

Abstract
Sensenbrenner syndrome/cranioectodermal dysplasia (CED) is an autosomal-recessive disease that is characterized by craniosynostosis and ectodermal and skeletal abnormalities. We sequenced the exomes of two unrelated CED patients and identified compound heterozygous mutations in WDR35 as the cause of the disease in each of the two patients independently, showing that it is possible to find the causative gene by sequencing the exome of a single sporadic patient. With RT-PCR, we demonstrate that a splice-site mutation in exon 2 of WDR35 alters splicing of RNA on the affected allele, introducing a premature stop codon. WDR35 is homologous to TULP4 (from the Tubby superfamily) and has previously been characterized as an intraflagellar transport component, confirming that Sensenbrenner syndrome is a ciliary disorder.
Main Text
Cranioectodermal dysplasia (CED; MIM 218330), also known as Sensenbrenner syndrome, is an autosomal-recessive disease that is characterized by sagittal craniosynostosis and facial, ectodermal, and skeletal anomalies.1,2 A proportion of cases have nephronophthisis, hepatic fibrosis, retinitis pigmentosa, and brain anomalies.3 This phenotype shows remarkable overlap with the ciliopathies, a spectrum of disorders associated with dysfunction of the cilium, a microtubule-based organelle that protrudes from the membrane in many vertebrate cell types.4 Furthermore, it has recently been shown that defects in the ciliary gene IFT122 (intraflagellar transport 122; MIM 606045) can be a cause of CED.5
Here we report on two unrelated Sensenbrenner patients with remarkably similar phenotypes (Figure 1; Table 1). These cases were previously screened diagnostically by using Affymetrix 250k arrays. Because neither pathogenic copy-number variants nor large homozygous regions were identified, we decided to use a different approach to identify the cause of disease in these two patients, hereafter referred to as patients 1 and 2. The current study was approved by the Medical Ethics Committee of the Radboud University Nijmegen Medical Centre. Written informed consent to participate in the study was obtained for both patients (and all other ciliopathy patients described in this paper), as well as informed consent to publish clinical photos for patients 1 and 2.
Figure 1.
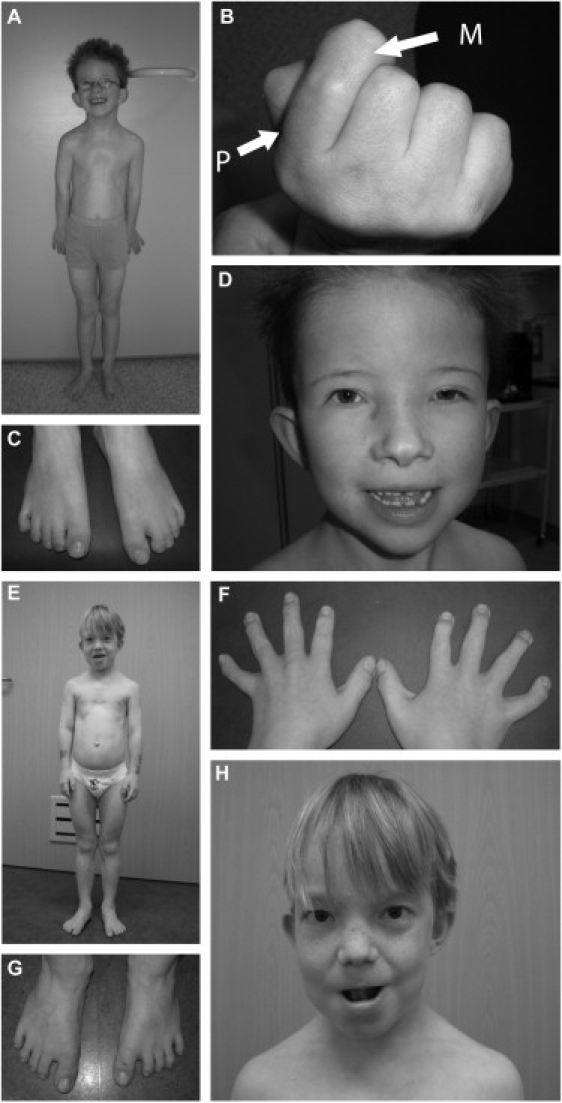
Two Patients with Sensenbrenner Syndrome for Whom Exome Sequencing Was Performed
(A) Patient 1: small thorax, pectus excavatum, rhizomelic shortening of limbs.
(B) Shortening of proximal second phalanx. The proximal phalanx is indicated by “P”; the middle phalanx is indicated by “M.”
(C) Syndactyly 2-3 right foot, 2-3-4 left foot.
(D) Hypertelorism, unilateral ptosis of left eye, low-set ears, everted lower lip.
(E) Patient 2: small thorax, pectus excavatum, rhizomelic shortening of limbs.
(F) Short, broad hands.
(G) Bilateral sandal gap between first and second toe.
(H) Hypertelorism, low-set simple ears, thin hair.
Table 1.
Clinical Details of Two Patients with Cranioectodermal Dysplasia
| Patient | 1 | 2 |
|---|---|---|
| Mutations DNA | c.1877A>G, c.25-2A>G | 1:c.2891delT, c.2623G>A |
| Mutations protein | p.E626G, p.I9TfsX7 | p.P964LfsX15, p.A875T |
| Age at diagnosis | 7 | 9 |
| Height | <2.5 standard deviations | <2.5 standard deviations |
| Dolichocephaly | + | + |
| Craniosynostosis | surgically corrected at age 1 | surgically corrected at age 1 |
| Frontal bossing | + | + |
| Macrocephaly | − | − |
| Sparse, fine hair | − | + |
| Narrow palpebral fissure | + | + |
| Telecanthus | + | + |
| Hypermetropia | + | − |
| Nystagmus | − | − |
| Ptosis | unilateral | − |
| Hypertelorism | + | + |
| Strabism | + | − |
| Low-implanted ears | + | + |
| Simple ears | + | + |
| Everted lower lip | + | + |
| Micrognathia | + | − |
| Widely spaced teeth | + | + |
| Hypoplastic teeth | + | + |
| Fused teeth | + | + |
| Short neck | + | + |
| Narrow thorax | + | + |
| Pectus excavatum | + | + |
| Short limbs | + | + |
| Brachydactyly | + | + |
| Webbing of fingers | + | + |
| Postaxial polydactyly | + | − |
| Restricted flexion of fingers | + | − |
| Syndactyly 2-3 (fourth) toe | + | − |
| Bilateral sandal gap | − | + |
| Joint laxity | + | + |
| Inguinal hernia (bilateral) | + | + |
| Renal disease | − | − |
| Hepatic disease | − | − |
| Recurrent lung infections | + | − |
| Intelligence | normal | normal |
| Behavior | happy, friendly | happy, friendly |
We applied a genome-wide approach and sequenced the exomes (targeting ∼18,000 genes) of both patients. We obtained 3.6 Gb and 3.4 Gb of mappable sequence data per patient by using a SureSelect human exome kit (Agilent, Santa Clara, CA, USA) in combination with one quarter of a SOLiD sequencing slide (Life Technologies, Carlsbad, CA, USA). Color space reads were mapped to the hg18 reference genome with SOLiD BioScope software version 1.0, which utilizes an iterative mapping approach. In total, 89% and 86% of bases originated from the targeted exome, resulting in a mean coverage of 67- and 59-fold (see Table S1 available online). Single-nucleotide variants were subsequently called by the DiBayes algorithm with a conservative call stringency. The DiBayes SNP caller requires at least two variant reads to call a SNP. We assumed a binomial distribution with probability 0.5 of sequencing the variant allele at a heterozygous position. At least ten reads are then required to obtain a 99% probability of having at least two reads containing the variant allele. More than 89% of the targeted exons were covered more than ten times. Small insertions and deletions were detected by using the SOLiD Small InDel Tool. Called SNP variants and indels were then combined and annotated by using a custom analysis pipeline.
On average, 12,736 genetic variants were identified per patient in the coding regions or the canonical dinucleotide of the splice sites, including 5,657 nonsynonymous changes (Table S2). A prioritization scheme was applied to identify the pathogenic mutation in each patient separately, similar to a recent study.6 We excluded known dbSNP130 variants as well as variants from our in-house variant database, reducing the number of candidates by more than 98%. The in-house database consists of data from in-house exome resequencing projects of patients with rare syndromes (548,103 variants), the 1000 Genomes Project, and published data from various studies7–9 (2,535,563 variants). For a recessive disease, it is possible that heterozygous variants found in healthy individuals have been reported as benign polymorphisms within dbSNP or our internal variant database. However, given the rare incidence of CED, it is almost impossible that heterozygous mutations occur at a reasonable frequency in the healthy population, and thus it is unlikely that they have been included in dbSNP.
Under the assumption of an autosomal-recessive disease model, we found three candidate genes with compound heterozygous variants in patient 1 (FLG, MFRP, and WDR35). The inheritance of the variants in the three candidate genes was determined by Sanger sequencing, showing that the two WDR35 and the two MFRP variants were inherited from different parents (Table S3). Based on evolutionary conservation score,10 both variants in WDR35 ranked at the top position among all variants of the three candidate genes (Table S3). Moreover, WDR35 was the only candidate with a ciliary function according to the Ciliary Proteome database11 (cutoff e value 30; 2,127 entries). In patient 2, we identified four candidate genes that harbored two or more variants. Sanger validation excluded two of these as candidate genes because the variants were inherited from a single parent. The remaining candidates, USH2A (MIM 608400) and WDR35, both had a putative ciliary function. The two conserved variants in USH2A were both inherited paternally, whereas a third nonconserved variant was not inherited paternally, which makes USH2A an unlikely candidate for CED. Furthermore, this patient had no signs of retinitis pigmentosa (MIM 608000) or Usher syndrome (MIM 276901). The WDR35 variants were inherited from both parents and affected base pairs with high evolutionary conservation (Table S3). In conclusion, we independently identified WDR35 as the most likely candidate disease gene in both patients.
In patient 1, we identified a canonical splice-site mutation 2 bp upstream of exon 2 (c.25-2A>G [p.I9TfsX7]) and a missense mutation in exon 17 (c.1877A>G [p.E626G]) (Figure 2; Figure S1). With RT-PCR, we demonstrated that splicing of WDR35 RNA (derived from Epstein-Barr virus cell lines) was indeed altered in patient 1 compared to an unrelated control individual (Figure 2). Sequencing of the bands revealed that the affected allele contained a 58 bp insertion that introduced a premature stop codon. The missense mutation in exon 17 was predicted to be “probably damaging” by PolyPhen.12 The mutated amino acid is highly conserved up to insects and nematodes (Figure S2). Because the C. elegans protein WDR35 ortholog (IFTA-1) is most distantly related to the human WDR35 protein (only 44.2% similar and 28.4% identical), the conservation of the glutamine provides a strong indication of the importance of this amino acid for WDR35/IFTA-1 function.
Figure 2.

Splice-Site Mutation in Patient 1
(A) Gene and protein structure of WDR35. WD domains are indicated by triangles; the box indicates a low-complexity region.
(B) Sequencing reads showing the heterozygous splice-site mutation at the splice-acceptor site of exon 2 as well as a nearby exonic polymorphism (rs1060742).
(C) Maternal inheritance of the splice-site mutation in patient 1 shown by Sanger sequencing.
(D) Effect of the splice-site mutation on the RNA shows two different products of equal intensity in lane 1 (523 bp and 465 bp for the upper and lower band, respectively). Lane 2 shows the product of an unrelated control (465 bp).
(E) Sequence of the two WDR35 RT-PCR products from Figure 2D, showing that the polymorphism (rs1060742) is predominantly present in the normal spliced product as a C (Figure 2D, lane 1, arrow a), whereas this is a T in the alternatively spliced product (Figure 2D, lane 1, arrow b). The sequence of the RT-PCR product of an unrelated individual with the same heterozygous SNP (rs1060742) is shown as a control (Figure 2D, lane 3, arrow c).
In patient 2, a deletion of a C nucleotide in exon 25 predicts a frameshift and a premature stop (1:c.2891 delC [p.P964LfsX15]) (Figure S1). On the second allele, a substitution in exon 23 (c.2623G>A [p.A875T]) leads to the change of a highly conserved alanine to a threonine (Figures S1 and S2). The amino acid substitution was classified as “potentially damaging” by PolyPhen. It is remarkable that the variants in both patients are a combination of a missense and a truncating mutation. In ciliary diseases, phenotypical severity is often determined by the combination of missense and nonsense mutations.13 This phenomenon could be used in the prioritization of variants from exome sequencing of other (ciliary) diseases, which in this case would have immediately identified WDR35.
None of the four identified variants in WDR35 were detected in 210 control alleles, indicating that the identified variants are uncommon in the Dutch population from which the patients originated, further supporting the thought that the WDR35 variants are pathogenic. Furthermore, complete loss of WDR35 function leads to a severe short-rib polydactyly syndrome, another ciliopathy (P.J. Lockhart, personal communication).
WDR35 contains 28 coding exons that encode at least four known protein isoforms (as determined in Ensembl). The WDR35 protein, which is part of the WD-repeat protein family, was first characterized in the green alga Chlamydomonas reinhardtii14. The Chlamydomonas ortholog (IFT121) is part of the intraflagellar transport complex A, together with at least five other proteins. One of them, IFT122/WDR10, also contains N-terminal WD repeats and has recently been shown to be involved in CED as well.5 Like other IFT-A proteins, WDR35 is instrumental for retrograde IFT (from the ciliary tip to the basal body) in mice.15 Studies in Drosophila and C. elegans also have demonstrated that these species’ WDR35 orthologs (Oseg4 and IFTA-1, respectively) localize to the cilium and act in IFT.16 It is thus most likely that cilium dysfunction due to disrupted (retrograde) IFT is underlying the CED phenotype of patients with mutations in WDR35.
To evaluate whether mutations in WDR35 are a common cause of CED, we performed mutation analysis in six additional CED patients. These patients presented with additional clinical phenotypes and did not show the striking phenotypic similarity observed between patients 1 and 2. In three of these patients, mutations in IFT122 had been excluded, whereas in two other patients, linkage regions were identified that did not contain IFT122 (or IFT121). We did not find any causative mutations in WDR35 in these patients. Thus, only 25% of our cohort (2 out of 8 CED patients) carried mutations in WDR35, further confirming that CED is a genetically heterogeneous disorder similar to other ciliopathies.5,13
Our results are consistent with the previously demonstrated importance of IFT in bone development; mutations in IFT80 (MIM 611177) and DYNC2H1 (MIM 603297) have been associated with Jeune syndrome (MIM 208500), a disorder with significant clinical overlap with CED.17,18 Based on this and the fact that many ciliary disease genes are associated with multiple ciliopathy syndromes,13 we screened WDR35 for mutations in 15 Jeune syndrome patients. No mutations were found in these patients, indicating that mutations in WDR35 are not a major cause of Jeune syndrome. So far, Sensenbrenner syndrome is the only ciliopathy that includes a craniosynostosis phenotype. Because conditional IFT knockout mouse models indicate that Sonic and Indian Hedgehog signaling is regulated by cilia during skeletal development, we hypothesize that disrupted ciliary Hedgehog signaling due to disrupted IFT is involved in the skeletal features, including craniosynostosis, in our CED patients. Wdr35 knockout mice do indeed display Hedgehog defects during limb development.15
By using several bioinformatic tools (i.e., the phylogenetics tree database TreeFam, Gene Tree from Ensembl, and NCBI BLAST), we found that TULP4 (a member of the Tubby superfamily) is homologous to WDR35. Although little is known about the function of TULP4, it is of interest that the Tubby family member Tulp3 is known to modulate Shh signaling during early embryonic development of the mouse,19 like WDR3515 and other IFT proteins. Moreover, because the phenotype of Tulp3 knockout mice as well as other mutants from the Tubby family shares features with the phenotypes of CED and other ciliopathies, we conclude that TULP4 is an excellent candidate gene for such disorders.19–23 Together, these findings also suggest that the IFT proteins and some members of the Tubby family of proteins, in particular TULP4, are functionally related.
In our study, the availability of two independent cases with a strikingly similar phenotype was obviously very useful for identifying the causative gene. For both individual patients, we identified only a small number of candidate disease genes. In each of the two cases, this was further reduced to a single candidate (WDR35) by segregation analysis and additional evidence on evolutionary conservation and the ciliome database. The finding that WDR35 is mutated in a family with short-rib polydactyly syndrome (MIM 263510) confirms that WDR35 is indeed involved in “skeletal” ciliopathies (P.J. Lockhart, personal communication). Interestingly, both patients presented with sagittal craniosynostosis, a birth defect characterized by premature closure of the skull sutures that occurs in 1.5 per 10.000 newborns. Familial recurrences and occasional concordant twins indicate the presence of genetic factors underlying sagittal synostosis, but these remain largely unknown. The involvement of WDR35 in this phenotype might provide insight into the underlying biology of sagittal craniosynostosis.
It should be noted that theoretically we could have missed additional variants that are relevant for the disease. Only high-quality whole-genome sequencing could have fully excluded disease-related genomic variants or modifiers. However, we believe that the combined genetic and functional data undoubtedly show the involvement of WDR35 in CED. In summary, our data indicate that for a rare recessive condition, it is possible to find the causative gene by sequencing the exome of a single sporadic patient. This is consistent with the results of a recent study of Perrault syndrome (MIM 233400) that also found the causative gene by sequencing the exome of a single affected individual.24
Acknowledgments
We thank the Sensenbrenner families for their participation. We also thank E.M. Bongers and P.L. Beales for supplying the additional CED patients, B.C. Hamel for the Jeune syndrome patients, and N. Wieskamp for bioinformatics support. We thank P. Mill, I. Jackson, and P.J. Lockhart for sharing their unpublished data with us. This research was supported by grants from the Dutch Kidney Foundation (KJPB09.009 to H.H.A.), the Netherlands Organization for Scientific Research (NWO/ZonMw Vidi 91786396 to R.R.), the Netherlands Organization for Health Research and Development (ZonMw 917-66-363 and 911-08-025 to J.A.V.), the European Union-funded TECHGENE project (Health-F5-2009-223143 to P.A. and J.A.V.), SYSCILIA (Health-F5-2010-241955 to R.R.), and the AnEUploidy project (LSHG-CT-2006-37627 to A.H., H.G.B., and J.A.V.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes project, http://www.1000genomes.org/
Ciliaproteome V3.0, http://v3.ciliaproteome.org/cgi-bin/index.php
Ensembl Gene Tree, http://www.ensembl.org/Homo_sapiens/Gene/Compara_Tree?g=ENSG00000118965
IGV browser, http://www.broadinstitute.org/igv
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
TreeFam, http://www.treefam.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Sensenbrenner J.A., Dorst J.P., Owens R.P. New syndrome of skeletal, dental and hair anomalies. Birth Defects Orig. Artic. Ser. 1975;11:372–379. [PubMed] [Google Scholar]
- 2.Levin L.S., Perrin J.C., Ose L., Dorst J.P., Miller J.D., McKusick V.A. A heritable syndrome of craniosynostosis, short thin hair, dental abnormalities, and short limbs: Cranioectodermal dysplasia. J. Pediatr. 1977;90:55–61. doi: 10.1016/s0022-3476(77)80764-6. [DOI] [PubMed] [Google Scholar]
- 3.Amar M.J., Sutphen R., Kousseff B.G. Expanded phenotype of cranioectodermal dysplasia (Sensenbrenner syndrome) Am. J. Med. Genet. 1997;70:349–352. doi: 10.1002/(sici)1096-8628(19970627)70:4<349::aid-ajmg3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 4.Baker K., Beales P.L. Making sense of cilia in disease: The human ciliopathies. Am. J. Med. Genet. C Semin. Med. Genet. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- 5.Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D.A., Emma F., Klingenberg C., Hennekam R.C., Torre G., Garshasbi M., Tzschach A. Cranioectodermal dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 2010;86:949–956. doi: 10.1016/j.ajhg.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoischen A., van Bon B.W., Gilissen C., Arts P., van Lier B., Steehouwer M., de Vries P., de Reuver R., Wieskamp N., Mortier G. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 7.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pushkarev D., Neff N.F., Quake S.R. Single-molecule sequencing of an individual human genome. Nat. Biotechnol. 2009;27:847–852. doi: 10.1038/nbt.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J., Wang W., Li R., Li Y., Tian G., Goodman L., Fan W., Zhang J., Li J., Zhang J. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gherman A., Davis E.E., Katsanis N. The ciliary proteome database: An integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 2006;38:961–962. doi: 10.1038/ng0906-961. [DOI] [PubMed] [Google Scholar]
- 12.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cardenas-Rodriguez M., Badano J.L. Ciliary biology: Understanding the cellular and genetic basis of human ciliopathies. Am. J. Med. Genet. C Semin. Med. Genet. 2009;151C:263–280. doi: 10.1002/ajmg.c.30227. [DOI] [PubMed] [Google Scholar]
- 14.Cole D.G. The intraflagellar transport machinery of Chlamydomonas reinhardtii. Traffic. 2003;4:435–442. doi: 10.1034/j.1600-0854.2003.t01-1-00103.x. [DOI] [PubMed] [Google Scholar]
- 15.Mill P., Hall E., Keighren M., Lawson K., Jackson I. Wdr35 is required for mammalian ciliogenesis and Hh responsiveness. Mech. Dev. 2009;126(Suppl. 1):S265. [Google Scholar]
- 16.Avidor-Reiss T., Maer A.M., Koundakjian E., Polyanovsky A., Keil T., Subramaniam S., Zuker C.S. Decoding cilia function: Defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- 17.Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 18.Dagoneau N., Goulet M., Geneviève D., Sznajer Y., Martinovic J., Smithson S., Huber C., Baujat G., Flori E., Tecco L. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 2009;84:706–711. doi: 10.1016/j.ajhg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cameron D.A., Pennimpede T., Petkovich M. Tulp3 is a critical repressor of mouse hedgehog signaling. Dev. Dyn. 2009;238:1140–1149. doi: 10.1002/dvdy.21926. [DOI] [PubMed] [Google Scholar]
- 20.Coleman D.L., Eicher E.M. Fat (fat) and tubby (tub): Two autosomal recessive mutations causing obesity syndromes in the mouse. J. Hered. 1990;81:424–427. doi: 10.1093/oxfordjournals.jhered.a111019. [DOI] [PubMed] [Google Scholar]
- 21.Ohlemiller K.K., Hughes R.M., Mosinger-Ogilvie J., Speck J.D., Grosof D.H., Silverman M.S. Cochlear and retinal degeneration in the tubby mouse. Neuroreport. 1995;6:845–849. doi: 10.1097/00001756-199504190-00005. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda S., Shiva N., Ikeda A., Smith R.S., Nusinowitz S., Yan G., Lin T.R., Chu S., Heckenlively J.R., North M.A. Retinal degeneration but not obesity is observed in null mutants of the tubby-like protein 1 gene. Hum. Mol. Genet. 2000;9:155–163. doi: 10.1093/hmg/9.2.155. [DOI] [PubMed] [Google Scholar]
- 23.Norman R.X., Ko H.W., Huang V., Eun C.M., Abler L.L., Zhang Z., Sun X., Eggenschwiler J.T. Tubby-like protein 3 (TULP3) regulates patterning in the mouse embryo through inhibition of Hedgehog signaling. Hum. Mol. Genet. 2009;18:1740–1754. doi: 10.1093/hmg/ddp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pierce S.B., Walsh T., Chisholm K.M., Lee M.K., Thornton A.M., Fiumara A., Opitz J.M., Levy-Lahad E., Klevit R.E., King M.C. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault syndrome. Am. J. Hum. Genet. 2010;87:282–288. doi: 10.1016/j.ajhg.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.