

Abstract
Band-like calcification with simplified gyration and polymicrogyria (BLC-PMG) is a rare autosomal-recessive neurological disorder showing highly characteristic clinical and neuroradiological features. Affected individuals demonstrate early-onset seizures, severe microcephaly, and developmental arrest with bilateral, symmetrical polymicrogyria (PMG) and a band of gray matter calcification on brain imaging; as such, the disorder can be considered as a “pseudo-TORCH” syndrome. By using autozygosity mapping and copy number analysis we identified intragenic deletions and mutations in OCLN in nine patients from six families with BLC-PMG. The OCLN gene encodes occludin, an integral component of tight junctions. Neuropathological analysis of an affected individual showed similarity to the mouse model of occludin deficiency with calcification predominantly associated with blood vessels. Both intracranial calcification and PMG are heterogeneous in etiology. Neuropathological and clinical studies of PMG have suggested that in utero ischemic or vascular insults may contribute to this common cortical abnormality. Tight junctions are functional in cerebral blood vessels early in fetal development and continue to play a vital role in maintenance of the blood-brain barrier during postnatal life. We provide evidence that the tight junction protein occludin (encoded by the OCLN gene) is involved in the pathogenesis of malformations of cortical development.
Main Text
Band-like calcification with simplified gyration and polymicrogyria (BLC-PMG) is a rare autosomal-recessive neurological condition demonstrating clinical and neuroradiological features that may be interpreted as sequelae of congenital infection, a so-called pseudo-TORCH syndrome (MIM 251290). We have previously described 12 affected children from 5 families with this disorder.1–3 Patients experienced early-onset seizures, severe progressive microcephaly, and developmental arrest. This patient cohort was collated on the basis of the pattern of gray matter calcification and cortical malformation. CT and MR imaging showed a prominent band of cortical gray matter calcification as well as calcification in the cerebellum and basal ganglia (Figures 1 and 2). Brain imaging also showed characteristic bilateral, symmetrical, predominantly fronto-parietal PMG. Intracranial calcification (ICC) is a finding common to a heterogeneous group of genetic syndromes, as well as a prominent manifestation of intrauterine infection, in particular with congenital cytomegalovirus (CMV). These phenotypes are typically defined, not by the pattern of ICC, but by the presence of other clinical features. The combination of ICC and PMG suggests congenital CMV infection during mid-gestation.4,5 However, in BLC-PMG, the ICC is seen in a uniform, semicontinuous ribbon or band on CT brain, unlike the patterning typical of CMV infection. PMG is an increasingly recognized and common malformation of cortical development associated with a growing number of syndromes and consistent cytogenetic abnormalities.6–8 Mutations in several genes have been identified as associated with PMG,9–19 underlining the heterogeneous etiology of this malformation. The importance of ischemic or vascular insults, occurring at around 5 months of gestation, in the pathogenesis of PMG has been suggested on the basis of animal models,20–23 twin studies,24–26 in utero insults,27 and case studies of affected patients.28 The site of PMG is most commonly within the territory of the middle cerebral artery, lending further weight to a vascular etiology.29–31 Here, we report mutations in the OCLN gene (MIM 602876) encoding the tight junction protein occludin in nine patients with BLC-PMG. Occludin is expressed as an integral component of the tight junction in all epithelia as well as endothelia in the brain.32,33 The Ocln knockout mouse model has a complex phenotype including abnormalities of salivary glands, gastric epithelium, bone, testes, and ICC.34 The human phenotype reported here is confined to the brain, suggesting, as in the mouse model, functional redundancy of occludin in other tissue types. We postulate that absence of occludin in the developing brain33 and subsequent abnormal blood-brain barrier (BBB) function35 results in cortical malformation.
Figure 1.
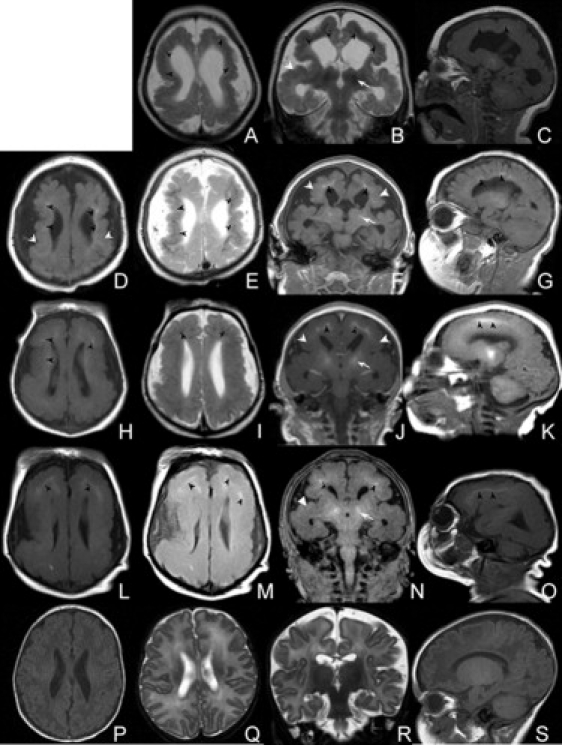
Selected MRI Images from Four Affected Individuals with BLC-PMG
(A–C) F085a2 age 7 months.
(D–G) F351 age 3 months.
(H–K) F386a1 age 4 days.
(L–O) F386a2 age 4 days.
(P–S) An age-appropriate control.
T1 (D, H, L) and T2 (A, E, I, M) weighted axial and T1/T2 coronal (B, F, J, N) and T1 sagittal (C, G, K, O) images show severe reduction in cerebral volume, simplified gyration, and bilateral fronto-parietal PMG (white arrowheads). A band of abnormal signal in both hemipheres in all images (black arrowheads) represents calcification in the deep cortical gray matter. Coronal and sagittal images (B, F, J, N) show calcification in the basal ganglia (arrows) that was also evident in the cerebellum and pons. Noticeable occipital scalp rugae are evident in F386a1 and F386a2.
Figure 2.
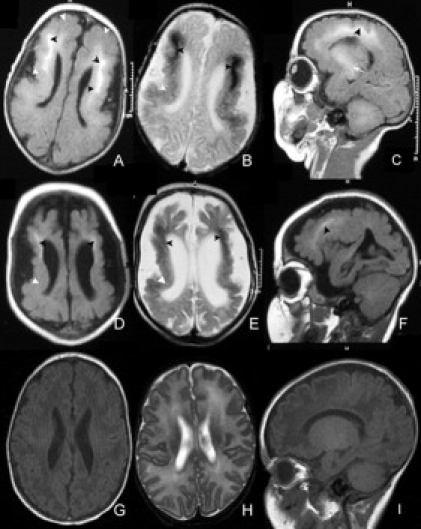
Selected MRI Images from a Single Affected Individual with BLC-PMG
Serial MRI images age 2 weeks (A–C) and 9 months (D–F) from F085a1 with an age-appropriate control (G–I) showing progressive cerebral atrophy. T1 (A, D) and T2 (B, E) weighted axial images also show the severe reduction in cerebral volume, deep cortical (black arrowheads) and basal ganglia (arrow) calcification, and bilateral fronto-parietal PMG (white arrowheads) seen in other affected individuals.
Affected individuals were recruited into our ongoing study of patients with ICC. Further patients were ascertained on the basis of highly concordant clinical and neuroradiological phenotypes. Written informed consent was obtained for all participants and the study has full ethical approval from the Leeds Multi-center Research Ethics Committee (Reference number 07/Q1206/7). Ten affected individuals from six families with the typical BLC-PMG phenotype are described (Table 1 and Table S1 available online). Four families, all from the Middle East, were consanguineous. In two other families, originating from the UK and from Mexico, the parents were not known to be related. The clinical details of affected individuals from families F275,3 F312,1 and F3751 have been previously reported. Information on the clinical phenotype for families F085, F351, and F386 are available in Table S1 including the sibling from F386 in whom genetic testing was not performed. In brief, affected individuals were severely microcephalic, developed seizures within 4 months of birth, and demonstrated minimal developmental progress and a spastic tetraparesis. Birth occipito-frontal circumference (OFC) ranged from +1 SD to −3 SD with early and sustained progression (−2.5 SD to −8 SD on review) in all patients in whom follow-up information was available1,3 (Table S1). CSF analysis performed on affected individuals from families F085, F312, F351, F375, and F386 was normal except for raised protein levels (Table 2). CSF interferon alpha levels were measured in a single patient and were found to be normal. One patient (F351) had mild hepatomegaly but testing for congenital infections was normal at birth. Further testing at 4 months showed CMV IgG in serum and positive CMV PCR in urine but not in blood or CSF. Her mother had raised serum CMV IgG. As previously reported,1 both siblings from F375 had positive CMV serology, indicative of postnatal infection in the elder sibling, but all remaining patients had negative neonatal CMV serology (Table S1). Mutation testing for the four known genes causing Aicardi-Goutieres syndrome (MIM 225750) was negative in F351. No health concerns were reported in any of the parents.
Table 1.
Details of Families with BLC-PMG and OCLN Mutations Described in This Report
| Family | Ancestry | Tested | Nucleotide Alteration | Exon | Amino Acid Alteration | Parental Consanguinity |
|---|---|---|---|---|---|---|
| F085 | Turkish | 2A | 46,XY, arr 5q13.2(68,839,890x2, 68,840,602-68,844,536x0, 68,852,777x2) | 3 (& 4?) | p.Lys18_Glu243? deletion of transmembrane domains |
yes |
| F275 | Egyptian | 1A | 46,XY, arr 5q13.2(68,839,890x2, 68,840,602-68,844,536x0, 68,852,777x2) | 3 (& 4?) | p.Lys18_Glu243? deletion of transmembrane domains |
yes |
| F312 | British | 2A | c.512 dupA | 3 | p.Tyr171X | no |
| M, F | c.656T>C | 3 | p.Phe219Ser | no | ||
| F351 | Saudi | 1A, M, F | c.1037+5G>A | intron 5-6 | alteration of donor splice site | yes |
| F375 | Turkish | 2A, M, F | c.171_193 del ATGGACCTCTCCTCCAGGAGTG | 3 | p.Trp58PhefsX9 deletion of first 8 amino acids of conserved Marvel domain |
yes |
| F386 | Mexican | 1A, M, F | c.51-?_730-?del | 3 | p.Lys18_Glu243? deletion of transmembrane domains |
no |
Nomenclature according to current ISCN70 and HGVS standards.
Abbreviations: A, affected; M, mother; F, father.
Table 2.
Results of Cerebrospinal Fluid Analysis for Five Children with BLC-PMG Including Three Measurements from F085a1
| Patient | F085a1 (1) | F085a1 (2) | F085a1 (3) | F085a2 | F312a1 | F375a1 | F386a1 |
|---|---|---|---|---|---|---|---|
| Age | 1 month | 3 months | 4 months | 6 months | 1 week | unknown | 3 days |
| White cell count | 9/μL (0–5/μL) | 4/μL (0–5/μL) | 2/μL (0-5/μL) | 1/μL (0-5/μL) | 1/μL (0-5/μL) | normal (value not available) | 2/μL (0-5/μL) |
| Protein | 1021 mg/L (100–200 mg/L) | 824 mg/L (100–200 mg/L) | 666 mg/L (100–200 mg/L) | 631 mg/L (100–200 mg/L) | 60 mg/L (40–400 mg/L) | 700 mg/L (100–200 mg/L) | 1260 mg/L (200–600 mg/L) |
| Glucose | 3 mmol/L (2.2–3.9 mmol/L) | 2.7 mmol/L (2.2–3.9 mmol/L) | - | 64 mg/dL (40–70 mg/dL) | - | - | 79 mg/dL (50–80 mg/dL) |
| Lactate | 1.1 mmol/L (<2.1 mmol/L) | 1.3 mmol/L (<2.1 mmol/L) | 1.5 mmol/L (<2.1 mmol/L) | 1.0 mmol/L (<2.1 mmol/L) | 2.2 mmol/L (<2.1 mmol/L) | - | - |
| CSF IFNα | normal | - | - | - | - | - | - |
| Serum IFNα | normal | - | - | - | - | - | - |
| CSF IgG | - | - | 26.6 mg/L | 37.8 mg/L | - | - | - |
| Oligoclonal bands | - | - | negative | negative | - | - | - |
Normal values for each measurement are in parentheses. Cerebrospinal fluid (CSF) protein levels were raised in 4 of 5 individuals. Serial measurement of CSF protein in F085a1 showed a reduction in protein concentration over a 3 month period. All other measurements were normal. IFNα, interferon alpha; IgG, immunoglobulin G.
The CT and MRI scans of affected individuals were reviewed for the presence of cortical, basal ganglia, and brainstem calcification and for cortical abnormality. Images from families F275, F312, and F375 were presented in the original case reports.1,3 Selected images of both siblings in F085, the affected child in F351, and two affected siblings in F386 are shown in Figures 1 and 2. All demonstrated bilateral symmetrical PMG in a perisylvian and temporal distribution with severe loss of cerebral volume, simplified gyration, and wide sylvian fissures. Calcification was present bilaterally in the deep cortical gray matter with variable calcification in the pons, thalami, and globus pallidus. Serial scans at 2 weeks and 9 months of age in a single affected individual from F085 show evidence of progressive atrophy (Figure 2).
Postmortem analysis of a single patient with BLC-PMG (F312a1) showed widespread gliosis, white matter loss, calcification, and perisylvian predominant PMG within the cerebral cortex as previously described.1 In light of the molecular findings, neuropathological samples from this patient were re-evaluated to determine the pattern of calcification (Figure 3). Mineral deposition, presumed to be calcification, was most prominent in the deep cerebral and cerebellar cortex, frequently surrounding small blood vessels. Calcification was noted in cells in close apposition to endothelia, which may represent pericytes or astrocytes. Unlike endothelial cells, brain-derived pericytes do not express the marker CD31 (PECAM-1).36 Figure 3 also shows sections from the cerebral cortex and cerebellum stained with CD31. These demonstrate endothelia adjacent to, but not incorporated within, areas of calcification. Blood vessels without associated calcification were also seen. No evidence of neuronal overmigration was apparent and the pial surface was intact. Widespread gliosis and centri-lobular sclerosis was also evident in the cerebellum, but the pattern of calcification differed from that seen in the cortex. Calcification surrounded blood vessels of varying sizes and was commonly globular rather than laminar in appearance. It was present in other cells closely allied to blood vessels, which may represent pericytes. Calcification was also present in Purkinje cells.
Figure 3.
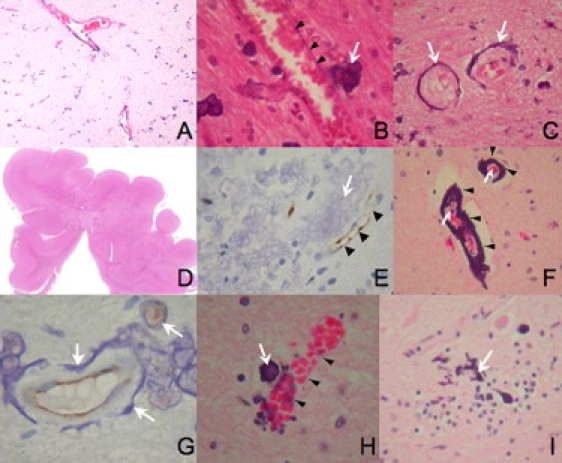
Sections from the Cerebral Cortex of Patient F312a1 with BLC-PMG
(A–C) H&E-stained sections. Low-power section (A) showing linear bands of calcification surrounding blood vessels and at higher power (B) showing calcification adjacent to (arrow), but not within, vessel walls (arrowheads). Rim (C) of calcification (arrows) surrounding two blood vessels in cross-section.
(D) Low-power view of section showing fused gyri indicative of PMG.
(E) CD31 stained section showing endothelium (arrowheads) staining adjacent to large region of calcification (arrow).
(F–I) H&E- (F, H, I) and CD31- (G) stained sections of cerebellum from F312a1.
(F) Calcification (arrowheads) surrounding two vessels of different sizes (arrows).
(G) Apparently intact endothelium (brown) within a ring of calcification (arrows).
(H) Calcification in a cell (arrow) in close apposition to a normal blood vessel (arrowheads).
(I) “Stag-horn” appearance suggesting calcification in a Purkinje cell (arrow).
Genomic DNA was extracted from lymphocytes from affected individuals, parents, and siblings by standard techniques. A genome-wide SNP microarray by means of the Affymetrix Human SNP Array 6.0 (Affymetrix, High Wycombe, Buckinghamshire, UK) was performed in six affected individuals according to the manufacturer's instructions. Copy number analysis was performed with Chromosome Analysis Suite (ChAS; Affymetrix). Regions of homozygosity were identified via AutoSNPa software37 with NCBI build 36 (hg18). Mutation analysis was performed by direct sequencing of purified genomic PCR products. Primers were designed for individual exons and intron boundaries of OCLN with Primer3Plus and the reference sequence NM_002358.2. PCR was performed on genomic DNA with Abgene ReddyMix PCR Mastermix and sequencing was performed with BigDye terminator cycle sequencer system v3.1 (primer sequences available in Table S2 and experimental conditions available on request). All eight coding exons and flanking intronic boundaries of OCLN were screened in affected individuals and the relevant exons were sequenced in both parents where available.
A 6.5 Mb region of shared homozygosity was identified in six affected individuals from consanguineous families (F085, F275, F351, and F375) on chromosome 5q13 (Figure 4). The region contained 1317 SNPs and was flanked by SNPs rs1423233 (position 67,827,934) and rs7711157 (position 74,275,250). The BLC-PMG critical interval contained approximately 65 genes.
Figure 4.
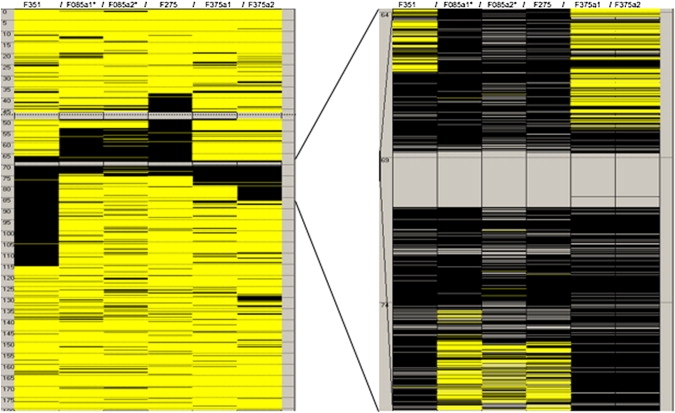
Region of Homozygosity on Chromosome 5 Common to Six Affected Individuals from Four Consanguineous Families
Black and yellow bars indicate homozygous and heterozygous SNPs, respectively. Isolated heterozygous SNPs within larger homozygous segments are likely to represent miss-calls. The SNP-barren interval (gray region in center of right image) represents a 500 kb inverted duplication on chromosome 5q13. The common region of homozygosity defining the critical interval lies between 67,789,350-74,259,390 base pairs. Information is derived from AutoSNPa.36 Listed in Table 1.
Detailed analysis of SNP and copy number probes within this region identified a deletion of three contiguous copy number probes: CN1139669 (position 68,840,602), CN1139670 (position 68,844,101), and CN303606 (position 68,844,536) in both affected individuals from family F085 (Figure 5). The probes were located within intron 2–3 and intron 3–4 of the OCLN gene. Probes on either side of this putative intragenic deletion (CN1139668 and CN1139671) were present on both alleles, as were 10 other probes (one SNP and nine copy number probes) mapping within the OCLN gene. A duplication of exons 6–9 of OCLN is located approximately 1.5 Mb downstream on 5q13. No SNP or copy number probes are annotated within this region.
Figure 5.
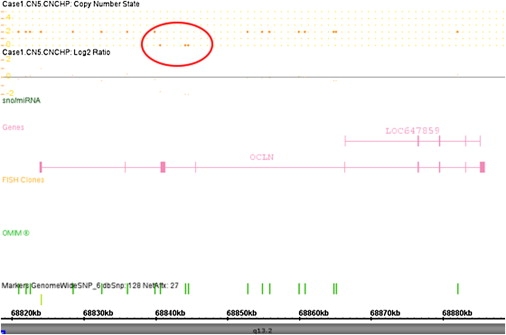
Screenshot from ChAS Software Showing Deletion of Three Contiguous Copy Number Probes on Chromosome 5q13 in F085a1 on Chromosome 5q13
The copy number probes (CN1139669, CN1139670, CN303606) fall within intron 2–3 and intron 3–4 of the OCLN gene.
Mutations in OCLN were identified in nine individuals from six families (Table 1 and Figures 5 and 6). Homozygous deletions of copy number probes within exons 3 and 4 of OCLN were detected by microarray analysis in three affected individuals from two consanguineous families (F085 and F275). These two families are not known to be related, one originating from Egypt and the other from Turkey. A homozygous deletion of exon 3 was found in a single affected individual from a nonconsanguineous native Mexican family via PCR and sequencing (F386). A full-length PCR product corresponding to exon 3 was obtained from DNA from both parents. DNA from his affected sibling was not available for analysis. A homozygous 22 base pair frameshift deletion of exon 3 (c.171_193 delATGGACCTCTCCTCCAGGAGTG) was found in two affected individuals from F375, a consanguineous Turkish family. Both parents from F375 were heterozygous for the same deletion found in their children. Compound heterozygous mutations (c.512 dupA and c.656T>C) in exon 3 were identified in two affected siblings from a nonconsanguineous British family (F312). The paternally inherited lesion c.512 dupA leads to the insertion of a premature stop codon (tyrosine to stop codon substitution at position p.171) (Figures 6 and 7), replacing the first amino acid of the third transmembrane domain. This change is predicted to result in nonsense-mediated decay of the protein. The maternally inherited missense mutation c.656T>C results in a substitution of phenylalanine for serine (p.Phe219Ser). This is a conserved amino acid in exon 3 in the extracellular loop lying between the third and fourth transmembrane domains. Analysis via in silico conservation prediction programs (Polyphen, SIFT, and AlignGVGD) suggests likely pathogenicity for this variant. Lymphoblastoid cell lines were unavailable for further analysis of the significance of these changes. A homozygous acceptor splice site change in the 5–6 intron was found in a single affected child (c.1037+5G>A) from family F351 (Figure 6). Both parents were heterozygous and an unaffected sibling was homozygous for the wild-type allele at the same nucleotide. By means of analysis software (Alamut, MaxEntScan, Drosophila BDG, Hbond, SROOGLE), this change was predicted to result in a severe splicing mutation leading to exon skipping. None of the above mutations were identified in 220 control chromosomes.
Figure 6.
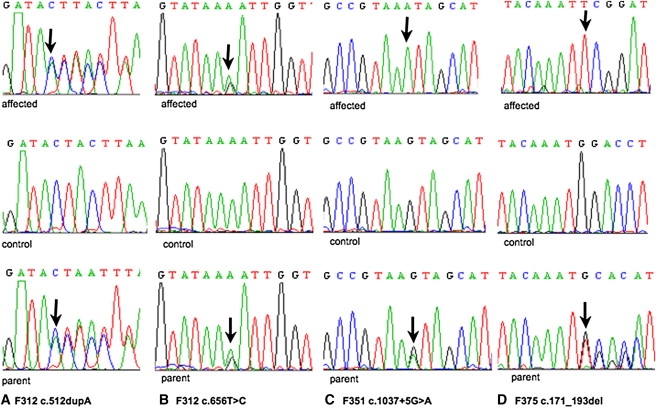
Demonstration of Four OCLN Variants
Top: Sequence chromatograms of affected individuals. Middle: Sequence chromatograms of control sample with wild-type allele. Bottom: Sequence chromatograms of parent with heterozygous mutation. Mutations are highlighted (arrow).
(A) Chromatogram of c.512 dupA in F312.
(B) Chromatogram of c.656T>C in F312.
(C) Chromatogram of splice site variant in F351.
(D) Chromatogram of 22 base pair deletion in F375.
Figure 7.
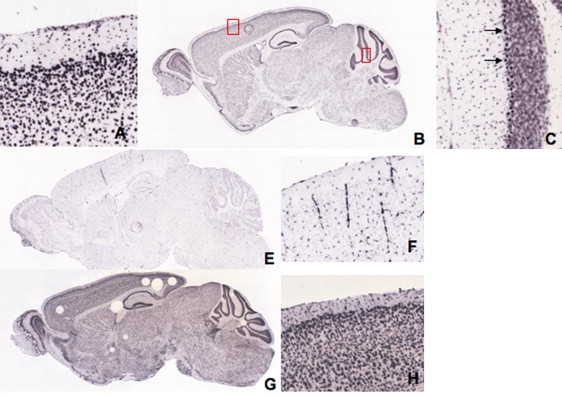
Occludin In Situ Hybridization Expression in Adult Mouse Brain
(A–C) Sagittal images from C57BL/6J mice at 55 days. Occludin is expressed throughout the brain, particularly in the cerebral cortex and cerebellum (B). Magnified images (boxed) of cerebral cortex (A) and cerebellum (C) showing marked occludin expression in layers 2 and 3 of the cerebral cortex and in the Purkinje cell layer in the cerebellum (arrows). Images from the Allen Mouse Brain Atlas, Section ID 79394328_12.
(E–H) Whole-brain sagittal images (E, G) and higher magnification of sections of cerebral cortex (F, H). Claudin 5 expression appears to be concentrated in cerebral blood vessels (E, F). Tubb2b is expressed throughout the brain and concentrated in the cerebral cortex and cerebellum (G, H) in a similar distribution to occludin (B, A). Images from the Allen Mouse Brain Atlas, Section IDs 79360268_14 (E, F) and 69854163_14 (G, H).
Occludin is highly conserved in vertebrates and contains two known functional domains, a Marvel domain and an occludin/ELL domain. A multiple alignment of occludin orthologs highlighting the mutations identified in the affected individuals and the position of the Marvel domain and all four transmembrane domains is shown in Figure S1 (generated by MUSCLE version 3.638 on NCBI). In four families (F085, F275, F375, and F386), intragenic deletion of exon 3 and/or exon 4 is predicted to result in removal of some or all of the Marvel domain in the absence of nonsense-mediated decay. Both mutations found in affected individuals in F312 are located within the Marvel domain and alter conserved amino acids.
We have shown that mutations in the OCLN gene cause BLC-PMG, a severe neurodevelopmental disorder demonstrating highly characteristic clinical and neuroradiological features. The OCLN gene in humans maps to two regions on 5q13.2, a full-length gene containing nine exons and a pseudogene (LOC647859) approximately 1.5 Mb away containing a copy of exons 6–9 within a 500 kb inverted duplication (Figure 4).
Full-length occludin is a ∼65 kDa protein with four transmembrane (TM) domains and two cytoplasmic domains.32 Marvel is a four transmembrane (TM) domain that has functional importance in proteins involved in membrane apposition (reviewed by Sanchez-Pulido et al., 2002).39 The tight junction Marvel protein family includes two other proteins, tricellulin and MarvelD3,40,41 which may contribute to noncerebral TJ function in patients with BLC-PMG. All three proteins have similar but nonredundant roles in epithelial barrier formation and maintenance.40,41 The Marvel domain itself is highly conserved (Figure S1), being present in related Caenorhabditis elegans and Drosophila melanogaster proteins (no OCLN ortholog has been identified in these organisms). Several splicing variants of occludin have been identified including forms lacking the Marvel domain encoded by exons 3 and 4.42–46 Proteins lacking the Marvel domain are located in the cytoplasm rather than the cell membrane and are therefore not incorporated into TJs.44,47,48 Five of six families reported here have deletions or mutations affecting the Marvel domain (Table 1 and Figure S1), and therefore we propose that these mutations will result in a protein product that fails to locate to the cell membrane and hence to TJs.
TJs are multiprotein cell-cell adhesion complexes in epithelia and endothelia that form a barrier to free paracellular transport between tissue compartments.49 Despite a wealth of research on tight junctions in different organs, the function of occludin has proven difficult to define. In vitro studies have shown that functional epithelial TJs can form in the absence of occludin. Other TJ proteins, primarily the claudins, are the major structural components of TJs to which occludin later becomes incorporated.34,48,50,51 Occludin is thought to be involved in the regulation of TJ function or intracellular signaling rather than TJ assembly per se.52 The phenotype of the Ocln knockout mouse34 demonstrates postnatal growth retardation, chronic gastritis, thinning of compact bone, abnormal salivary striatal duct, and atrophy of the seminiferous ducts of the testes with sparing of the Sertoli cells. Examination showed no evidence of a systemic abnormality of calcium metabolism on blood and urine testing, and tight junction morphology was unaffected in intestinal, kidney, and liver epithelia. Of note, the brain of occludin-deficient mice showed progressive cerebellar and basal ganglia calcification, particularly along the walls of capillaries and postcapillary venules. Measurement of BBB permeability was not reported. These mice were viable at birth and, although homozygous mutant males were unable to mate and females unable to suckle, they showed none of the severe neurodevelopmental aspects (seizures, developmental arrest) of the human phenotype we report here. Conversely, in the patients we describe there was no evidence of the extracerebral abnormalities seen in the Ocln knockout mouse. Occludin expression in wild-type adult mouse is prominent in layers 2/3 of the cerebral cortex and in Purkinje cells of the cerebellum (Figure 7), reflecting the pattern of brain calcification in patients with BLC-PMG (Figures 1 and 2). This compares to claudin 5, the major claudin subtype in cerebral TJs, which, unlike occludin, appears to be confined to distribution along blood vessels (Figure 7).
Occludin is ubiquitously expressed in epithelial TJs throughout the body. In the developing human brain, occludin is expressed early in neuroepithelium, but expression is lost during the transition from epithelia to neuronal cell types.53,54 Occludin, along with other components of the TJ such as claudin-5 and JAM-1, are highly expressed in neurovascular TJs during angiogenesis and in mature cerebral (but not noncerebral) endothelia that form the BBB.33,55 Prior to this report a cerebral phenotype has been reported in knockout models of only two TJ proteins (claudin-5 and claudin-11),56,57 neither of which includes cerebral malformation. The mouse model of claudin-5 has a size-specific defect in TJ permeability in the BBB but shows no other alterations in structure or function, leaving the early postnatal mortality of these mice unexplained. Occludin has a putative role in the regulation of paracellular transport. Paracellular transport of macromolecules across the BBB is blocked by TJs, whereas transmigration of leucocytes via TJs is permitted (reviewed in Abbott et al., 2010).58 Leucocytes have been shown to cause neuronal injury in response to inflammatory stimuli59 and therefore a BBB lacking functional occludin may increase the vulnerability of the brain to immune cell-mediated tissue damage. The window of PMG development suggests that such an insult occurs by, or during, a critical interval in fetal development.
The calcification in BLC-PMG is found adjacent to endothelium comprising small blood vessels (Figure 3) in a pattern highly similar to that reported in Ocln null mice (Figure 7 in Saitou et al., 2000).34 Pericytes are multipotential cells present in almost all tissues and organ systems, selectively associated with the microvasculature. Together with astrocytes they encapsulate cerebral microvessels and are separated from them by a basal lamina. Pericytes are reported to have roles in both normal fetal cerebral angiogenesis60 and ectopic vascular calcification.61 Pericytes have the ability to transform into osteoblasts and are thus an obvious candidate cell type for aberrant mineral deposition in these patients. The cortical malformations in BLC-PMG may reflect disordered signaling between cells of the neurovascular unit as well as increased permeability of the BBB.
Mutations in a number of genes encoding components or regulators of the microtubule network have been reported in patients with PMG and pachygyria.19,62–65 The cerebral distribution of the expression of one of these proteins, Tubb2b, is similar to occludin (Figure 7), but, unlike PMG associated with mutations in TUBB2B64 (MIM 610031), no neuronal overmigration through the pial membrane was seen in brain tissue from a patient with BLC-PMG. Few studies have examined the relationship between microtubules and tight junction complexes. Elements of the microtubular complex mediate assembly and disassembly of the apical junctional complex (adherens and tight junctions) in epithelia,66,67 but this process has not been described in endothelia. Targeting and maintenance of occludin at the cell membrane is dependent on both microtubules and the actin cytoskeleton.68 More recently, involvement of occludin in organization and orientation of the microtubular network and actin cytoskeletion during cell migration has been demonstrated in epithelia.69 This process has not been explored during neuronal migration.
The neurodevelopmental phenotype in human we report, and to a lesser extent that seen in the Ocln null mouse, reflects the functional importance of occludin in the developing brain, and its redundancy in other tissues. The BLC-PMG phenotype may be mediated by more than one cell type involved in the neurovascular unit and implicates TJs and BBB dysfunction in malformations of cortical development. Finally, we note that several of the patients included in this report were originally referred to us as possible cases of Aicardi-Goutières syndrome, after the identification of intracranial calcification on brain imaging. This report demonstrates the utility of the classification of neurological diseases on the basis of the pattern and distribution of intracranial calcification, taken in clinical context and association with other neuroimaging features, as a tool to gene discovery.
Acknowledgments
We sincerely thank the families for their involvement in this work. Y.J.C. acknowledges the Manchester NIHR Biomedical Research Centre and the Manchester Academic Health Sciences Centre. M.C.O'D. is funded by the Manchester NIHR Biomedical Research Centre and the Wellcome Trust. We are grateful to Dr. E. Hellen in the Manchester National Genetics Reference Laboratory at the University of Manchester and Dr. Diana Baralle at the University of Southampton for their assistance with mutation analysis.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
AlignGVGD, http://agvgd.iarc.fr/agvgd_input.php
Allen Mouse Brain Atlas, http://mouse.brain-map.org/
AutoSNPa, http://dna.leeds.ac.uk/autosnpa
Chromas software, http://www.technelysium.com.au/chromas.html
Chromosome Analysis Suite, http://www.affymetrix.com/
Drosophila BDG, http://www.fruitfly.org/seq_tools/splice.html
Hbond, http://www.uni-duesseldorf.de/rna/html/hbond_score.php
Human Genome Variation Society, http://www.hgvs.org/mutnomen/
MaxEntScan, http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html
National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
Primer3Plus, http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi
SROOGLE, http://sroogle.tau.ac.il/
University of California, Santa Cruz (UCSC) Genome Browser, http://www.genome.ucsc.edu
References
- 1.Briggs T.A., Wolf N.I., D'Arrigo S., Ebinger F., Harting I., Dobyns W.B., Livingston J.H., Rice G.I., Crooks D., Rowland-Hill C.A. Band-like intracranial calcification with simplified gyration and polymicrogyria: A distinct “pseudo-TORCH” phenotype. Am. J. Med. Genet. A. 2008;146A:3173–3180. doi: 10.1002/ajmg.a.32614. [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Salam G.M., Zaki M.S., Saleem S.N., Gaber K.R. Microcephaly, malformation of brain development and intracranial calcification in sibs: Pseudo-TORCH or a new syndrome. Am. J. Med. Genet. A. 2008;146A:2929–2936. doi: 10.1002/ajmg.a.32549. [DOI] [PubMed] [Google Scholar]
- 3.Abdel-Salam G.M., Zaki M.S. Band-like intracranial calcification (BIC), microcephaly and malformation of brain development: A distinctive form of congenital infection like syndromes. Am. J. Med. Genet. A. 2009;149A:1565–1568. doi: 10.1002/ajmg.a.32894. [DOI] [PubMed] [Google Scholar]
- 4.Boppana S.B., Fowler K.B., Vaid Y., Hedlund G., Stagno S., Britt W.J., Pass R.F. Neuroradiographic findings in the newborn period and long-term outcome in children with symptomatic congenital cytomegalovirus infection. Pediatrics. 1997;99:409–414. doi: 10.1542/peds.99.3.409. [DOI] [PubMed] [Google Scholar]
- 5.Noyola D.E., Demmler G.J., Nelson C.T., Griesser C., Williamson W.D., Atkins J.T., Rozelle J., Turcich M., Llorente A.M., Sellers-Vinson S., Houston Congenital CMV Longitudinal Study Group Early predictors of neurodevelopmental outcome in symptomatic congenital cytomegalovirus infection. J. Pediatr. 2001;138:325–331. doi: 10.1067/mpd.2001.112061. [DOI] [PubMed] [Google Scholar]
- 6.Jansen A., Andermann E. Genetics of the polymicrogyria syndromes. J. Med. Genet. 2005;42:369–378. doi: 10.1136/jmg.2004.023952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobyns W.B., Mirzaa G., Christian S.L., Petras K., Roseberry J., Clark G.D., Curry C.J., McDonald-McGinn D., Medne L., Zackai E. Consistent chromosome abnormalities identify novel polymicrogyria loci in 1p36.3, 2p16.1-p23.1, 4q21.21-q22.1, 6q26-q27, and 21q2. Am. J. Med. Genet. A. 2008;146A:1637–1654. doi: 10.1002/ajmg.a.32293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robin N.H., Taylor C.J., McDonald-McGinn D.M., Zackai E.H., Bingham P., Collins K.J., Earl D., Gill D., Granata T., Guerrini R. Polymicrogyria and deletion 22q11.2 syndrome: Window to the etiology of a common cortical malformation. Am. J. Med. Genet. A. 2006;140:2416–2425. doi: 10.1002/ajmg.a.31443. [DOI] [PubMed] [Google Scholar]
- 9.Roll P., Rudolf G., Pereira S., Royer B., Scheffer I.E., Massacrier A., Valenti M.P., Roeckel-Trevisiol N., Jamali S., Beclin C. SRPX2 mutations in disorders of language cortex and cognition. Hum. Mol. Genet. 2006;15:1195–1207. doi: 10.1093/hmg/ddl035. [DOI] [PubMed] [Google Scholar]
- 10.Baala L., Briault S., Etchevers H.C., Laumonnier F., Natiq A., Amiel J., Boddaert N., Picard C., Sbiti A., Asermouh A. Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat. Genet. 2007;39:454–456. doi: 10.1038/ng1993. [DOI] [PubMed] [Google Scholar]
- 11.Passemard S., Titomanlio L., Elmaleh M., Afenjar A., Alessandri J.L., Andria G., de Villemeur T.B., Boespflug-Tanguy O., Burglen L., Del Giudice E. Expanding the clinical and neuroradiologic phenotype of primary microcephaly due to ASPM mutations. Neurology. 2009;73:962–969. doi: 10.1212/WNL.0b013e3181b8799a. [DOI] [PubMed] [Google Scholar]
- 12.Valente E.M., Marsh S.E., Castori M., Dixon-Salazar T., Bertini E., Al-Gazali L., Messer J., Barbot C., Woods C.G., Boltshauser E. Distinguishing the four genetic causes of Jouberts syndrome-related disorders. Ann. Neurol. 2005;57:513–519. doi: 10.1002/ana.20422. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell T.N., Free S.L., Williamson K.A., Stevens J.M., Churchill A.J., Hanson I.M., Shorvon S.D., Moore A.T., van Heyningen V., Sisodiya S.M. Polymicrogyria and absence of pineal gland due to PAX6 mutation. Ann. Neurol. 2003;53:658–663. doi: 10.1002/ana.10576. [DOI] [PubMed] [Google Scholar]
- 14.Brooks A.S., Bertoli-Avella A.M., Burzynski G.M., Breedveld G.J., Osinga J., Boven L.G., Hurst J.A., Mancini G.M., Lequin M.H., de Coo R.F. Homozygous nonsense mutations in KIAA1279 are associated with malformations of the central and enteric nervous systems. Am. J. Hum. Genet. 2005;77:120–126. doi: 10.1086/431244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keren B., Suzuki O.T., Gérard-Blanluet M., Brémond-Gignac D., Elmaleh M., Titomanlio L., Delezoide A.L., Passos-Bueno M.R., Verloes A. CNS malformations in Knobloch syndrome with splice mutation in COL18A1 gene. Am. J. Med. Genet. A. 2007;143A:1514–1518. doi: 10.1002/ajmg.a.31784. [DOI] [PubMed] [Google Scholar]
- 16.Graham J.M., Jr., Hennekam R., Dobyns W.B., Roeder E., Busch D. MICRO syndrome: An entity distinct from COFS syndrome. Am. J. Med. Genet. A. 2004;128A:235–245. doi: 10.1002/ajmg.a.30060. [DOI] [PubMed] [Google Scholar]
- 17.Hevner R.F. The cerebral cortex malformation in thanatophoric dysplasia: Neuropathology and pathogenesis. Acta Neuropathol. 2005;110:208–221. doi: 10.1007/s00401-005-1059-8. [DOI] [PubMed] [Google Scholar]
- 18.Geerdink N., Rotteveel J.J., Lammens M., Sistermans E.A., Heikens G.T., Gabreëls F.J., Mullaart R.A., Hamel B.C. MECP2 mutation in a boy with severe neonatal encephalopathy: Clinical, neuropathological and molecular findings. Neuropediatrics. 2002;33:33–36. doi: 10.1055/s-2002-23598. [DOI] [PubMed] [Google Scholar]
- 19.Abdollahi M.R., Morrison E., Sirey T., Molnar Z., Hayward B.E., Carr I.M., Springell K., Woods C.G., Ahmed M., Hattingh L. Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am. J. Hum. Genet. 2009;85:737–744. doi: 10.1016/j.ajhg.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dvorák K., Feit J. Migration of neuroblasts through partial necrosis of the cerebral cortex in newborn rats-contribution to the problems of morphological development and developmental period of cerebral microgyria. Histological and autoradiographical study. Acta Neuropathol. 1977;38:203–212. doi: 10.1007/BF00688066. [DOI] [PubMed] [Google Scholar]
- 21.Ferrer I. A Golgi analysis of unlayered polymicrogyria. Acta Neuropathol. 1984;65:69–76. doi: 10.1007/BF00689830. [DOI] [PubMed] [Google Scholar]
- 22.Ferrer I., Catalá I. Unlayered polymicrogyria: Structural and developmental aspects. Anat. Embryol. (Berl.) 1991;184:517–528. doi: 10.1007/BF01236058. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki M., Choi B.H. Repair and reconstruction of the cortical plate following closed cryogenic injury to the neonatal rat cerebrum. Acta Neuropathol. 1991;82:93–101. doi: 10.1007/BF00293950. [DOI] [PubMed] [Google Scholar]
- 24.Barth P.G., van der Harten J.J. Parabiotic twin syndrome with topical isocortical disruption and gastroschisis. Acta Neuropathol. 1985;67:345–349. doi: 10.1007/BF00687825. [DOI] [PubMed] [Google Scholar]
- 25.Graff-Radford N.R., Bosch E.P., Stears J.C., Tranel D. Developmental Foix-Chavany-Marie syndrome in identical twins. Ann. Neurol. 1986;20:632–635. doi: 10.1002/ana.410200513. [DOI] [PubMed] [Google Scholar]
- 26.Van Bogaert P., Donner C., David P., Rodesch F., Avni E.F., Szliwowski H.B. Congenital bilateral perisylvian syndrome in a monozygotic twin with intra-uterine death of the co-twin. Dev. Med. Child Neurol. 1996;38:166–170. doi: 10.1111/j.1469-8749.1996.tb12088.x. [DOI] [PubMed] [Google Scholar]
- 27.Richman D.P., Stewart R.M., Caviness V.S., Jr. Cerebral microgyria in a 27-week fetus: An architectonic and topographic analysis. J. Neuropathol. Exp. Neurol. 1974;33:374–384. doi: 10.1097/00005072-197407000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Williams R.S., Ferrante R.J., Caviness V.S., Jr. The cellular pathology of microgyria. A Golgi analysis. Acta Neuropathol. 1976;36:269–283. doi: 10.1007/BF00685371. [DOI] [PubMed] [Google Scholar]
- 29.Reutens D.C., Berkovic S.F., Kalnins R.M., McKelvie P., Saling M.M., Fabinyi G.C. Localised neuronal migration disorder and intractable epilepsy: A prenatal vascular aetiology. J. Neurol. Neurosurg. Psychiatry. 1993;56:314–316. doi: 10.1136/jnnp.56.3.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raybaud C., Di Rocco C. Brain malformation in syndromic craniosynostoses, a primary disorder of white matter: A review. Childs Nerv. Syst. 2007;23:1379–1388. doi: 10.1007/s00381-007-0474-7. [DOI] [PubMed] [Google Scholar]
- 31.McBride M.C., Kemper T.L. Pathogenesis of four-layered microgyric cortex in man. Acta Neuropathol. 1982;57:93–98. doi: 10.1007/BF00685375. [DOI] [PubMed] [Google Scholar]
- 32.Furuse M., Hirase T., Itoh M., Nagafuchi A., Yonemura S., Tsukita S., Tsukita S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Virgintino D., Errede M., Robertson D., Capobianco C., Girolamo F., Vimercati A., Bertossi M., Roncali L. Immunolocalization of tight junction proteins in the adult and developing human brain. Histochem. Cell Biol. 2004;122:51–59. doi: 10.1007/s00418-004-0665-1. [DOI] [PubMed] [Google Scholar]
- 34.Saitou M., Furuse M., Sasaki H., Schulzke J.D., Fromm M., Takano H., Noda T., Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell. 2000;11:4131–4142. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirase T., Staddon J.M., Saitou M., Ando-Akatsuka Y., Itoh M., Furuse M., Fujimoto K., Tsukita S., Rubin L.L. Occludin as a possible determinant of tight junction permeability in endothelial cells. J. Cell Sci. 1997;110:1603–1613. doi: 10.1242/jcs.110.14.1603. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu F., Sano Y., Maeda T., Abe M.A., Nakayama H., Takahashi R., Ueda M., Ohtsuki S., Terasaki T., Obinata M., Kanda T. Peripheral nerve pericytes originating from the blood-nerve barrier expresses tight junctional molecules and transporters as barrier-forming cells. J. Cell. Physiol. 2008;217:388–399. doi: 10.1002/jcp.21508. [DOI] [PubMed] [Google Scholar]
- 37.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 38.Edgar R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sánchez-Pulido L., Martín-Belmonte F., Valencia A., Alonso M.A. MARVEL: A conserved domain involved in membrane apposition events. Trends Biochem. Sci. 2002;27:599–601. doi: 10.1016/s0968-0004(02)02229-6. [DOI] [PubMed] [Google Scholar]
- 40.Steed E., Rodrigues N.T., Balda M.S., Matter K. Identification of MarvelD3 as a tight junction-associated transmembrane protein of the occludin family. BMC Cell Biol. 2009;10:95. doi: 10.1186/1471-2121-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raleigh D.R., Marchiando A.M., Zhang Y., Shen L., Sasaki H., Wang Y., Long M., Turner J.R. Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol. Biol. Cell. 2010;21:1200–1213. doi: 10.1091/mbc.E09-08-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghassemifar M.R., Sheth B., Papenbrock T., Leese H.J., Houghton F.D., Fleming T.P. Occludin TM4(-): An isoform of the tight junction protein present in primates lacking the fourth transmembrane domain. J. Cell Sci. 2002;115:3171–3180. doi: 10.1242/jcs.115.15.3171. [DOI] [PubMed] [Google Scholar]
- 43.Gu J.M., Lim S.O., Park Y.M., Jung G. A novel splice variant of occludin deleted in exon 9 and its role in cell apoptosis and invasion. FEBS J. 2008;275:3145–3156. doi: 10.1111/j.1742-4658.2008.06467.x. [DOI] [PubMed] [Google Scholar]
- 44.Kohaar I., Ploss A., Korol E., Mu K., Schoggins J.W., O'Brien T.R., Rice C.M., Prokunina-Olsson L. Splicing diversity of the human OCLN gene and its biological significance for hepatitis C virus entry. J. Virol. 2010;84:6987–6994. doi: 10.1128/JVI.00196-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mankertz J., Waller J.S., Hillenbrand B., Tavalali S., Florian P., Schöneberg T., Fromm M., Schulzke J.D. Gene expression of the tight junction protein occludin includes differential splicing and alternative promoter usage. Biochem. Biophys. Res. Commun. 2002;298:657–666. doi: 10.1016/s0006-291x(02)02487-7. [DOI] [PubMed] [Google Scholar]
- 46.Muresan Z., Paul D.L., Goodenough D.A. Occludin 1B, a variant of the tight junction protein occludin. Mol. Biol. Cell. 2000;11:627–634. doi: 10.1091/mbc.11.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y., Merzdorf C., Paul D.L., Goodenough D.A. COOH terminus of occludin is required for tight junction barrier function in early Xenopus embryos. J. Cell Biol. 1997;138:891–899. doi: 10.1083/jcb.138.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saitou M., Fujimoto K., Doi Y., Itoh M., Fujimoto T., Furuse M., Takano H., Noda T., Tsukita S. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J. Cell Biol. 1998;141:397–408. doi: 10.1083/jcb.141.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Förster C. Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 2008;130:55–70. doi: 10.1007/s00418-008-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Furuse M., Sasaki H., Fujimoto K., Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 1998;143:391–401. doi: 10.1083/jcb.143.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balda M.S., Whitney J.A., Flores C., González S., Cereijido M., Matter K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J. Cell Biol. 1996;134:1031–1049. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rao R. Occludin phosphorylation in regulation of epithelial tight junctions. Ann. N Y Acad. Sci. 2009;1165:62–68. doi: 10.1111/j.1749-6632.2009.04054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aaku-Saraste E., Hellwig A., Huttner W.B. Loss of occludin and functional tight junctions, but not ZO-1, during neural tube closure—remodeling of the neuroepithelium prior to neurogenesis. Dev. Biol. 1996;180:664–679. doi: 10.1006/dbio.1996.0336. [DOI] [PubMed] [Google Scholar]
- 54.Hay E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. (Basel) 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 55.Ballabh P., Hu F., Kumarasiri M., Braun A., Nedergaard M. Development of tight junction molecules in blood vessels of germinal matrix, cerebral cortex, and white matter. Pediatr. Res. 2005;58:791–798. doi: 10.1203/01.PDR.0000180535.14093.FB. [DOI] [PubMed] [Google Scholar]
- 56.Gow A., Southwood C.M., Li J.S., Pariali M., Riordan G.P., Brodie S.E., Danias J., Bronstein J.M., Kachar B., Lazzarini R.A. CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell. 1999;99:649–659. doi: 10.1016/s0092-8674(00)81553-6. [DOI] [PubMed] [Google Scholar]
- 57.Nitta T., Hata M., Gotoh S., Seo Y., Sasaki H., Hashimoto N., Furuse M., Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abbott N.J., Patabendige A.A., Dolman D.E., Yusof S.R., Begley D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 59.Scholz M., Cinatl J., Schädel-Höpfner M., Windolf J. Neutrophils and the blood-brain barrier dysfunction after trauma. Med. Res. Rev. 2007;27:401–416. doi: 10.1002/med.20064. [DOI] [PubMed] [Google Scholar]
- 60.Virgintino D., Girolamo F., Errede M., Capobianco C., Robertson D., Stallcup W.B., Perris R., Roncali L. An intimate interplay between precocious, migrating pericytes and endothelial cells governs human fetal brain angiogenesis. Angiogenesis. 2007;10:35–45. doi: 10.1007/s10456-006-9061-x. [DOI] [PubMed] [Google Scholar]
- 61.Collett G.D., Canfield A.E. Angiogenesis and pericytes in the initiation of ectopic calcification. Circ. Res. 2005;96:930–938. doi: 10.1161/01.RES.0000163634.51301.0d. [DOI] [PubMed] [Google Scholar]
- 62.des Portes V., Pinard J.M., Billuart P., Vinet M.C., Koulakoff A., Carrié A., Gelot A., Dupuis E., Motte J., Berwald-Netter Y. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 63.Gleeson J.G., Allen K.M., Fox J.W., Lamperti E.D., Berkovic S., Scheffer I., Cooper E.C., Dobyns W.B., Minnerath S.R., Ross M.E., Walsh C.A. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 64.Jaglin X.H., Poirier K., Saillour Y., Buhler E., Tian G., Bahi-Buisson N., Fallet-Bianco C., Phan-Dinh-Tuy F., Kong X.P., Bomont P. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet. 2009;41:746–752. doi: 10.1038/ng.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keays D.A., Tian G., Poirier K., Huang G.J., Siebold C., Cleak J., Oliver P.L., Fray M., Harvey R.J., Molnár Z. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128:45–57. doi: 10.1016/j.cell.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ivanov A.I., McCall I.C., Babbin B., Samarin S.N., Nusrat A., Parkos C.A. Microtubules regulate disassembly of epithelial apical junctions. BMC Cell Biol. 2006;7:12. doi: 10.1186/1471-2121-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shultz T., Shmuel M., Hyman T., Altschuler Y. Beta-tubulin cofactor D and ARL2 take part in apical junctional complex disassembly and abrogate epithelial structure. FASEB J. 2008;22:168–182. doi: 10.1096/fj.06-7786com. [DOI] [PubMed] [Google Scholar]
- 68.Subramanian V.S., Marchant J.S., Ye D., Ma T.Y., Said H.M. Tight junction targeting and intracellular trafficking of occludin in polarized epithelial cells. Am. J. Physiol. Cell Physiol. 2007;293:C1717–C1726. doi: 10.1152/ajpcell.00309.2007. [DOI] [PubMed] [Google Scholar]
- 69.Du D., Xu F., Yu L., Zhang C., Lu X., Yuan H., Huang Q., Zhang F., Bao H., Jia L. The tight junction protein, occludin, regulates the directional migration of epithelial cells. Dev. Cell. 2010;18:52–63. doi: 10.1016/j.devcel.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 70.Shaffer L.G., Slovak M.L., Campbell L.J. S Karger; Basel: 2009. An International System for Human Cytogenetic Nomenclature. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.