

Abstract
Polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract (PHARC) is a neurodegenerative disease marked by early-onset cataract and hearing loss, retinitis pigmentosa, and involvement of both the central and peripheral nervous systems, including demyelinating sensorimotor polyneuropathy and cerebellar ataxia. Previously, we mapped this Refsum-like disorder to a 16 Mb region on chromosome 20. Here we report that mutations in the ABHD12 gene cause PHARC disease and we describe the clinical manifestations in a total of 19 patients from four different countries. The ABHD12 enzyme was recently shown to hydrolyze 2-arachidonoyl glycerol (2-AG), the main endocannabinoid lipid transmitter that acts on cannabinoid receptors CB1 and CB2. Our data therefore represent an example of an inherited disorder related to endocannabinoid metabolism. The endocannabinoid system is involved in a wide range of physiological processes including neurotransmission, mood, appetite, pain appreciation, addiction behavior, and inflammation, and several potential drugs targeting these pathways are in development for clinical applications. Our findings show that ABHD12 performs essential functions in both the central and peripheral nervous systems and the eye. Any future drug-mediated interference with this enzyme should consider the potential risk of long-term adverse effects.
Main Text
Inherited neurodegenerative diseases affecting both the peripheral and central nervous systems and the eye can be caused by a variety of metabolic disturbances. Mitochondrial dysfunction is a potent cause,1,2 arising either from mutation in the mitochondrial genome—e.g., neuropathy, ataxia, retinitis pigmentosa (NARP, MIM 551500) and Kearns-Sayre syndrome (ophthalmoplegia, retinal pigmentation, ataxia, and frequently peripheral neuropathy, MIM 530000)—or from a mutated nuclear gene. Friedreich ataxia (MIM 229300) and POLG-related diseases (MIM 174763) are examples of the latter. Defects involving peroxisomal metabolism, such as Refsum disease (MIM 266500) and alpha-methylacyl-CoA racemase (AMACR; MIM 604489) deficiency, also give rise to similar phenotypes.3
Recently, in a Norwegian family we described a progressive, autosomal-recessive, neurodegenerative disease that we ascertained initially as a phenocopy for Refsum disease (Figures 1A–1E). We named the disorder polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract, or PHARC4 (MIM 612674). The disease is slowly progressive, with recognition of the first symptoms typically in the late teens. Although the condition has similarities to Refsum disease, patients do not have anosmia and both phytanic acid levels and peroxisomal function are normal. We mapped the disease to a 16 Mb region on chromosome 20.4 Subsequently, additional affected individuals in four countries were identified, and we used homozygosity mapping to identify candidate regions for the mutated gene, followed by sequencing of candidate genes.
Figure 1.
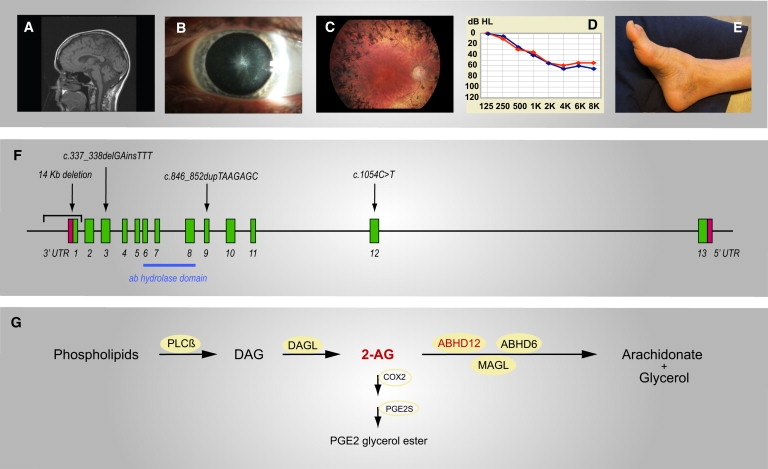
Phenotype and Genotype of PHARC Patients with Genetic Disruption of the 2-AG-Hydrolyzing Enzyme ABHD12
(A–E) Main symptoms in PHARC patients are shown. A summary of the symptoms and findings in each case is given in Table 1.
(A) MRI scan of an American female aged 50 (case 7.1) showing cerebellar atrophy.
(B) Star-shaped cataract of the posterior pole of the lens in a Norwegian male aged 24 (case 4.1).
(C) Fundus of a Norwegian male aged 56 (case 1.2) showing bone-spicule-shaped pigment deposits in the mid-periphery, pallor of the optic disc, attenuation of retinal vessels, and maculopathy.
(D) Audiogram of a Norwegian male aged 16 (case 5.1) showing sensorineural hearing loss of both right (red curve) and left (blue curve) ear, around 60 dB in the higher frequencies.
(E) Signs of peripheral neuropathy with pes cavus and hammertoes in a Norwegian male aged 56 (case 1.2).
(F) The ABHD12 gene is located 25,223,379–25,319,477 bp from pter on the reverse strand of chromosome 20 (NCBI build 36.3). Two isoforms containing the α/β-hydrolase domain have been identified, differing only in the last exon. The positions of the homozygous mutations found in the families from the Emiraties (14 Kb deletion), Norway (c.337_338 delGAinsTTT), Algeria (c.846_852 dupTAAGAGC), and USA (c.1054C>T) are indicated.
(G) 2-AG is formed nearly exclusively by the hydrolysis of diacylglycerol (DAG), catalyzed by DAG lipases (α or β). The main pathway for formation of DAG from phospholipids is catalyzed by phospholipase Cß (PLCß). Several enzymes are responsible for the breakdown of 2-AG to arachidonate and glycerol. Although MAGL is responsible for 85% of the 2-AG hydrolysis in the mouse brain, ABHD12 and ABHD6 may be important for hydrolysis in specific cell types and/or cellular compartments. 2-AG is also a substrate for the inducible enzyme cyclooxygenase-2 (COX2), which is involved in neuroinflammation. COX2 converts 2-AG to the corresponding hydroperoxy derivative, which is further metabolized to prostaglandin E2 glycerol ester by prostaglandin E2 glycerol ester synthase (PGE2S).
For the present study, DNA was obtained from 19 persons affected with PHARC disease and from healthy siblings and parents. The patients (10 females and 9 males) had a mean age of 32.5 years (range 6–62 years) and originated from Norway (n = 8), Algeria (n = 7), the United Arab Emirates (n = 3), and the USA (n = 1) (Table 1). In the previously published Norwegian family, individuals 1.1 and 1.2 are siblings and 1.3 is their third cousin. There are two affected siblings in families 2, 8, 9, and 10, and three affected in family 6. The adults gave informed consent to the investigation and publication of the results. The healthy individuals were not subject to clinical investigation, whereas the affected individuals have all been examined by neurologists, ophthalmologists, and otologists (Table 1). The study was approved of by the Regional Ethics Committee of Western Norway and by the local ethics committees of the University Hospitals of Bonn, Constantine, and Algiers.
Table 1.
Clinical Findings and Results of Investigations in the 19 Patients with PHARC Disease
| Family/Case | Age (yr) and Sex | Sensory and Motor Neuropathy | Neurography and EMG | Sensorineural Hearing Loss | Ataxia | MR/CT of Brain | Pyramidal Tract Signs | Retinitis Pigmentosa | ERG | Cataract |
|---|---|---|---|---|---|---|---|---|---|---|
| Norway mutation: c.337_338delGAinsTTT [p.Asp113PhefsX15] | ||||||||||
| 1.1 | 62 F | 38 years; pes cavus; sensory loss; absent ankle reflexes | Demyelinating polyneuropathy |
Twenties | No | Normal | No | 38 years | Rod-cone dystrophy | 28 years |
| 1.2 | 56 M | 37 years; pes cavus from childhood | Demyelinating polyneuropathy |
Thirties | 37 years; gait ataxia | Normal | Extensor plantar response at lower limbs; spasticity; hyperreflexia | 37 years | Rod-cone dystrophy | 37 years |
| 1.3 | 46 M | 38 years; no pes cavus; sensory loss distally | Demyelinating polyneuropathy |
From childhood | 43 years; gait ataxia; upper limb intention tremor | Cerebellar atrophy | Extensor plantar response at lower limbs; spasticity; hyperreflexia | 46 years | Rod-cone dystrophy | 25 years |
| 2.1 | 58 M | 51 years; pes cavus; sensory loss; reduced tendon reflexes | Demyelinating/axonal polyneuropathy |
Twenties | No | Cerebellar atrophy | Extensor plantar response at lower limbs | 35 years | Rod-cone dystrophy | 26 years |
| 2.2 | 54 F | 53 years; pes cavus; normal sensibility; reduced tendon reflexes | ND | Twenties | No | ND | No | 25 years | Flat | 25 years |
| 3.1 | 36 F | Pes cavus; normal sensibility; reduced tendon reflexes in lower limbs | Demyelinating polyneuropathy |
Deaf by the age of 10 | Yes | Atrophy of vermis and medulla oblongata | Extensor plantar response at right side; spasticity | 36 years | Rod-cone dystrophy | 32 years |
| 4.1 | 24 M | Pec cavus; hammertoes; reduced tendon reflexes in upper and lower limbs | Demyelinating polyneuropathy |
Late in teens | No | Slight ventricular assymmetry. No cerebellar atrophy |
Indifferent plantar response | No | Normal | 15 years |
| 5.1 | 16 M | Pes cavus; reduced sensibility; reduced tendon reflexes in upper limbs, absent in lower limbs | Demyelinating polyneuropathy |
13 years | No | Normal | No | No | Normal | 16 years (slight) |
| The Emirates mutation: 14 Kb deletion removing exon 1 | ||||||||||
| 6.1 | 24 M | Pec cavus from childhood; absent tendon reflexes | Abnormal | Deaf by the age of 14 | Mild | Normal | Indifferent plantar response | Twenties | ND | 15 years |
| 6.2 | 20 M | Pes cavus from age 4; absent tendon reflexes | Demyelinating polyneuropathy |
6 years | 2 years; gait, limb, and speech ataxia; wheelchair-bound from age 10 |
Cerebellar atrophy (age 3) | Extensor plantar response | Yes | ND | Yes |
| 6.3 | 6 F | Absent tendon reflexes | ND | Yes | Speech and limb | Cerebellar atrophy | Indifferent plantar response | No | ND | Yes |
| USA mutation: c.1054C>T [p.Arg352X] | ||||||||||
| 7.1 | 50 F | 34 years; pes cavus; hammertoes; sensibility slightly reduced | Abnormal | 17 years | 18 years; dysarthria; gait ataxia; jerky eye movements; tremor in hands | Cerebellar atrophy Increased signal in periventricular white matter. | Flexor plantar response; spasticity; preserved reflexes | Twenties | ND | 22 years |
| Algeria mutation: c.846_852dupTAAGAGC [p.His285fsX1] | ||||||||||
| 8.1 | 11 M | Absent tendon reflexes and moderate muscle weakness of lower limbs; normal sensibility | ND | No | 3-4 years; limb and gait ataxia; horizontal nystagmus; dysarthria; dysmetria upper and lower limbs; delayed walking at 15 month; action and intention tremor | Cerebellar atrophy | Extensor plantar response at lower limbs | No | ND | No |
| 8.2 | 10 F | Absent tendon reflexes of lower limbs; normal sensibility | ND | No | 4–5 years; gait ataxia | Vermian atrophy | Extensor plantar response at lower limbs | No | ND | No |
| 9.1 | 44 M | Pes cavus; sensory loss; absent tendon reflexes at lower limbs; scoliosis | Demyelinating polyneuropathy |
Yes | 7–10 years; gait and limb ataxia; cerebellar dysarthria; dysmetria at upper limbs with adiadocokinesia; head titubation | Vermian atrophy | Extensor plantar response at lower limbs; macroglossia | amblyopia | ND | |
| 9.2 | 26 F | Pes cavus; sensory loss; reduced tendon reflexes at upper limbs, and absent at lower limbs | Severe demyelinating polyneuropathy |
Deaf | 4–9 years; gait and limb ataxia; horizontal nystagmus; moderate dysarthria; dysmetria at upper and lower limbs | Vermian atrophy | Extensor plantar response at lower limbs; tongue fasciculations | Yes | ND | Yes |
| 10.1 | 26 F | Pes cavus; sensory loss; absent tendon reflexes | Severe demyelinating polyneuropathy on nerve biopsy |
6 years | 6–12 years; gait and limb ataxia |
Normal | Indifferent plantar response | No | ND | No |
| 10.2 | 19 F | 12 years; pes cavus; sensory loss; absent tendon reflexes at upper and lower limbs | ND | No | ND | ND | ||||
| 11.1 | 32 F | Pes cavus; sensory loss and absent tendon reflexes at lower limbs | Axonal polyneuropathy | Yes | 16–20 years; gait ataxia; dysarthria; dysmetria at upper limbs | Cerebellar atrophy | Extensor plantar response at lower limbs | Decreased visual acuity and amblyopia | ND | No |
Data on patients from four different countries (11 families) are shown. All individuals in one family are siblings, except for 1.3, who is the third cousin of 1.1 and 1.2. All adult patients have polyneuropathy of demyelinating type and sensorineural hearing loss (three patients are deaf), and nearly all adult patients have developed cataracts. Retinitis pigmentosa is typically recognized in the twenties or thirties. Ataxia is present in about half of the patients, with cerebellar atrophy and pyramidal tract signs like spasticity and extensor plantar response. The onset of ataxia is highly variable, starting particularly early in the families from the Emirates and Algeria.
From the same region as the original Norwegian family (family 1, Table 1),4 we ascertained a further five, apparently unrelated, patients (including a brother and sister, family 2) with suspected PHARC disease (family 2-5, Table 1). Homozygosity mapping was performed with GeneChip 250K NspI arrays (GEO accession number GSE23151). The data were exported and treated for further analysis by the programs GTYPE and Progeny Lab. Regions of homozygosity were identified with the PLINK program5. All eight Norwegian patients from five families were homozygous for overlapping parts of the previously published 16 Mb region on chromosome 20 (Figure S1, available online), indicating distant relationship. The inclusion of these five additional patients enabled us to refine the candidate region to approximately 6.4 Mb (23,553,833–29,936,849 bp from pter, NCBI build 36.3). Twenty-three of approximately 60 genes in this region were sequenced, and a homozygous indel mutation in exon 3 in the ABHD12 gene (c.337_338 delGAinsTTT; Figure 1F, Figure S2) was identified in all eight patients. The reference sequence for ABHD12 was NM_001042472.1. This frameshift mutation predicts the replacement of an asparagine at codon 113 with phenylalanine leading to a downstream premature stop codon (p.Asp113PhefsX15). The mutation segregated fully with the disease in these families. We screened 190 local healthy blood donors and found two heterozygous carriers of this mutation, corresponding to a disease incidence of approximately 1/36,000 in this population. This indicates that the frequency of PHARC in Western Norway is comparable to, or may be even higher than, relevant differential diagnoses like Friedreich ataxia and Refsum disease.
Concurrent mapping studies in one family from the United Arab Emirates and four families from Algeria were performed with Genechip 10K XbaI arrays followed by analysis on selected individuals with the GeneChip 6.0 array (Affymetrix, Santa Clara, USA). Regions of homozygosity were identified with the HomoSNP software (Figure S3). These patients, initially diagnosed with recessive ataxia, defined a 5.5 Mb linkage interval in the 20p11.21-q12 region on chromosome 20 (24,393,550–29,940,293 bp from pter, NCBI build 36.3, Figure S1). Twelve of the 29 genes of this region were sequenced, and a 14 Kb deletion (g.25,312,257_25,326,263 del14007insGG, NCBI Ref.Seq: NC_000020.10) in ABHD12, encompassing the promoter region and exon 1 of the gene (Figure 1F, Figures S4A–S4C), was identified in the family from the Emirates. No copy-number variations in this region have been reported to the Database of Genomic Variants (hg 18). The seven patients in the four Algerian families were homozygous for a 7 bp duplication in exon 9 (c.846_852 dupTAAGAGC) in ABHD12 (Figure S2), which directly replaces the histidine residue at codon 285 with a stop codon (p.His285fsX1). Also in these families the mutation segregated fully with the disease. Finally, a patient from the USA of French-Canadian heritage with suspected PHARC disease was found to be homozygous for a nonsense mutation (c.1054C>T) in exon 12 in ABHD12 (Figure 1F, Figures S1 and S2), leading to a predicted stop codon in position 352 in the protein (p.Arg352X). The finding of four different deleterious ABHD12 mutations in a total of 19 patients with PHARC disease from four countries clearly supports a causal genotype-phenotype relationship.
The addition of several new families requires refinement of our earlier clinical description.4 The essential clinical features are summarized in Figures 1A–1E and Table 1. PHARC in the Norwegian patients, and in the single American patient, appears to be a slowly progressive disease with recognition of the first symptoms typically in the teens. Cataracts, hearing loss, and a predominantly demyelinating peripheral neuropathy are present in all adult patients (Table 1), whereas the presence and extent of ataxia is variable. Retinitis pigmentosa typically presents in young adult life (twenties or thirties), and electroretinograms in most patients show a rod-cone dysfunction. The disorder in families from Algeria and the Emirates shows an earlier onset of ataxia that has both central and peripheral characteristics (Table 1). No evidence of behavioral disturbances or abnormalities related to appetite was detected in our adult patients. Cerebral cortical function appears to be spared, with only one patient having mental retardation (case 9.1) and another epilepsy (case 7.1, myoclonic seizures). Adult heterozygous carriers of ABHD12 mutations do not have an obvious phenotype, implying that their residual enzyme activity is sufficient to avoid clinical symptoms.
Each of the four different ABHD12 mutations is interpreted as a null mutation that would either abolish or severely reduce the activity of the encoding enzyme, α/β-hydrolase 12 (ABHD12). PHARC may, therefore, be considered a human ABHD12 knockout model. The question also arises whether less detrimental mutations may cause various incomplete phenotypes. The serious and progressive disease seen in our patients suggests that ABHD12 performs an essential function in the peripheral and central nervous systems and in the eye. This is supported by the high expression of ABHD12 in the brain, with a striking enrichment in microglia (Figure 2), as shown by our replotting of data from GNF Mouse Gene Atlas V3. Expression is also high in macrophages. Currently, the only known substrate for ABHD12 is the main endocannabinoid 2-arachidonoyl glycerol (2-AG) (Figure 1G). This compound has important functions in synaptic plasticity6,7 and neuroinflammation.8,9 In acute ischemia and/or excitotoxicity, 2-AG appears to have neuroprotective properties,9–11 but the effects of long-term increased levels of this metabolite have not been investigated.
Figure 2.
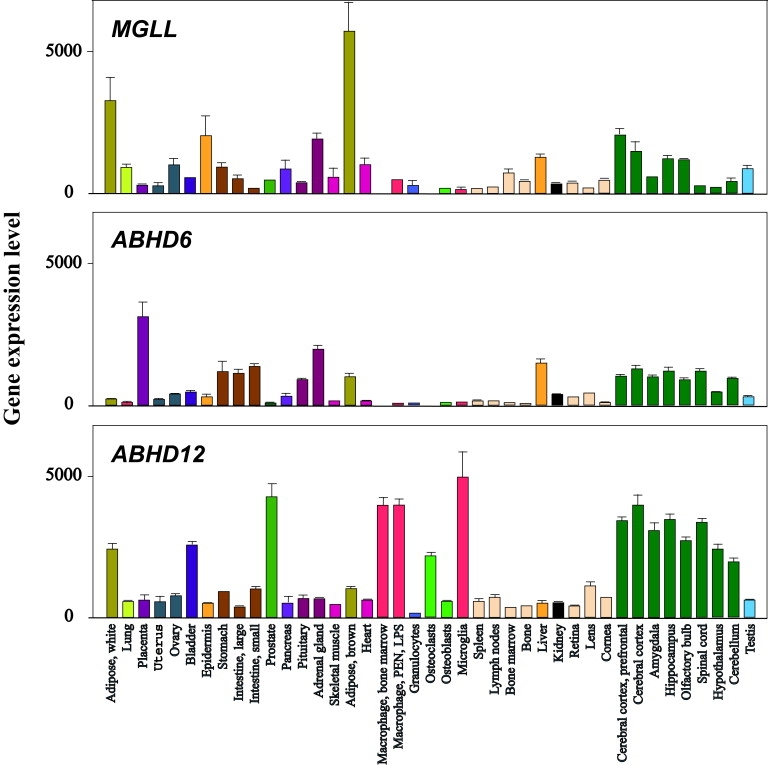
Gene Expression of ABHD12, ABHD6, and MGLL in Mouse Tissues
This is a replot of a subset of the GNF Mouse GeneAtlas V3 data, provided by Lattin et al.23 The data are published online in the BioGPS database under the alias GeneAtlas MOE430, and the NCBI GEO accession number is GSE10246. There is high expression of ABHD12, ABHD6, and MGLL (encoding MAGL) in different brain tissues (dark green bars). The highest level of ABHD12 is found in microglia (red bar, lower panel), and the expression is also high in the related cell types macrophages (red bars) and osteoclasts (light green bar). There is scarce expression of both ABHD6 (mid panel) and MGLL (upper panel) in microglia, macrophages, and osteoclasts. Bars represent the mean of two biological replicates (RNA from two separate pools from independent mice), and error bars show standard error of the mean. Regarding eye tissue, however, bars are the mean of two technical replicates (RNA from the same pool was split for two amplifications).
The endocannabinoid signaling system is the focus of increasing scientific interest, in part because of the potential for developing novel therapeutic agents.11–13 The system is tightly regulated and appears to be important for many physiological processes including neurotransmission, pain appreciation, appetite, mood, addiction behavior, body temperature, and inflammation.11 Key players in these pathways are the G protein-coupled cannabinoid receptors CB1 and CB2 and their endogenous ligands, endocannabinoids, as well as enzymes that synthesize or hydrolyze these ligands.14 The most abundant endocannabinoid, 2-AG, (Figure 1G) is formed on demand from the membrane lipid diacylglycerol (by diacylglycerol lipase α or β).14 Endocannabinoids act locally as lipid transmitters and are rapidly cleared by hydrolysis. Interestingly, our patients did not show overt cannabinomimetic effects.
Several enzymes are involved in 2-AG hydrolysis15,16 (Figure 1G), and there is evidence that these enzymes are differentially expressed in various cell types17 and cellular compartments.7,16,17 In the mouse brain, monoacylglycerol lipase (MAGL) accounts for 85% of the hydrolase activity,11,17 with additional contributions from ABHD12 and α/β-hydrolase 6 (ABHD6).16 The apparent paradox of a purported minor role of ABHD12 in 2-AG hydrolysis versus the serious PHARC phenotype in the brain and eye suggests either that ABHD12 is of crucial importance only in certain cell types12 or that it is also acting on a hitherto unknown substrate other than 2-AG. The finding that microglial cells have a particularly high expression of ABHD12, but very low levels of MGLL (encoding MAGL) and ABHD6 (Figure 2), indicates that the former alternative of differential cellular expression exists. Moreover, microglia dysfunction is known to be involved in neurodegenerative diseases18 as well as in retinal dystrophies.19 Whether ABHD12 acts on more than one substrate is currently unknown, but many hydrolases have overlapping functions, including MAGL, which is involved in lipolysis20 as well as in hydrolyzing 2-AG.
Despite great interest in manipulating 2-AG hydrolysis in vivo,8,21 knockout animal models have not yet been developed, and only recently a blocker of MAGL with substantial effect in vivo was reported.22 Notwithstanding this, inhibition of endocannabinoid hydrolases, including ABHD12, has been suggested as a potential therapy for neurodegenerative diseases such as multiple sclerosis.21 However, the consequences of irreversible loss of ABHD12 function, as seen in our patients with PHARC, may serve as a cautionary reminder that any potential drug inhibiting this enzyme be thoroughly evaluated with respect to the potential risk of severe long-term adverse effects.
In conclusion, mutations in the ABHD12-gene causes PHARC, a disease with serious dysfunction of the central and peripheral nervous systems, as well as hearing loss and impaired vision. Our findings have implications for clinicians working with both children and adults and suggest disrupted endocannabinoid metabolism as a cause of neurodegenerative disease.
Acknowledgments
This work was supported by grants from Helse Vest (Western Norway Regional Health Authority, 911308, to P.K., T.F., H.B., V.M.S, and B.I.H.) and from the Agence Nationale pour la Recherche-Maladies Rares (ANR-05-MRAR-013-01, France, to M.K.). D.H.-B.B. was supported by the French association Connaître les Syndromes Cérébelleux. M.A. was supported by a BDI fellowship from the Centre National de la Recherche Scientifique (CNRS). We thank John Walker and Andrew Su for the kind permission to replot (Figure 2) gene expression data from GNF Mouse Gene Atlas V3. The technical assistance of Jorunn Skeie Bringsli, Guri Matre, Hilde Rusaas, Sigrid Erdal, Paal Borge, Christine Stansberg, Bård Kjersem, Christelle Thibault, Serge Vicaire, Jone Vignes, and Ingrid Bauer was highly appreciated. We thank the patients and their families for participating in this study.
Contributor Information
Torunn Fiskerstrand, Email: torunn.fiskerstrand@helse-bergen.no.
Michel Koenig, Email: mkoenig@igbmc.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BioGPS database, http://biogps.gnf.org
Database of Genomic Variants, http://projects.tcag.ca/variation/?source=hg18
NCBI Build 36.3, http://www.ncbi.nlm.nih.gov/mapview
NCBI Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Accession Numbers
Microarray data have been deposited in NCBI's Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE23151.
References
- 1.Tucker E.J., Compton A.G., Thorburn D.R. Recent advances in the genetics of mitochondrial encephalopathies. Curr. Neurol. Neurosci. Rep. 2010;10:277–285. doi: 10.1007/s11910-010-0112-8. [DOI] [PubMed] [Google Scholar]
- 2.Finsterer J. Mitochondrial ataxias. Can. J. Neurol. Sci. 2009;36:543–553. doi: 10.1017/s0317167100008027. [DOI] [PubMed] [Google Scholar]
- 3.Wanders R.J., Komen J.C. Peroxisomes, Refsum's disease and the alpha- and omega-oxidation of phytanic acid. Biochem. Soc. Trans. 2007;35:865–869. doi: 10.1042/BST0350865. [DOI] [PubMed] [Google Scholar]
- 4.Fiskerstrand T., Knappskog P., Majewski J., Wanders R.J., Boman H., Bindoff L.A. A novel Refsum-like disorder that maps to chromosome 20. Neurology. 2009;72:20–27. doi: 10.1212/01.wnl.0000333664.90605.23. [DOI] [PubMed] [Google Scholar]
- 5.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makara J.K., Mor M., Fegley D., Szabó S.I., Kathuria S., Astarita G., Duranti A., Tontini A., Tarzia G., Rivara S. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat. Neurosci. 2005;8:1139–1141. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- 7.Straiker A., Hu S.S., Long J.Z., Arnold A., Wager-Miller J., Cravatt B.F., Mackie K. Monoacylglycerol lipase limits the duration of endocannabinoid-mediated depolarization-induced suppression of excitation in autaptic hippocampal neurons. Mol. Pharmacol. 2009;76:1220–1227. doi: 10.1124/mol.109.059030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J., Chen C. Endocannabinoid 2-arachidonoylglycerol protects neurons by limiting COX-2 elevation. J. Biol. Chem. 2008;283:22601–22611. doi: 10.1074/jbc.M800524200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kreutz S., Koch M., Böttger C., Ghadban C., Korf H.W., Dehghani F. 2-Arachidonoylglycerol elicits neuroprotective effects on excitotoxically lesioned dentate gyrus granule cells via abnormal-cannabidiol-sensitive receptors on microglial cells. Glia. 2009;57:286–294. doi: 10.1002/glia.20756. [DOI] [PubMed] [Google Scholar]
- 10.Panikashvili D., Simeonidou C., Ben-Shabat S., Hanus L., Breuer A., Mechoulam R., Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- 11.Di Marzo V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 12.Marrs W., Stella N. 2-AG + 2 new players = forecast for therapeutic advances. Chem. Biol. 2007;14:1309–1311. doi: 10.1016/j.chembiol.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Kinsey S.G., Long J.Z., O'Neal S.T., Abdullah R.A., Poklis J.L., Boger D.L., Cravatt B.F., Lichtman A.H. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J. Pharmacol. Exp. Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J., Ueda N. Biology of endocannabinoid synthesis system. Prostaglandins Other Lipid Mediat. 2009;89:112–119. doi: 10.1016/j.prostaglandins.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Marrs W., Stella N. Measuring endocannabinoid hydrolysis: Refining our tools and understanding. AAPS J. 2009;11:307–311. doi: 10.1208/s12248-009-9109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blankman J.L., Simon G.M., Cravatt B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muccioli G.G., Xu C., Odah E., Cudaback E., Cisneros J.A., Lambert D.M., López Rodríguez M.L., Bajjalieh S., Stella N. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J. Neurosci. 2007;27:2883–2889. doi: 10.1523/JNEUROSCI.4830-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landreth G.E. Microglia in central nervous system diseases. J. Neuroimmune Pharmacol. 2009;4:369–370. doi: 10.1007/s11481-009-9173-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebert S., Weigelt K., Walczak Y., Drobnik W., Mauerer R., Hume D.A., Weber B.H., Langmann T. Docosahexaenoic acid attenuates microglial activation and delays early retinal degeneration. J. Neurochem. 2009;110:1863–1875. doi: 10.1111/j.1471-4159.2009.06286.x. [DOI] [PubMed] [Google Scholar]
- 20.Guzmán M. A new age for MAGL. Chem. Biol. 2010;17:4–6. doi: 10.1016/j.chembiol.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Stella N. Endocannabinoid signaling in microglial cells. Neuropharmacology. 2009;56(Suppl 1):244–253. doi: 10.1016/j.neuropharm.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long J.Z., Li W., Booker L., Burston J.J., Kinsey S.G., Schlosburg J.E., Pavón F.J., Serrano A.M., Selley D.E., Parsons L.H. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lattin J.E., Schroder K., Su A.I., Walker J.R., Zhang J., Wiltshire T., Saijo K., Glass C.K., Hume D.A., Kellie S., Sweet M.J. Expression analysis of G Protein-Coupled Receptors in mouse macrophages. Immunome Res. 2008;4:5. doi: 10.1186/1745-7580-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.