 (34.47–87.29) |
p. Arg596X p. Arg596X |
| MOL0784 | 2 | 2:2 | Syrian Jew | 27.13 (52.85–79.98) |
p. Arg523X p. Arg523X |
| MOL0786 | 1 | none | Moroccan Jew | 5.93 (59.18–65.11) |
p.Thr452SerfsX3 p.Thr452SerfsX3 |
| TB21 | 4 | 2:1 | Moroccan Jew | 19.07 (49.26–68.33) |
p.Thr452SerfsX3 p.Thr452SerfsX3 |
The table includes only families in which mutations were found on both alleles. Excluded from the table are two patients who were heterozygous for either the c.1355_6delCA or the c.1567C>T mutation with no identifiable mutation on the counter allele. na, not analyzed.
Level of consanguinity is measured by the number of generations separating the spouse from the common ancestor (e.g., 2:2 designates first cousins, 2:1 designates marriage between an uncle and his niece).
M, maternal; P, paternal.
To assess the frequency of the identified FAM161A mutations in the Israeli and Palestinian populations, we screened a set of 206 index patients with inherited retinal degeneration (mainly with arRP) for the three null mutations by restriction analysis (Table S2) or by sequencing of the corresponding exons on the basis of the patient's origin. This analysis revealed patients from 12 additional families who were homozygous for the c.1355_6delCA mutation and patients from two families with a homozygous c.1567C>T mutation (Table 1 and Figure S1). In addition, we identified patients from three families who were compound heterozygous for c.1355_6delCA and c.1567C>T (Table 1 and Figure S1). On the basis of haplotype data, these two mutations (c.1355_6delCA and c.1567C>T) are founder mutations in the Israeli Jewish population. Whereas such founder mutations in this population are usually restricted to a specific ethnic group (e.g., the USH3A [MIM ∗606397] p.N48K mutation in Ashkenazi Jews, or the CERKL [MIM ∗608381] c.238+1G>A mutation among patients of Jewish Yemenite origin),16,17 it is interesting to note that both FAM161A mutations were identified in three different ethnic groups (Table 1). Although the c.1355_6delCA mutation was identified mainly in families of North African Jewish origin, an Ashkenazi Jewish origin was reported in two of the families and a Bulgarian Jewish origin in one family. The c.1567C>T mutation was identified in two Syrian Jewish families, three North African Jewish families, and one family of Bulgarian Jewish origin. The occurrence of a mutation in different Jewish subpopulations, from different diasporas, may indicate a relatively ancient origin of these mutations. To assess the carrier frequency of each identified mutation, we screened ethnically matched healthy controls for each of the mutations. The p.Thr452SerfsX3 mutation was identified heterozygously in four out of 127 controls of North African Jewish ancestry (an estimated carrier frequency of 1:32), and the mutation was not identified in 108 Ashkenazi Jewish controls. The p.Arg523X mutation was not identified in 108 North African Jewish controls, and the p.Arg596X mutation was not identified in 105 Arab Muslim controls.
In total, FAM161A mutations were identified in 20 (out of 172; 11.6%) arRP families (including 41 patients) in our cohort, and they perfectly cosegregated with the disease (Table 1 and Figure S1). Until now, founder mutations in two arRP genes were found to be a relatively frequent cause of disease in the Israeli population: EYS (MIM ∗612424, responsible for 5.8% of cases)18 and CERKL (responsible for 4.7%; D.S. and T.B.Y. unpublished data and ref. 16). The data that we present here suggest that FAM161A mutations are currently the most common cause of RP in our set of patients, representing the Israeli and Palestinian populations (Figure S2). Moreover, the location of FAM161A within the RP28 locus might indicate a more general involvement of this gene as the cause of arRP in other populations as well. Screening FAM161A in other populations will indicate what percentage of arRP is caused by this gene worldwide. Although the first nonsyndromic retinal degeneration gene was identified about 20 years ago,19 it is interesting to note that some of the newly identified genes, mainly CEP290 (MIM ∗610142) and EYS, also account for a relatively large proportion of cases.18,20–22
Aiming to clinically characterize patients with FAM161A mutations, we performed a full ophthalmologic examination, full-field electroretinography (ERG), optical coherence tomography (OCT), autofluorescence (AF), Goldmann visual fields, and color vision testing as previously described.23 Clinical evaluation of 28 patients showed a spectrum of findings (Table S3, Figure 2). The majority of patients were myopic, visual acuity ranged from no light perception to 1.0, and mild lens opacities were observed in many patients (Table S3). Fundus findings were relatively mild and often intermingled with myopic changes. Pallor of the optic disc and attenuation of retinal blood vessels were almost always present, but bone-spicule-like pigmentation was observed to only a limited degree in most patients (perhaps related to the coexistent myopia) (Figures 2A–2K). OCT imaging showed marked thinning of the outer nuclear layer with relative preservation under the fovea itself (Figures 2L and 2M). Full-field ERG responses were extinguished in most patients (Table S3). A comparison of cone flicker ERG responses in arRP patients (Figure S3) with FAM161A mutations, EYS mutations, or other genetic causes revealed significantly lower amplitudes in the FAM161A (mean of 4.7 μV) and EYS (0.7 μV) groups compared to patients with arRP of other causes (17.8 μV). Impaired night vision and constriction of visual fields showed a wide spectrum of severity (Figure S4). In summary, similar to patients with mutations in other retinal genes,10,16,17,24,25 patients with FAM161A mutations show a variable retinal phenotype that may be due to modifier genes and/or environmental factors.
Figure 2.
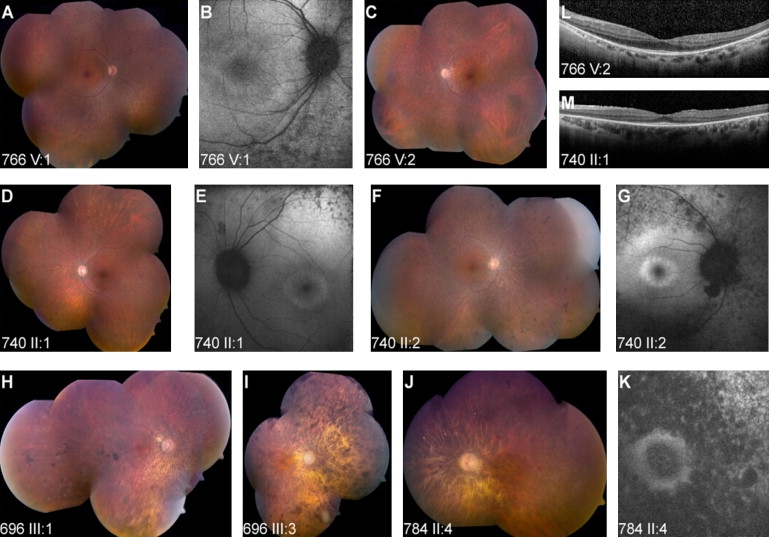
The Spectrum of Fundus Findings among Different Families with Mutations in FAM161A
(A–I) Imaging of three pairs of siblings from families with the p.Thr452SerfsX3 mutation, showing different severity of funduscopic changes: bone-spicule-like pigmentation was observed to only a limited degree in most patients (A, C, D, F as compared to H, I), but many patients showed grayish spots extending from the arcades to the periphery. These correlated with spots of hypofluorescence on AF imaging (B, E, G). In addition, a ring of hyperfluoresence around the fovea could be observed in many cases (B, E, G).
(J and K). Color fundus mosaic (J) and AF imaging of the macular area (K) in a patient with the R523X mutation.
(L and M) OCT imaging shows marked thinning of the outer nuclear layer, with relative preservation under the fovea itself. Fine wrinkling of the retina can be seen in one of the scans (M), related to an epiretinal membrane.
The FAM161A gene contains seven coding exons along a genomic region of ∼30 Kb. An analysis of FAM161A transcripts available at NCBI revealed a number of variants, two of which have an intact open reading frame (ORF) (Figure 3A): a more common transcript in which exon 4 is skipped (FAM161A-001 corresponding to ENST00000405894 at Ensemble) with an ORF of 1983 bp (a total of seven clones, including one from the human retina) and a less common transcript (a total of two clones, none from the human retina) that includes all coding exons (FAM161A-005; ENST00000404929 with an ORF of 2151 bps). In both variants, the translation-initiation codon might lie either within exon 1 or 109 codons downstream within exon 2 (FAM161A-201, ENSP00000397336; Figure 3B). For verification of the expression of FAM161A-005, RNA was isolated from the human retina with TRI Reagent (Sigma-Aldrich) and cDNA was synthesized with the Verso cDNA Kit (Thermo) in accordance with the manufacturer's protocol. PCR-specific primers (Table S1) were designed with Primer3 and RT-PCR analysis was performed, followed by sequence analysis of the PCR products. The analysis confirmed that both variants are transcribed in the human retina, with FAM161A-001 being the more common transcript (Figure 3A). In silico analysis of FAM161A expression in human tissues (using expressed sequence tags [ESTs] and serial analysis of gene expression [SAGE] data) revealed the highest expression level in the eye (10 out of 211,510 ESTs; 47 per million), whereas lower expression levels were observed in a few other tissues, including the brain, testis, liver, and lung. To better characterize the distribution of FAM161A in human tissues, we performed RT-PCR analysis using four primer sets on cDNAs that were synthesized from RNA derived from 20 human tissues (Clontech; catalog no. 636643, lot no. 8101369A) and the human retina (Figures 3B and 3C). The human gene PGM1 (MIM ∗171900) was used as an internal control. Three of the four primer sets showed retina-specific expression (Figure 3C, rows 1, 2, and 4). The primer set designed to exclusively amplify the transcript lacking exon 4 (FAM161A-001) showed a wider distribution, including the cerebellum, fetal brain, testis, and thyroid gland, which had cDNA amplifications that were not as intense as the retinal cDNA amplification (Figure 3C, row 3).
Figure 3.
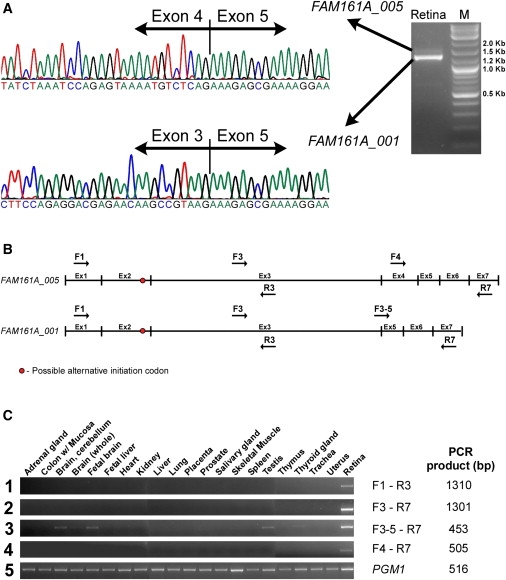
RT-PCR Analysis of FAM161A
(A) RT-PCR analysis of retinal cDNA (right panel) revealed two FAM161A transcripts. The major one, FAM161A_001, does not contain exon 4, and the corresponding chromatogram is depicted in the lower sequencing panel. The less common variant FAM161A_005 contains exon 4, and the corresponding chromatogram is depicted in the upper sequencing panel.
(B) The structure of the two transcripts and the location of the primers used to amplify the different mRNA regions that are shown in (C). The red circle represents a possible alternative initiation codon. The black arrows above (forward) and below (reverse) the transcript scheme represent the four primer sets that were used to analyze FAM161A transcripts in different human tissues.
(C) RT-PCR analysis in 21 different human tissues using the four sets of FAM161A primers (panels 1–4) and a control gene, PGM1 (panel 5). The primer names (corresponding to B) and the PCR product length are shown on the right.
To determine the spatial and temporal expression pattern of Fam161a, we performed in situ hybridization (ISH) on sections of embryonic and postnatal mouse eyes. ISH was performed with the use of a mouse clone corresponding to the splice variant lacking exon 4 (FAM161A_001) and conducted as previously described.26 In brief, tissues were fixed for 24 hr in 4% paraformaldehyde, embedded in OCT medium, and sectioned at 16 μm. Hybridization was conducted overnight at 65°C with digoxigenin-labeled probes (∼5 μg/ml). The slides were then treated with RNaseA, washed, blocked with 10% normal goat serum (NGS), and incubated with sheep anti-Digoxigenin Fab fragments conjugated to alkaline phosphatase (1:250, Roche) in MABT (Maleic Acid Buffer, 0.1% Tween-20) with 20% NGS overnight at 4°C, then washed and incubated in BM Purple (Roche). Probes used were Fam161a (Open Biosystem clone MMM1013-7512149) and Crx.27 During embryogenesis (embryonic day 12.5 [E12.5]–E16.5), low levels of Fam161a expression were detected in the retinal progenitor cells (RPCs) of the optic cup as well as in the posterior compartment of the lens (Figures 4A–4C, antisense control in Figures 4M–4R). Interestingly, Fam161a was not elevated in photoreceptor precursors, identified by the expression of the cone rod homeobox gene, Crx (Figures 4G–4I). In the postnatal retina, however, Fam161a expression was barely detected at postnatal day 1 (P1) but was elevated at P5 in the postmigratory Crx+ photoreceptor precursors at the apical side of the outer nuclear layer (ONL; Figures 4D, 4E, 4J, and 4K). At P10, when all retinal cell types and layers have been generated, Crx was detected in the photoreceptor layer and in the inner nuclear layer (Figure 4L),28 whereas the expression of Fam161a was confined to the photoreceptor layer (Figure 4F). The restricted expression of the mouse ortholog to the ONL postnatally is supported by SAGE and in situ hybridization data of a previous report.29 In addition, Fam161a is expressed by RPCs during embryogenesis, but this expression in the progenitors is downregulated close to birth. In mice, photoreceptor precursors are generated throughout embryogenesis, with a peak around birth. The differentiation of these precursors is completed only after birth, when the outer segment is formed and genes involved in phototransduction are expressed.29 The gradual accumulation of Fam161a in the photoreceptor layer in mice after birth seems to parallel the temporal pattern of expression of other genes that characterize mature photoreceptors.29 Together, the expression pattern of Fam161a in the developing and postnatal retina implicates an involvement during embryogenesis in the retinal progenitors, whereas after birth its activity is restricted to mature photoreceptors. It should be noted, however, that our human patients with RP due to FAM161A mutations did not have any ocular developmental abnormalities. It is therefore possible that in human embryogenesis the paralogous protein, FAM161B, has redundant activity with FAM161A. Interestingly, a high percentage did develop myopia, and many had high myopia of over −6 diopters.
Figure 4.
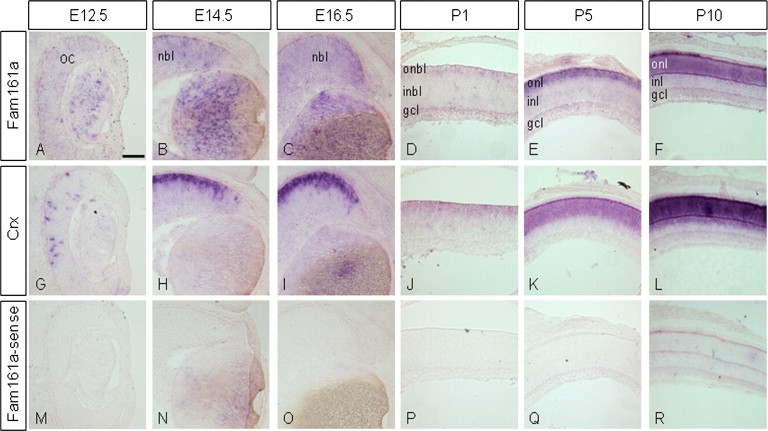
Fam161a Is Expressed throughout the Optic Cup during Mouse Embryogenesis, and Its Expression Is Restricted to the Photoreceptor Layer in the Postnatal Mouse Retina
Characterization of Fam161a and Crx expression by in situ hybridization during embryogenesis and postnatal development. Fam161a expression was detected throughout the retinal neuroblastic layer (NBL) during embryonic stages (E12.5, E14.5, E16.5; A–C), whereas Crx expression was restricted to the photoreceptor precursors (G–I). At postnatal stages, Fam161a expression became restricted to the photoreceptors located at the outer rim of the outer neuroblastic layer (ONBL) (D–F). This expression was low at P1 (D) and increased at P5 (E). At this stage, Crx was highly expressed in the ONBL and in some cells of the inner neuroblastic layer (INBL) (J–L). At P10, Fam161a was detected in the ONL similar to Crx (F). Fam161a sense probe served as a negative control and failed to produce any nonspecific staining at all stages tested (M–R). Abbreviations are as follows: OC, optic cup; NBL, neuroblastic layer; ONBL, outer neuroblastic layer; INBL, inner neuroblastic layer; ONL, outer nuclear layer; INL, inner nuclear layer. Scale bar in (A) represents 100 μm.
FAM161A encodes at least two protein isoforms composed of 660 (due to skipping of exon 4) or 716 amino acids. In addition, two translation-initiation sites might be active, producing proteins that differ in the 109 N terminus amino acids. The encoded proteins contain a single domain, Pfam UPF0564 (included in 15 proteins and with an average length of 277 amino acids), of unknown function. FAM161A has one paralog in the human genome, FAM161B, which also contains the UPF0564 domain. The conserved UPF0564 sequence is also found in proteins of “lower” organisms and mainly in the unicellular ciliate protozoa Paramecium tetraurelia. Within UPF0564, FAM161A contains three conserved α-helical coiled-coil structural motifs. These motifs can be found in many proteins of all organisms and might mediate oligomerization or protein-protein interactions or are perhaps important for protein structure and function. Only one protein is currently known to interact with FAM161A: a large-scale effort aimed at creating a human protein-protein interaction network using the two-hybrid system resulted in the identification of an interaction between FAM161A and PPM1F,30 a ubiquitously expressed protein acting as a Ser/Thr protein phosphatase. Comprehensive functional analyses are needed in order to gain more information about the function of FAM161A in the retina and the mechanism by which lack or dysfunction of this protein causes retinal degeneration.
FAM161A orthologs can be found in mammals, chicken, and zebrafish, but not in Drosophila. To gain insight into protein regions that are preserved along evolution, we performed a sliding window analysis comparing the human protein sequence to selected orthologs (Figure 5A). Amino acid sequences of FAM161A orthologs were extracted from the Homologene database at NCBI. Amino acid sequences were aligned with the ClustalW2 multiple alignment tool at EBI. The sliding window analysis was performed, with a 30 amino acid interval, between the human FAM161A sequence and each representative ortholog. A very limited percentage of amino acid identity was found in the C terminus as well as in the N terminus, but two areas showing a relatively high degree of preservation were identified: a region preserved in all sequences at amino acids 290–360, and a region conserved in mammalians orthologs at amino acids 535–595. Both regions are located within the UPF0564 domain. Notably, one of these regions is encoded by the alternatively spliced exon 4, which is present in only a low percentage of FAM161A transcripts. The sliding window analysis indicated a relatively low level of preservation of FAM161A along evolution. To compare the level of amino acid preservation between FAM161A and 25 other arRP proteins (CDHR1, CERKL, CNGA1, CNGB1, CRB1, EYS, IDH3B, LRAT, MERTK, NR2E3, NRL, PDE6A, PDE6B, PRCD, PROM1, RBP3, RGR, RHO, RLBP1, RP1, RPE65, SAG, SPATA7, TULP1, USH2A), we calculated the amino acid identity levels between each human protein sequence and its orthologs in six species (Figure 5B). The analysis showed that FAM161A is one of the least preserved proteins in five of the six available comparisons. Phylogenetic analysis of the two FAM161 human paralogs (FAM161A and FAM161B) sequences was performed with the use of the neighbor joining procedure, with the chicken FAM161B sequence serving as an outgroup. The analysis revealed that the two human paralogs are distantly related, with a faster evolutionary rate (indicated by the length of each branch) of FAM161A (Figure 5C).
Figure 5.
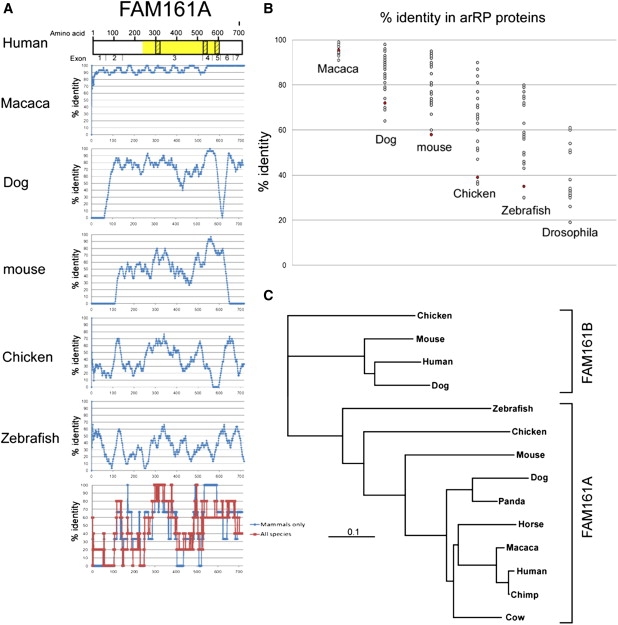
Evolutionary Analysis of FAM161A
(A) A scheme representing the human protein FAM161A is depicted at the top, with identified domains highlighted in yellow (UPF0564) or dashed boxes (coiled-coil sequences). The exon boundaries are shown below. An amino acid sliding window (length of 30 amino acids) comparing the human protein sequence to selected orthologs (macaca, dog, mouse, chicken, and zebrafish) is shown. x axis: amino acid number; y axis: percentage of amino acid identity in a 30 amino acid window. The lower graph represents a summary of the data in mammalian sequences (blue) versus all five sequences (red), with a cumulative sliding window analysis of data points that are above the average percentage of amino acid identity for each studied sequence (macaca 95%, dog 72%, mouse 58%, chicken 39%, zebrafish 35%).
(B) A comparison of amino acid identity levels (comparing the human protein sequence to its ortholog in each of the six species) of all 25 known arRP proteins. The results obtained for FAM161A are marked in red. Note that there is no ortholog for FAM161A in Drosophila.
(C) A phylogenetic tree of FAM161A and FAM161B. Note the relatively long distances separating the orthologous FAM161A protein sequences.
In conclusion, we report here that mutations in the FAM161A gene, which is included in the RP28 region, can cause arRP. FAM161A is currently the most common arRP gene in our Israeli cohort of patients. It is expressed mainly in the retina, but the function of the encoded protein is as yet unknown.
Acknowledgments
This study was financially supported mainly by the Foundation Fighting Blindness USA (grant BR-GE-0607-0395-HUJ to D.S.), as well as by the Legacy Heritage Biomedical Program of the Israeli Science Foundation (grant 612/09 to D.S. and T.B.Y.), a Yedidut 1 research grant (to E.B.), the Israel Science Foundation (to R.A.P.), the Israel Ministry of Science (to R.A.P.), and the Ziegler Foundation (to R.A.P.). We thank the patients and their families for their participation in the study. The authors thank Inbar Erdinest, Devora Marks-Ohana, Malka Sela, and Israel Barzel for excellent technical and imaging assistance.
Contributor Information
Eyal Banin, Email: banine@cc.huji.ac.il.
Dror Sharon, Email: dror.sharon1@gmail.com.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ClustalW2 multiple alignment tool at the European Bioinformatics Institute, http://www.ebi.ac.uk/Tools/clustalw2/index.html
Ensembl, http://www.ensembl.org/index.html
HomoloGene, http://www.ncbi.nlm.nih.gov/homologene/
The Israeli National Genetic Database, http://www.goldenhelix.org/server/israeli/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3, http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi
UCSC Genome Bioinformatics, http://genome.ucsc.edu/index.html?org=Human&db=hg19&hgsid=161902362
UniGene, http://www.ncbi.nlm.nih.gov/unigene/
References
- 1.Rosenberg T. Epidemiology of hereditary ocular disorders. Dev. Ophthalmol. 2003;37:16–33. doi: 10.1159/000072036. [DOI] [PubMed] [Google Scholar]
- 2.Bundey S., Crews S.J. A study of retinitis pigmentosa in the City of Birmingham. II Clinical and genetic heterogeneity. J. Med. Genet. 1984;21:421–428. doi: 10.1136/jmg.21.6.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bunker C.H., Berson E.L., Bromley W.C., Hayes R.P., Roderick T.H. Prevalence of retinitis pigmentosa in Maine. Am. J. Ophthalmol. 1984;97:357–365. doi: 10.1016/0002-9394(84)90636-6. [DOI] [PubMed] [Google Scholar]
- 4.Hartong D.T., Berson E.L., Dryja T.P. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 5.Gu S., Kumaramanickavel G., Srikumari C.R., Denton M.J., Gal A. Autosomal recessive retinitis pigmentosa locus RP28 maps between D2S1337 and D2S286 on chromosome 2p11-p15 in an Indian family. J. Med. Genet. 1999;36:705–707. [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar A., Shetty J., Kumar B., Blanton S.H. Confirmation of linkage and refinement of the RP28 locus for autosomal recessive retinitis pigmentosa on chromosome 2p14-p15 in an Indian family. Mol. Vis. 2004;10:399–402. [PubMed] [Google Scholar]
- 7.Rio Frio T., Panek S., Iseli C., Di Gioia S.A., Kumar A., Gal A., Rivolta C. Ultra high throughput sequencing excludes MDH1 as candidate gene for RP28-linked retinitis pigmentosa. Mol. Vis. 2009;15:2627–2633. [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H., den Hollander A.I., Moayedi Y., Abulimiti A., Li Y., Collin R.W., Hoyng C.B., Lopez I., Abboud E.B., Al-Rajhi A.A. Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am. J. Hum. Genet. 2009;84:380–387. doi: 10.1016/j.ajhg.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.den Hollander A.I., McGee T.L., Ziviello C., Banfi S., Dryja T.P., Gonzalez-Fernandez F., Ghosh D., Berson E.L. A homozygous missense mutation in the IRBP gene (RBP3) associated with autosomal recessive retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2009;50:1864–1872. doi: 10.1167/iovs.08-2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collin R.W.J., Littink K.W., Klevering B.J., van den Born L.I., Koenekoop R.K., Zonneveld M.N., Blokland E.A., Strom T.M., Hoyng C.B., den Hollander A.I., Cremers F.P. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am. J. Hum. Genet. 2008;83:594–603. doi: 10.1016/j.ajhg.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collin R.W., Safieh C., Littink K.W., Shalev S.A., Garzozi H.J., Rizel L., Abbasi A.H., Cremers F.P., den Hollander A.I., Klevering B.J., Ben-Yosef T. Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010;86:783–788. doi: 10.1016/j.ajhg.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zlotogora J., van Baal S., Patrinos G.P. Documentation of inherited disorders and mutation frequencies in the different religious communities in Israel in the Israeli National Genetic Database. Hum. Mutat. 2007;28:944–949. doi: 10.1002/humu.20551. [DOI] [PubMed] [Google Scholar]
- 13.Zlotogora J. Autosomal recessive diseases among Palestinian Arabs. J. Med. Genet. 1997;34:765–766. doi: 10.1136/jmg.34.9.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zlotogora J., Bach G., Munnich A. Molecular basis of mendelian disorders among Jews. Mol. Genet. Metab. 2000;69:169–180. doi: 10.1006/mgme.2000.2969. [DOI] [PubMed] [Google Scholar]
- 15.Rozen S., Skaletsky H.J. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S., Misener S., editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press; Totowa, NJ: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 16.Auslender N., Sharon D., Abbasi A.H., Garzozi H.J., Banin E., Ben-Yosef T. A common founder mutation of CERKL underlies autosomal recessive retinal degeneration with early macular involvement among Yemenite Jews. Invest. Ophthalmol. Vis. Sci. 2007;48:5431–5438. doi: 10.1167/iovs.07-0736. [DOI] [PubMed] [Google Scholar]
- 17.Ness S.L., Ben-Yosef T., Bar-Lev A., Madeo A.C., Brewer C.C., Avraham K.B., Kornreich R., Desnick R.J., Willner J.P., Friedman T.B., Griffith A.J. Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J. Med. Genet. 2003;40:767–772. doi: 10.1136/jmg.40.10.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bandah-Rozenfeld D., Littink K.W., Ben-Yosef T., Strom T.M., Chowers I., Collin R.W., den Hollander A.I., van den Born I., Zonneveld M.N., Merin S. Novel null mutations in the EYS gene are a frequent cause of autosomal recessive retinitis pigmentosa in the Israeli population. Invest. Ophthalmol. Vis. Sci. 2010 doi: 10.1167/iovs.09-4732. in press. [DOI] [PubMed] [Google Scholar]
- 19.Dryja T.P., McGee T.L., Reichel E., Hahn L.B., Cowley G.S., Yandell D.W., Sandberg M.A., Berson E.L. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- 20.den Hollander A.I., Koenekoop R.K., Yzer S., Lopez I., Arends M.L., Voesenek K.E., Zonneveld M.N., Strom T.M., Meitinger T., Brunner H.G. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abd El-Aziz M.M., O'Driscoll C.A., Kaye R.S., Barragan I., El-Ashry M.F., Borrego S., Antiñolo G., Pang C.P., Webster A.R., Bhattacharya S.S. Identification of Novel Mutations in the Ortholog of Drosophila Eyes Shut Gene (EYS) Causing Autosomal Recessive Retinitis Pigmentosa. Invest. Ophthalmol. Vis. Sci. 2010;51:4266–4272. doi: 10.1167/iovs.09-5109. [DOI] [PubMed] [Google Scholar]
- 22.Audo I., Sahel J.A., Mohand-Saïd S., Lancelot M.E., Antonio A., Moskova-Doumanova V., Nandrot E.F., Doumanov J., Barragan I., Antinolo G. EYS is a major gene for rod-cone dystrophies in France. Hum. Mutat. 2010;31:E1406–E1435. doi: 10.1002/humu.21249. [DOI] [PubMed] [Google Scholar]
- 23.Beit-Ya'acov A., Mizrahi-Meissonnier L., Obolensky A., Landau C., Blumenfeld A., Rosenmann A., Banin E., Sharon D. Homozygosity for a novel ABCA4 founder splicing mutation is associated with progressive and severe Stargardt-like disease. Invest. Ophthalmol. Vis. Sci. 2007;48:4308–4314. doi: 10.1167/iovs.07-0244. [DOI] [PubMed] [Google Scholar]
- 24.Bandah D., Merin S., Ashhab M., Banin E., Sharon D. The spectrum of retinal diseases caused by NR2E3 mutations in Israeli and Palestinian patients. Arch. Ophthalmol. 2009;127:297–302. doi: 10.1001/archophthalmol.2008.615. [DOI] [PubMed] [Google Scholar]
- 25.Audo I., Michaelides M., Robson A.G., Hawlina M., Vaclavik V., Sandbach J.M., Neveu M.M., Hogg C.R., Hunt D.M., Moore A.T. Phenotypic variation in enhanced S-cone syndrome. Invest. Ophthalmol. Vis. Sci. 2008;49:2082–2093. doi: 10.1167/iovs.05-1629. [DOI] [PubMed] [Google Scholar]
- 26.Jensen A.M., Wallace V.A. Expression of Sonic hedgehog and its putative role as a precursor cell mitogen in the developing mouse retina. Development. 1997;124:363–371. doi: 10.1242/dev.124.2.363. [DOI] [PubMed] [Google Scholar]
- 27.Furukawa T., Morrow E.M., Cepko C.L. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell. 1997;91:531–541. doi: 10.1016/s0092-8674(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 28.Bibb L.C., Holt J.K., Tarttelin E.E., Hodges M.D., Gregory-Evans K., Rutherford A., Lucas R.J., Sowden J.C., Gregory-Evans C.Y. Temporal and spatial expression patterns of the CRX transcription factor and its downstream targets. Critical differences during human and mouse eye development. Hum. Mol. Genet. 2001;10:1571–1579. doi: 10.1093/hmg/10.15.1571. [DOI] [PubMed] [Google Scholar]
- 29.Blackshaw S., Harpavat S., Trimarchi J., Cai L., Huang H., Kuo W.P., Weber G., Lee K., Fraioli R.E., Cho S.H. Genomic analysis of mouse retinal development. PLoS Biol. 2004;2:E247. doi: 10.1371/journal.pbio.0020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stelzl U., Worm U., Lalowski M., Haenig C., Brembeck F.H., Goehler H., Stroedicke M., Zenkner M., Schoenherr A., Koeppen S. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.