

Abstract
Breast cancer metastasis suppressor gene-1 (BRMS1) mRNA and protein expression are significantly decreased in non-small cell lung cancer (NSCLC) and this is a poor prognostic indicator. Given that the BRMS1 promoter region contains a promoter-associated CpG island (CGI) that encompasses the transcriptional start site, we hypothesized that decreased BRMS1 mRNA and protein levels in NSCLC was secondary to increased BRMS1 promoter methylation. Methylation-specific PCR (MSP) of the two known CGIs (−3477 to −2214 and −531 to +608) in the BRMS1 genome was performed in NSCLC cells. This demonstrated a robust increase in methylation of the promoter-associated CGI (−531 to +608) but not of the upstream CGI (−3477 to −2214). To experimentally verify that methylation contributes to BRMS1 transcriptional repression, we cloned the BRMS1 promoter region, including the promoter-associated CGI, into a luciferase reporter gene and found that BRMS1 promoter activity was dramatically inhibited under methylated conditions. We then assessed the BRMS1 methylation profile with MSP and bisulphite-sequencing PCR in human NSCLC adenocarcinoma (n = 20) and squamous cell carcinoma (n = 20) relative to adjacent non-cancerous bronchial epithelium. There was a significant increase in BRMS1 promoter methylation in all NSCLC specimens relative to non-cancerous tissues, with the most dramatic difference in squamous cell cancer histology. Subsequent immunostaining demonstrated that nuclear BRMS1 expression is reduced in lung cancer specimens compared to normal bronchial epithelium. The association between BRMS1 promoter methylation and specific clinical and histopathological variables was examined using a general linear model. Pathological tumour stage was associated with increased BRMS1 methylation in squamous cell cancers. These observations demonstrate that methylation of the promoter-associated CGI in BRMS1 results in its transcriptional repression, and highlight the potential clinical relevance of this methylation event with respect to NSCLC tumour histology and pathological stage.
Keywords: lung cancer, BRMS1, methylation, methylation-specific PCR, immunohistochemistry, pathological stage, squamous cell cancer
Introduction
Lung cancer remains a common cause of cancer-related deaths for men and women in the Western world [1]. Early stage, node-negative lung cancer that has not metastasized has a more favourable prognosis (50% 5-year survival) compared to advanced-stage disease with lymph nodal or other organ dissemination (21% and 3% 5-year survival, respectively) [1]. Therefore, understanding the mechanisms through which lung cancer metastasizes is important if new therapies are to be designed to better treat lung cancer patients.
Welch and colleagues initially identified and described breast cancer metastasis suppressor 1 (BRMS1), a gene shown to suppress metastasis in human breast cancer cell lines without affecting primary tumour growth [2], and whose reduced expression has been correlated with poor prognosis in breast cancer patients [3,4]. There are several proposed mechanisms of action for BRMS1 and its role in the regulation of tumour metastasis; these include suppression of NF-κB signalling via (a) inhibition of IκBα or (b) functioning as a co-repressor for the transcriptionally active subunit of NF-κB, RelA/p65 [5–7]. Other studies have shown that BRMS1 reduces phosphoinositide signalling [8] and plays a role in cell–cell communication [9–11]. A recent study in breast cancer showed that of the two CpG islands (CGI) in the BRMS1 genome, only the upstream CGI (−3477 to −2214) was methylated. Importantly, correlation with this CGI methylation and BRMS1 protein expression were noted, but no direct link to BRMS1 transcription was described, leaving open the question of the transcriptional significance of this methylation event [12]. Moreover, it is known that CGI methylation profiles are tissue-specific, suggesting that analysis of BRMS1 promoter methylation profiles in other tissues and cancers merits further investigation [13].
Until recently, limited data existed on the role of BRMS1 in non-small cell lung cancer (NSCLC). We have shown that BRMS1 mRNA and protein expression are decreased in human NSCLC specimens [6]. Moreover, we have demonstrated that BRMS1 does function as a metastasis suppressor in a mouse model of lung cancer, and that reduced NSCLC BRMS1 protein expression strongly correlated with a worse clinical prognosis [14]. Given these observations, we hypothesized that BRMS1 transcription is decreased in NSCLC through increased BRMS1 promoter methylation, specifically methylation of the promoter-associated CGI (−527 to +612), which encompasses the transcriptional start site and exon 1 in the BRMS1 promoter. We also sought to determine whether BRMS1 promoter methylation correlated with distinct NSCLC histologies and specific clinicopathological variables.
Methods
Cell culture, surgical specimens, and reagents
Human NSCLC cell lines (NCI-H157, NCI-A549, NCI-H1299), normal bronchial human bronchial epithelial cell line (NHBE) and human embryonic kidney cells (HEK 293T) [15] were obtained from the American Type Culture Collection (Manassas, VA, USA) and were grown as previously described [6]. Human NSCLC specimens and adjacent non-cancerous lung were obtained from our tissue bank of NSCLC specimens from patients undergoing surgery at the University of Virginia. Informed patient consent and Human Investigations Committee approval was obtained on all cases. The antibodies used in this study were BRMS1 (Abnova Corp., Taiwan) [6,14] and α-tubulin (Sigma Aldrich, St. Louis, MO, USA).
Western blotting analysis
Whole cell lysates were obtained and western blots were performed as previously described [6]. The primary antibodies were used at 1 : 1000 dilution and secondary antibodies (Promega, Madison, WI, USA) were used at 1 : 5000 dilution.
DNA extraction, sodium bisulphite modification, and bisulphite genomic sequencing
Genomic DNA was extracted using DNeasy Blood and Tissue Kit (Qiagen, Austin, TX, USA) and modified by sodium bisulphite (EZ DNA Methylation Kit, Zymo Research, Orange, CA, USA). The modified DNA was amplified by PCR (primers: forward, 5′-ATTAAATTGTTTAATTGTGAGTATTTT-3′; reverse, 5′-AAAACTAAAACCTCTAACCTCAC-3′) and cloned into pCR2.1 (TOPO TA Cloning Kit, Invitrogen, Carlsbad, CA, USA). The constructs were then sequenced (DNA Sciences Core, University of Virginia, Charlottesville, VA, USA).
Methylation-specific PCR (MSP)
Bisulphite-treated DNA was used as a template for PCR with primers designed using MethPrimer (www.urogen.org/methprimer) that were specific to the downstream CGI region (−527 to +612) of BRMS1. Quantitative MSPs were performed using iCycler real-time PCR (Bio-Rad iCycler IQ5, Hercules, CA, USA) with SYBR green detection dye (Invitrogen) at an annealing temperature of 57 °C. The internal control was measured using GAPDH. The methylation state of each gene was represented as relative methylation. Primers for the upstream CGI region (−3477 to −2214) were designed as previously described [12]; primers for the downstream CGI region (−527 to +612) of BRMS1 were: methylated region, (−43 from transcription start site, TSS) 5′-TTTTGATGACGTATACGGAAGTATC-3′ (forward) and (111 from TSS) 5′-GAAAACTAAAACCTCTAACCTCACG-3′ (reverse); unmethylated region, (−42 from TSS) 5′-TTTGATGATGTATATGG AAGTATTGA-3′ (forward) and (112 from TSS) 5′-CAAAAACTAAAACCTCTAACCTCACA-3′ (reverse). In some experiments, the PCR products were separated using 2% agarose gel electrophoresis.
Total RNA isolation and quantitative reverse transcriptase PCR (RT–PCR)
Total RNA isolation and quantitative RT–PCR was performed as previously described [6].
Plasmid construction and antibodies
Human BRMS1 promoter sequences were amplified by PCR from genomic DNA, using primers (forward) 5′-GAAGAAGATCTCGGGCTGGGCTGGGTAT-3′ and (reverse) 5′-CCCAAGCTTCACTGTTAGTATATTCA CTTTG-3′ (restriction sites are underlined) located respectively at positions −500 and +601 from the transcription start site. The amplified product was cloned into the BglII/HindIII (New England Biolabs, Ipswich, MA)-digested pGL3 basic reporter gene (Promega) to create the BRMS1–luc construct.
Transfection and luciferase assays
Plasmids and reporter genes were transiently transfected using Polyfect reagent (Qiagen) and luciferase reporter activity assays were performed as previously described [6]. CMV-β-galactosidase activities were analysed as controls for the efficacy of transfection.
In vitro methylation assay
The BRMS1–luc reporter plasmid (20 μg) was incubated with CpG methlytransferase M.SssI (20 U; New England Biolabs, Beverly, MA, USA) or buffer only, following the manufacturer’s instructions. The efficiency of CpG methylation was tested as follows: plasmid DNA (2 μg) was digested with restriction enzymes BglII/HindIII (New England Biolabs). The completeness of methylation was proven by the inhibition of the digestion with 5-methyl-cytosine-sensitive restriction enzyme BstUI (New England Biolabs).
Tissue microarray
Thirty-six cases of non-small cell lung carcinoma had formalin-fixed, paraffin-embedded tissue available for a tissue microarray. Haematoxylin and eosin (H&E)-stained slides were reviewed and areas of viable, well-preserved tumour were selected. Four 0.4 mm cores were taken and set into a single tissue array block (Beecher Instruments, Woodland, USA). Array blocks were incubated (37 °C) for 30 min. The blocks were cut with a microtome. Sections were reviewed and showed tumour in all cores. Control normal lung parenchyma was also used.
Immunohistochemistry
Immunohistochemistry (IHC) was performed using a DAKO Autostainer. Pressure/heat antibody retrieval was performed with DAKO TRS9 buffer. A DAKO dual endogenous enzyme block was used (10 min). Primary BRMS1 monoclonal antibody (1 : 200) was then applied (30 min). The detection agent (DAKO Envision Dual Link) was then applied (30 min). DAKO DAB+ substrate chromagen was used with a haematoxylin counterstain. Scoring was performed by a pathologist blinded to BRMS1 promoter methylation status and other clinicopathological variables, as previously described [14]. Briefly, the IHC scoring index was a product of the percentage staining (0–4) and the relative intensity (1–3).
Statistical analysis
The results of all experiments represent the mean ± SD of three separate experiments performed in triplicate. Wilcoxon rank sum and Kruskal–Wallis tests were used to determine significance between variables of two patient populations separated by tumour histology (squamous cell or adenocarcinoma) and to evaluate the methylation profile between the two tumour histologies. A general linear model was used to compare the level of BRMS1 promoter methylation, BRMS1 protein expression and specific clinicopathological variables in human NSCLC specimens [16]. p < 0.05 was considered significant.
Results
BRMS1 promoter methylation decreases BRMS1 transcription
To explore the methylation profile of the BRMS1 promoter region, NSCLC cell lines H157 (squamous cell carcinoma) and A549 (adenocarcinoma) along with NHBE cells were treated or not with 5-aza-2′ deoxycytidine (5 μM), a global demethylating agent, for 5 consecutive days. Using methylation-specific PCR (MSP) primers for the promoter-associated CGI region (−527 to +612) of BRMS1 (Figure 1A), semi-quantitative MSP (lower panel) and quantitative MSP (upper panel) demonstrated that NSCLC cell lines had increased levels of BRMS1 methylation at baseline compared to NHBE cells (Figure 1B). After treatment with the demethylating agent, both NSCLC cell lines exhibited methylation levels similar to the NHBE cells. Having identified the promoter-associated CGI (−527 to +612) to be hypermethylated in all NSCLC cell lines, we next examined the upstream CGI (−3477 to −2214) in both the NHBE and NSCLC cells and found no evidence of enhanced methylation of this CGI region (see Supporting information, Figure S1). Collectively, this demonstrates that the BRMS1 promoter is differentially methylated in normal lung tissue and lung cancer cells. Moreover, a different methylation profile for BRMS1 appears to exist in lung cancer cells compared to prior studies with breast cancer cells [12], confirming the well-documented observation of CGI tissue specificity.
Figure 1.

BRMS1 promoter activity, mRNA and protein levels are regulated by promoter methylation. (A) Schematic illustration of the CpG island in BRMS1 promoter. Arrow, transcription start site (TSS); empty box, exon 1; small bars, positions of primers for methylation-specific PCR (MSP). (B) Lung cancer (H157 and A549) and NHBE cells were treated or not with 5-Aza (5 μM) for 5 days. Genomic DNAs were extracted. Quantitative MSP was done by using methylated (M)- and unmethylated (U)-specific primers in the CpG island of BRMS1 promoter. (C) H157, A549 and NHBE cells were treated or not with 5-Aza (5 μM) for 5 days; (lower panels) BRMS1 mRNA levels determined by QRT–PCR; (upper panel) BRMS1 protein levels detected by western blot. (D) Methylation inhibits BRMS1 promoter activity. (Left panel) 20 μg BRMS1 promoter reporter was treated with CpG methyltransferase M. SssI (20 U) and digested with BglII/HindIII, as described in Materials and methods. The completeness of methylation was visualized by 2% agarose gel electrophoresis after digestion with BstUI. (Centre panel) pGL3 basic empty vector and pGL3–BRMS1 were treated with SssI or buffer only, as described above, and then were transiently transfected into 293T cells. Luciferase activity was determined. (Right panel) pGL3–BRMS1 was treated with SssI or buffer only, as described above, and then was transiently transfected into H157 and H1299 NSCLC cells. Luciferase activity was determined.
To determine the effects of BRMS1 promoter methylation on BRMS1 transcription and translation, the experiments described above were repeated and BRMS1 mRNA and protein levels assessed. As shown in Figure 1C, there is a robust rescue of the BRMS1 mRNA compared to baseline, as measured by quantitative RT–PCR, as well as BRMS1 protein levels in both NHBE and NSCLC cells following 5-aza-2′ deoxycytidine exposure. In order to verify that methylation of the promoter-associated CGI (−527 to +612) contributes to decreased BRMS1 promoter activity, we cloned this specific region of the BRMS1 promoter into a luciferase reporter gene, pGL3 basic. The ability of our cloned BRMS1 promoter to be methylated in vitro was confirmed following treatment with the CpG methyltransferase SssI, as described in Materials and methods (Figure 1D, left panel). The pGL3 basic empty vector and pGL3–BRMS1 were then treated with SssI or buffer, transiently transfected into 293T cells and luciferase activity determined. This demonstrates a dramatic four-fold reduction in BRMS1 promoter activity following methylation of the CGI (−527 to +612) (Figure 1D, centre panel). To confirm the activity of our cloned BRMS1 promoter construct in NSCLC cells, identical experiments were carried our in H157 and H1299 NSCLC cells (Figure 1D, right panel). Both cell lines demonstrated a two-fold reduction in BRMS1 promoter activity following CGI (−527 to +612) methylation.
Collectively, the experiments described here confirm that methylation of the promoter-associated CGI (−527 to +612) contributes to BRMS1 transcriptional repression in NSCLC cells. Inhibition of this methylation event results in a robust increase in BRMS1 transcript and protein expression levels.
Hypermethylation of the BRMS1 promoter occurs in human NSCLC and is more predominant in squamous cell cancer histology
To confirm that a distinct and differential methylation profile of the BRMS1 promoter methylation was also present in human NSCLC specimens, we obtained 40 (20 adenocarcinoma and 20 squamous cell) human NSCLCs and their patient-matched adjacent non-cancerous tissues. Patient demographics are shown in Table 1. Using quantitative MSP, both adenocarcinoma and squamous cell histologies demonstrated a significant increase in the amount of BRMS1 promoter methylation when comparing tumour specimen to adjacent non-cancerous tissue for both tumour histologies. Representative quantitative MSP results are shown for eight of the 40 matched patient specimens (Figure 2A). Interestingly, the BRMS1 promoter region was significantly more methylated in squamous cell cancer histologies compared to adenocarcinoma (Table 2).
Table 1.
Patient demographics
| Variables | Adenocarcinoma (n = 20) | Squamous cell (n = 20) | p | |
|---|---|---|---|---|
| Age (years) | Range | 46–82 | 54–80 | 0.986 |
| Median | 69 | 67 | ||
| Gender | Male | 10 | 9 | 1.000 |
| Female | 10 | 11 | ||
| Race | Caucasian | 19 | 19 | 0.368 |
| African-American | 0 | 1 | ||
| Asian | 1 | 0 | ||
| Albumin | Mean | 4.2 ± 0.33 | 4.3 ± 0.36 | 0.320 |
| Smoking history | Yes | 17 | 20 | 0.231 |
| No | 3 | 0 | ||
| Packs/year | Range | 0–100 | 25–100 | 0.556 |
| Median | 50 | 48 | ||
| Pathological stage | IA | 8 | 6 | 0.586 |
| IB | 7 | 5 | ||
| IIA | 1 | 1 | ||
| IIB | 1 | 4 | ||
| IIIA | 1 | 3 | ||
| IIIB | 2 | 1 | ||
| Recurrence | No | 10 | 12 | 0.792 |
| Yes | 8 | 6 | ||
| Unknown | 2 | 2 |
Figure 2.

BRMS1 promoter is hypermethylated in lung cancer tissue when compared to normal adjacent tissue. (A) BRMS1 is hypermethylated in lung cancer tissues. Quantitative MSP for BRMS1 promoter in tumour tissues (T) and patient-matched adjacent lung tissues (N). The data are plotted as the fold over patient-matched adjacent non-cancerous lung tissues (N), where results from N were normalized to 1 for each patient. (B) The BRMS1 promoter presents higher methylation status in lung cancer compared with adjacent non-cancerous tissue. (Upper panel) Schematic illustration of the bisulphite sequence PCR target sequence in the BRMS1 promoter. The 18 bars represent CpG dinucleotides; the arrow represents the transcription start site. (Lower panel) Methylation status of the BRMS1 5′ CpG island in lung cancer samples and adjacent non-cancerous tissues. Each horizontal row of circles represents one patient sample. Each CpG dinucleotide bar from the schematic is depicted by a circle, the fill pattern of which indicates the methylated cytosine and the blank indicates the unmethylated cytosine in CpG dinucleotide. T, tumour; n = adjacent non-cancerous tissue.
Table 2.
Quantitative MSP for BRMS1 promoter using human lung cancer specimens versus patient-matched adjacent non-cancerous tissues
| Histology | No. of patients | Methylation (%) |
p (tumour versus adjacent) | |
|---|---|---|---|---|
| Adjacent tissue | Tumour tissue* | |||
| Adenocarcinoma | 20 | 8.1 ± 7.7 | 27.3 ± 14.4 | 0.0001 |
| Squamous cell | 20 | 8.6 ± 5.9 | 41.0 ± 19.6 | 0.0114 |
p = 0.03, squamous cell carcinoma group compared to adenocarcinoma group.
A potential criticism of methylation-specific PCR is its dependence on annealing temperatures to detect the completely methylated or unmethylated state of these CpG sites. Additionally, as it is PCR-based, small changes in the methylation state of a few CpG sites can occasionally result in large changes in this methylation assay. To experimentally address this concern, we assessed the methylation of endogenous BRMS1 in human lung cancer specimens using bisulphite sequencing PCR (BSP). Fifteen NSCLC specimens and their matched adjacent non-cancerous lung tissues underwent BSP analysis with primers constructed across the promoter-associated CGI region of BRMS1 (Figure 2B). Compared to adjacent non-cancerous lung tissues (N), the methylation status of BRMS1 promoter in tumour (T) exhibited a marked increase in squamous cell and adenocarcinoma lung cancer histologies (squamous cell, T 38.2 ± 7.2% versus N 6.3 ± 3.2%, p < 0.001; adenocarcinoma: T 24.6 ± 4.1% versus N 4.8 ± 2.8%, p < 0.001). Squamous cell cancers also had significantly more methylation of the BRMS1 promoter compared to adenocarcinoma (p = 0.006).
Finally, to experimentally address the possibility that mutations may exist in the downstream CGI region (−527 to +612) of the BRMS1 promoter in human NSCLC, we sequenced this region in all 15 patient NSCLC samples. Analysis of these results revealed no mutations of this region of the BRMS1 promoter.
BRMS1 promoter methylation correlates with decreased BRMS1 protein expression
Immunohistochemistry for BRMS1 nuclear and cytosolic staining was performed using our NSCLC tissue microarray (see Supporting information, Table S1). We chose to examine the cytosolic staining of BRMS1, given the recent study by Frolova and colleagues [17]. Figure 3 illustrates that BRMS1 expression is reduced in lung cancer specimens compared to normal bronchial epithelium. Normal bronchial epithelium demonstrated strong and diffuse nuclear immunoreactivity (Figure 3A) when compared to both adenocarcinoma (moderately diffuse, Figure 3B) and squamous cell carcinoma (nearly absent, Figure 3C). Analysis of BRMS1 promoter methylation and the intensity of nuclear BRMS1 protein expression demonstrated a strong trend toward a significant correlation (p = 0.06).
Figure 3.
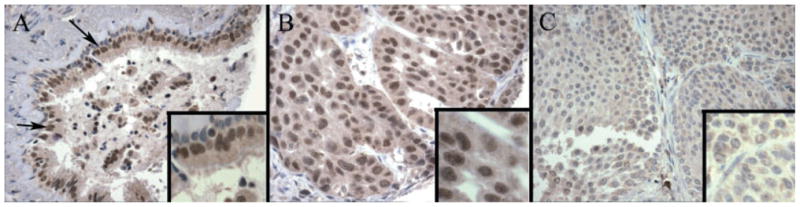
Tissue micro-arrays (TMAs) of nuclear staining for BRMS1 in normal bronchial epithelium and human lung cancer specimens. (A) TMA of human normal bronchial epithelium (arrows) stained with BRMS1 antibody, as described in Materials and methods, demonstrates strong and diffuse nuclear immunoreactivity. (B) TMA of human lung adenocarcinoma tissue stained with BRMS1 antibody, as described in Materials and methods, demonstrates moderate, diffuse nuclear immunoreactivity. (C) TMA of human lung squamous cell carcinoma tissue stained with BRMS1 antibody, as described in Materials and methods, demonstrates an absence of staining.
BRMS1 promoter hypermethylation and clinicopathological variables
A general linear model was created to analyse patient (age, gender, smoking packs/year and cancer recurrence) and tumour (pathological tumour stage) variables with respect to NSCLC and tumour histology (Table 3). We then stratified the 40 patient samples by histology and found that none of the variables had an association with BRMS1 promoter methylation for the adenocarcinoma histology. However, when examining the squamous cell histology, we found that smoking trended towards significance (p = 0.06) and pathological tumour stage was significant (p = 0.04) with respect to BRMS1 promoter methylation.
Table 3.
General linear model illustrating the relationship between patient variables and BRMS1 CGI (−527 to +612) methylation in human NSCLC
| Variable | Main effect* | Main effect stratified by tumour histology* |
|
|---|---|---|---|
| Adenocarcinoma | Squamous cell | ||
| Age | 0.68 | 0.29 | 0.67 |
| Gender | 0.56 | 0.83 | 0.46 |
| Smoking pack/year | 0.29 | 0.74 | 0.06 |
| Pathological tumour stage | 0.42 | 0.80 | 0.04 |
| Cancer recurrence | 0.45 | 0.15 | 0.44 |
p value.
Discussion
CGIs are comprised of CpG dinucleotides clustered in or near promoter regions of nearly half of all protein-coding genes in DNA [18]. Tumour suppressor genes (TSGs) are a well-studied class of genes in which aberrant hypermethylation of normally unmethylated cytosine nucleotides in the CGI of these genes leads to loss of expression and enhanced tumour growth [19,20]. Multiple studies have shown a reduced expression of TSGs in lung cancer secondary to aberrant hypermethylation of CGI, including p16I N K4a, retinoic acid receptor-β (RARβ), E-cadherin (CDH1) and the fragile histidine triad gene (FHIT) [21–24]. Unlike TSGs, metastasis suppressor genes (MSGs), such as BRMS1, function to decrease the formation of spontaneous, macroscopic metastasis [25]. Despite the extensive study of hypermethylation as a mechanism of silencing TSGs in lung cancer, no studies in lung cancer examining the mechanisms through which MSGs, in particular BRMS1, are silenced [2,6,14].
In this report we demonstrate that methylation of the promoter-associated CGI in BRMS1 results in its transcriptional repression, and that this occurs more commonly in NSCLC compared to normal bronchial epithelium tissues. We also show, in lung tissue (cancer and NHBE), that the promoter-associated CGI, not the upstream CGI of BRMS1, is preferentially methylated. A previous study involving breast cancer cell lines had likewise identified these two CpG islands (−3477 to −2214 and −531 to +608) in BRMS1. In that study, the researchers were unable to identify methylation of specific amplicons in the promoter-associated (−531 to +608) CGI, but were able to demonstrate methylated amplicons across all metastatic breast cancer cell lines for the upstream CGI (−3477 to −2214) [12].
Recently, methylation-specific gene expression arrays or ‘methylomes’ have shown that CGI methylation can localize to genomic regions distal to the promoters [26], although the majority of evidence supports that it is methylation of the promoter-associated CGI that directly affects gene transcription [18,27,28]. Interestingly, approximately 40% of CGIs display a tissue-specific expression profile [29,30] and differential methylation of specific CGIs can and do occur across different tissue types. Thus, the different methylation profiles observed in our study, compared to that of Metge and colleagues [12], is likely explained by the tissue specificity of these CGI methylation profiles. Another possibility for the difference between studies could be related to MSP primer design. Given that MSP primer design for the distal CGI was identical in both studies, and that the primers used in this study for the promoter-associated CGI region of BRMS1 were encompassed by primers used in the Metge study, this seems highly unlikely.
To confirm that methylation of promoter-associated CGI (−527 to +612) is integral for BRMS1 promoter activity, we cloned the BRMS1 promoter and performed a luciferase assay that demonstrated methylation dramatically inhibits BRMS1 promoter activity (Figure 1D). In light of the evidence of the relatively poor correlation between CGI hypermethylation and the transcriptional status of associated genes [31], these studies provide an important, mechanistic link between BRMS1 promoter-associated CGI methylation and its transcriptional repression.
Recent studies have identified that promoter hypermethylation of specific genes correlate to lung cancer recurrence and overall outcome [32]. Prior to this report, no studies had investigated the correlation between BRMS1 promoter methylation profiles and clinicopathological variables in NSCLC patients. Examination of our study cohort revealed that BRMS1 promoter methylation was significantly more robust in squamous cell compared to adenocarcinoma histologies (Table 2). This observation was confirmed using the more stringent BSP analysis, where identical results were obtained. In agreement with our findings, other studies have shown differential methylation profile expression based on tumour histology, particularly with p16I N K4a [24]. This difference between histologies may be attributed to the resultant inflammation [33] commonly associated with heavy smokers and the squamous cell cancer histology. Finally, using our general linear model we found that when stratified by tumour histology, pathological stage correlated significantly and that smoking trended towards significance with squamous cell histology only (Table 3). These results are supported by prior studies from our group demonstrating a direct correlation between smoking status and BRMS1 protein expression in NSCLC patients [14]. Smoking is also associated with a loss of both mRNA and protein expression of RARβ [34–36] and with aberrant promoter hypermethylation of CGIs in p16I N K4a [37] and TSCL1/IGSF4 [38].
In conclusion, we have demonstrated that BRMS1 promoter-associated CGI methylation results in decreased BRMS1 promoter activity and transcriptional repression in both NSCLC cells and human NSCLC specimens. Further analysis of BRMS1 methylation in 40 human NSCLC specimens identified a significant increase in BRMS1 promoter methylation compared to adjacent non-cancerous tissue. This difference in BRMS1 promoter methylation was most pronounced in squamous cell cancer histology, where it also correlated with advanced pathological tumour stage and smoking history.
Supplementary Material
The CGI (−3477 to −2214) upstream of the BRMS1 promoter does not exhibit enhanced methylation in NSCLC. Genomic DNAs were extracted from lung cancer (A549 and H157) and normal bronchial epithelial cells (NHBE) cells. Quantitative MSP was done as described
Both unmethylated and methylated luciferase constructs demonstrate equivalent ectopic copy numbers in NSCLC cell lines. The pGL3–BRMS1 construct was treated with SssI or buffer only, as described in Materials and methods, and then was transiently transfected into H157 and H1299 NSCLC cells and 293T cells. The ectopic copy number of the luciferase construct was checked by real-time PCR, using the following primers: forward: 5′-GTGTTGGGCGCGTTATTTAT-3′; reverse: 5′-CATCGACTGAAATCCCTGGT-3′. A total of 15 ng DNA/PCR reaction was used at an annealing temperature of 55 °C. Internal control was measured using HPRT
Associated BRMS1 methylation profiles, IHC scores and histopathological variables
Acknowledgments
This work was supported by both a NIH Grant No. 1R01 CA136705 (to DRJ) and the Thoracic Surgery Foundation for Research and Education (TSFRE) Research Fellowship (to ASN), and also in part by a gift provided to the University of Virginia Tobacco Research Program by Philip Morris USA (to DRJ). The review and approval process was overseen by an independent National External Advisory Board without any affiliation with the University, PM USA, or any other tobacco company. Funding for this project was determined by an independent panel of experts within the University.
Footnotes
No conflicts of interest were declared.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000;60:2764–2769. [PubMed] [Google Scholar]
- 3.Stark AM, Tongers K, Maass N, Mehdorn HM, Held-Feindt J. Reduced metastasis-suppressor gene mRNA expression in breast cancer brain metastases. J Cancer Res Clin Oncol. 2005;131:191–198. doi: 10.1007/s00432-004-0629-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Z, Yamashita H, Toyama T, Yamamoto Y, Kawasoe T, Iwase H. Reduced expression of the breast cancer metastasis suppressor 1 mRNA is correlated with poor progress in breast cancer. Clin Cancer Res. 2006;12:6410–6414. doi: 10.1158/1078-0432.CCR-06-1347. [DOI] [PubMed] [Google Scholar]
- 5.Cicek M, Fukuyama R, Welch DR, Sizemore N, Casey G. Breast cancer metastasis suppressor 1 inhibits gene expression by targeting nuclear factor-κB activity. Cancer Res. 2005;65:3586–3595. doi: 10.1158/0008-5472.CAN-04-3139. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Smith PW, Jones DR. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol Cell Biol. 2006;26:8683–8696. doi: 10.1128/MCB.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samant RS, Clark DW, Fillmore RA, Cicek M, Metge BJ, Chandramouli KH, et al. Breast cancer metastasis suppressor 1 (BRMS1) inhibits osteopontin transcription by abrogating NF-κB activation. Mol Cancer. 2007;6:6. doi: 10.1186/1476-4598-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeWald DB, Torabinejad J, Samant RS, Johnston D, Erin N, Shope JC, et al. Metastasis suppression by breast cancer metastasis suppressor 1 involves reduction of phosphoinositide signaling in MDA-MB-435 breast carcinoma cells. Cancer Res. 2005;65:713–717. [PubMed] [Google Scholar]
- 9.Samant RS, Seraj MJ, Saunders MM, Sakamaki TS, Shevde LA, Harms JF, et al. Analysis of mechanisms underlying BRMS1 suppression of metastasis. Clin Exp Metast. 2000;18:683–693. doi: 10.1023/a:1013124725690. [DOI] [PubMed] [Google Scholar]
- 10.Saunders MM, Seraj MJ, Li Z, Zhou Z, Winter CR, Welch DR, et al. Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res. 2001;61:1765–1767. [PubMed] [Google Scholar]
- 11.Shevde LA, Samant RS, Goldberg SF, Sikaneta T, Alessandrini A, Donahue HJ, et al. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp Cell Res. 2002;273:229–239. doi: 10.1006/excr.2001.5452. [DOI] [PubMed] [Google Scholar]
- 12.Metge BJ, Frost AR, King JA, Dyess DL, Welch DR, Samant RS, et al. Epigenetic silencing contributes to the loss of BRMS1 expression in breast cancer. Clin Exp Metast. 2008;25:753–763. doi: 10.1007/s10585-008-9187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Illingworth RS, Bird AP. CpG islands—‘a rough guide’. FEBS Lett. 2009;583:1713–1720. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 14.Smith PW, Liu Y, Siefert SA, Moskaluk CA, Petroni GR, Jones DR. Breast cancer metastasis suppressor 1 (BRMS1) suppresses metastasis and correlates with improved patient survival in non-small cell lung cancer. Cancer Lett. 2009;276:196–203. doi: 10.1016/j.canlet.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan W, Shao R. Transduction of a mesenchyme-specific gene periostin into 293T cells induces cell invasive activity through epithelial–mesenchymal transformation. J Biol Chem. 2006;281:19700–19708. doi: 10.1074/jbc.M601856200. [DOI] [PubMed] [Google Scholar]
- 16.McCullagh P, Nelder J. Generalized Linear Models. 2. Chapman and Hall/CRC; Boca Raton, FL: 1989. [Google Scholar]
- 17.Frolova N, Edmonds MD, Bodenstine TM, Seitz R, Johnson MR, Feng R, et al. A shift from nuclear to cytoplasmic breast cancer metastasis suppressor 1 expression is associated with highly proliferative estrogen receptor-negative breast cancers. Tumour Biol. 2009;30:148–159. doi: 10.1159/000228908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–174. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- 19.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 20.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 21.Sato M, Shames DS, Gazdar AF, Minna JD. A translational view of the molecular pathogenesis of lung cancer. J Thorac Oncol. 2007;2:327–343. doi: 10.1097/01.JTO.0000263718.69320.4c. [DOI] [PubMed] [Google Scholar]
- 22.Tsou JA, Hagen JA, Carpenter CL, Laird-Offringa IA. DNA methylation analysis: a powerful new tool for lung cancer diagnosis. Oncogene. 2002;21:5450–5461. doi: 10.1038/sj.onc.1205605. [DOI] [PubMed] [Google Scholar]
- 23.Zochbauer-Muller S, Fong KM, Maitra A, Lam S, Geradts J, Ashfaq R, et al. 5′ CpG island methylation of the FHIT gene is correlated with loss of gene expression in lung and breast cancer. Cancer Res. 2001;61:3581–3585. [PubMed] [Google Scholar]
- 24.Zochbauer-Muller S, Fong KM, Virmani AK, Geradts J, Gazdar AF, Minna JD. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001;61:249–255. [PubMed] [Google Scholar]
- 25.Yoshida BA, Sokoloff MM, Welch DR, Rinker-Schaeffer CW. Metastasis-suppressor genes: a review and perspective on an emerging field. J Natl Cancer Inst. 2000;92:1717–1730. doi: 10.1093/jnci/92.21.1717. [DOI] [PubMed] [Google Scholar]
- 26.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyes J, Bird A. Repression of genes by DNA methylation depends on CpG density and promoter strength: evidence for involvement of a methyl–CpG binding protein. EMBO J. 1992;11:327–333. doi: 10.1002/j.1460-2075.1992.tb05055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsieh CL. Dependence of transcriptional repression on CpG methylation density. Mol Cell Biol. 1994;14:5487–5494. doi: 10.1128/mcb.14.8.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsen F, Gundersen G, Lopez R, Prydz H. CpG islands as gene markers in the human genome. Genomics. 1992;13:1095–1107. doi: 10.1016/0888-7543(92)90024-m. [DOI] [PubMed] [Google Scholar]
- 30.Zhu J, He F, Hu S, Yu J. On the nature of human housekeeping genes. Trends Genet. 2008;24:481–484. doi: 10.1016/j.tig.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 31.Oakes CC, La Salle S, Smiraglia DJ, Robaire B, Trasler JM. A unique configuration of genome-wide DNA methylation patterns in the testis. Proc Natl Acad Sci USA. 2007;104:228–233. doi: 10.1073/pnas.0607521104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo M, Ames S, et al. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med. 2008;358:1118–1128. doi: 10.1056/NEJMoa0706550. [DOI] [PubMed] [Google Scholar]
- 33.Seifart C, Lin HM, Seifart U, Plagens A, DiAngelo S, von Wichert P, et al. Rare SP-A alleles and the SP-A1–6A(4) allele associate with risk for lung carcinoma. Clin Genet. 2005;68:128–136. doi: 10.1111/j.1399-0004.2005.00470.x. [DOI] [PubMed] [Google Scholar]
- 34.Gebert JF, Moghal N, Frangioni JV, Sugarbaker DJ, Neel BG. High frequency of retinoic acid receptor-β abnormalities in human lung cancer. Oncogene. 1991;6:1859–1868. [PubMed] [Google Scholar]
- 35.Geradts J, Chen JY, Russell EK, Yankaskas JR, Nieves L, Minna JD. Human lung cancer cell lines exhibit resistance to retinoic acid treatment. Cell Growth Differ. 1993;4:799–809. [PubMed] [Google Scholar]
- 36.Xu XC, Sozzi G, Lee JS, Lee JJ, Pastorino U, Pilotti S, et al. Suppression of retinoic acid receptor-β in non-small-cell lung cancer in vivo: implications for lung cancer development. J Natl Cancer Inst. 1997;89:624–629. doi: 10.1093/jnci/89.9.624. [DOI] [PubMed] [Google Scholar]
- 37.Yanagawa N, Tamura G, Oizumi H, Takahashi N, Shimazaki Y, Motoyama T. Promoter hypermethylation of tumor suppressor and tumor-related genes in non-small cell lung cancers. Cancer Sci. 2003;94:589–592. doi: 10.1111/j.1349-7006.2003.tb01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kikuchi S, Yamada D, Fukami T, Maruyama T, Ito A, Asamura H, et al. Hypermethylation of the TSLC1/IGSF4 promoter is associated with tobacco smoking and a poor prognosis in primary non-small cell lung carcinoma. Cancer. 2006;106:1751–1758. doi: 10.1002/cncr.21800. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The CGI (−3477 to −2214) upstream of the BRMS1 promoter does not exhibit enhanced methylation in NSCLC. Genomic DNAs were extracted from lung cancer (A549 and H157) and normal bronchial epithelial cells (NHBE) cells. Quantitative MSP was done as described
Both unmethylated and methylated luciferase constructs demonstrate equivalent ectopic copy numbers in NSCLC cell lines. The pGL3–BRMS1 construct was treated with SssI or buffer only, as described in Materials and methods, and then was transiently transfected into H157 and H1299 NSCLC cells and 293T cells. The ectopic copy number of the luciferase construct was checked by real-time PCR, using the following primers: forward: 5′-GTGTTGGGCGCGTTATTTAT-3′; reverse: 5′-CATCGACTGAAATCCCTGGT-3′. A total of 15 ng DNA/PCR reaction was used at an annealing temperature of 55 °C. Internal control was measured using HPRT
Associated BRMS1 methylation profiles, IHC scores and histopathological variables