

Abstract
In the motor system, one specific movement is generated, and, simultaneously, other possible movements are suppressed; a process called surround inhibition. Focal hand dystonia (FHD) is a movement disorder characterized by a loss of surround inhibition. In order to explain the deficit in surround inhibition induced by volitional movement in FHD patients, we examined the inhibitory circuit activated by afferent stimulation at “long latency”. We studied 14 patients (age 48.9 ± 13.2 years, 3 female, 11 male) with idiopathic task-related focal hand dystonia. To measure long-latency afferent inhibition (LAI), TMS was applied to the affected hemisphere for FHD patients and to the dominant hemisphere for 17 healthy volunteers. Motor evoked potentials (MEPs) were recorded over abductor digiti minimi (ADM) and first dorsal interosseous (FDI) during rest and during voluntary phasic flexion of the second digit. Subjects were given electrical stimulation to either their fifth digit (homotopic to ADM, heterotopic to FDI) or their second digit (heterotopic to FDI, homotopic to ADM) at twice sensory perceptual threshold 180ms prior to TMS application. Additionally, F-waves were recorded from ADM. At rest, we found a significant decrease in ADM MEP amplitudes with both homotopic and heterotopic stimulation compared to the corresponding non-stimulated trials. There was a trend towards less LAI in FHD patients. During movement, LAI was significantly decreased in both patients and controls. There was no significant group effect. The results for LAI in FDI were similar to those from ADM. F-wave area in ADM was greater during movement for both homo- and heterotopic stimulation. We found no difference in F-wave area between patients and healthy volunteers. Our results indicate that LAI is unlikely to be an underlying mechanism that contributes to the generation of normal surround inhibition in healthy volunteers or in the disruption of surround inhibition in FHD.
Keywords: Dystonia, Transcranial magnetic stimulation, Afferent inhibition, Surround inhibition
Introduction
Dystonia is a neurological disorder characterized by abnormal posturing due to sustained muscle contractions, which interferes with the performance of motor tasks (Hallett 2004). Dystonia can be classified by age at onset, by distribution, and by cause (Tarsy and Simon 2006). When the dystonia is restricted to a limb it is called focal limb dystonia (such as in a foot or hand) with an approximate prevalence of 68.9 per million persons (Nutt, Muenter et al. 1988). The pathophysiology of focal hand dystonia (FHD) is characterized by abnormal sensorimotor integration (Abbruzzese and Berardelli 2003), loss of plasticity (Quartarone, Rizzo et al. 2005; Weise, Schramm et al. 2006) and loss of inhibition—both in the motor system (for review please see Hallett et al (Hallett 2004)) and in the somatosensory system (Tinazzi, Rosso et al. 2003).
For patients with focal hand dystonia, it is unclear whether abnormal sensorimotor integration manifests due to an aberrant brain response to afferent stimulation or whether the afferent stimulation is a trigger itself (Rosenkranz, Altenmuller et al. 2000; Abbruzzese, Marchese et al. 2001; Abbruzzese and Berardelli 2003). The phenomenon of sensory afferent inhibition is seen when peripheral electrical stimulation is delivered to a digit or peripheral nerve prior to the administration of transcranial magnetic stimulation (TMS) to the contralateral primary motor cortex (M1) and the resulting motor evoked potential (MEP) is reduced. This sensory input can produced inhibition only when appropriately timed. When the interstimulus interval between peripheral electrical stimulation and TMS is approximately 200 ms, this motor inhibitory effect is known as long-latency afferent inhibition (LAI) (Chen, Corwell et al. 1999; Classen, Steinfelder et al. 2000; Sailer, Molnar et al. 2003). The role that LAI plays during voluntary movement in FHD patients is not known. In healthy volunteers, there was evidence from a study done by Voller et al that LAI contributes to a form of functional inhibition, namely surround inhibition (Voller, St Clair Gibson et al. 2005). They showed that LAI increased in a hand muscle not involved in index finger flexion, suggesting that LAI enhanced surround inhibition, leading to the suppression of movement in the non-moving muscles.
Surround inhibition is a well-known brain mechanism in visual and sensory systems and likely plays a role in the motor system. The ability to selectively activate particular muscles to perform a specific task likely is generated by suppressing the excitability of a neural network surrounding an activated network (Sohn and Hallett 2004). Among other instances of loss of inhibition in FHD (Stinear and Byblow 2005; Tinazzi, Farina et al. 2005), there is evidence that surround inhibition is disturbed in FHD (Sohn and Hallett 2004). However, the mechanisms responsible for this phenomenon are not well understood. The role LAI plays in surround inhibition in FHD patients has not been examined.
In this study, we sought to evaluate the role of LAI during voluntary movement in patients with FHD to determine if a disruption in this sensory inhibitory pathway contributes to the abnormal surround inhibition in FHD. We hypothesized that LAI supports surround inhibition by acting on fingers not involved in the active movement, and that this inhibition will be diminished in patients with FHD.
Methods
Patient characteristics
We studied 14 patients (age 48.9± 13.2 years, 3 female, 11 male) with idiopathic task-related focal dystonia of their hand (writer’s cramp and musician’s cramp) without any other peripheral or central neurological disorders. All patients except one were right-handed; in four patients dystonia was present in their non-dominant hand. None of the patients received centrally acting medication at the time of the investigation. Patients on treatment with botulinum toxin were studied at least 3 months after the last injection. Seventeen healthy, age-matched (age 44.6 ± 18 years, 7 female, 10 male) volunteers served as a control group. All except one were right-handed. The protocol was approved by the National Institute of Neurological Disorders and Stroke Institutional Review Board, and all subjects gave their written informed consent.
TMS measurements
Surface electromyography (EMG) activity was recorded from ADM (the target muscle), FDI, flexor digitorum superficialis (FDS), and extensor indicis propius (EIP) using Dantec disposable silver-silver chloride surface EMG electrodes placed in a bipolar montage. The EMG activity in FDS and EIP was collected for monitoring the index finger movement. The EMG was amplified using a conventional EMG machine (Viking IV, Nicolet Biomedical, Madison, Wisconsin) with bandpass between 10 and 2000 Hz. The signal was digitized at a frequency of 5 kHz and stored on a personal computer for further off-line analysis.
Focal TMS was performed with the target muscle at complete rest, which is defined as the absence of any EMG activity exceeding a background noise level of 25 μV. The motor cortex was stimulated with a figure 8-shaped coil (each loop 70 mm in diameter) connected to a Magstim 200 magnetic stimulator (Magstim, Whitland, Dyfed, UK). The intersection of the coil was placed tangentially to the scalp with the handle pointing backwards and laterally at a 45-degree angle away from the midline. The “hotspot” for ADM was identified with a suprathreshold stimulus, and this location was marked on the scalp.
At this optimal position of the coil, motor threshold for producing a MEP in resting muscle was assessed. Resting motor threshold (RMT) was determined to the nearest 1% of the maximal stimulator output and was defined as the minimal stimulus intensity required to produce motor evoked potentials (MEPs) of > 50 μV in at least 5 of 10 consecutive trials. MEP size was determined by averaging peak-to-peak amplitudes over 24 single trials for each session at stimulus intensity of 140% of RMT.
In order to get subjects to make a reaction time movement at an exact time, we used a series of four equally spaced tones of different frequencies. A Master-8 pulse generator (A.M.P.I, Jerusalem, Israel) was programmed to trigger an auditory click/tone generator (model S10CTCM, Grass-Telefactor, an Astro-Med, Inc. Product Group, West Warwick, Rhode Island) at a rate of 1 Hz. Subjects were trained to flex the second finger on the fourth tone. The accuracy of movement initiation was determined by the onset of EMG activity in FDS during finger flexion on the fourth tone. At the end of the training session, all subjects had EMG initiation within 40 ms prior to or 20 ms after the fourth tone in at least 25 consecutive trials. MEP size was first measured at rest. Then, using a LabVIEW program (National Instruments, Austin, Texas) to control the timing, a digital electrical stimulus could be given 160 ms prior to expected movement onset, and TMS was given 20 ms after the movement onset, resulting in an interstimulus interval between electrical stimulation and TMS administration of 180 ms (Figure 1). TMS stimulus intensity was adjusted during the movement condition to match the MEP size at rest. Peak-to-peak MEP amplitudes obtained during the movement condition with peripheral stimulation and during rest condition with peripheral stimulation were compared.
FIG. 1.

EMG tracings from one subject recorded over abductor digiti minimi (ADM) and flexor digitorum superficialis (FDS). Digital electrical stimulation occurs at point A (0 ms). Voluntary index finger flexion occurs with the 4th auditory tone at point B (160 ms). TMS administration follows electrical stimulation after an interval of 180 ms (point C).
Peripheral stimulation
Peripheral cutaneous stimulation was performed using ring electrodes around the proximal phalanges of digits 2 and 5 and delivered by a Grass S88 (Grass-Telefactor, An Astro-Med, Inc. Product Group, West Warwick, Rhode Island). The stimulation was applied at 200% percent of sensory perceptual threshold with a pulse duration of 0.2 ms (Classen, Steinfelder et al. 2000). The stimuli were applied at 180 ms prior to the onset of TMS. We used the interval of 180 ms instead of 200 ms due to evidence that inhibition was stronger at intervals just under 200 ms (Classen, Steinfelder et al. 2000; Voller, St Clair Gibson et al. 2005). Stimuli were applied randomly both at rest and during flexion of FDS. For each condition, 25 trials with peripheral stimuli (stimulated trials) and 25 trials without peripheral stimuli (non-stimulated trials) were administered. During both stimulated and nonstimulated trials, TMS was administered.
For this study, a stimulus was defined as being homotopic if the peripheral stimulation was applied to the finger from which the MEP was being measured, and heterotopic if the stimulus was applied to the finger distant to the digit from which the MEP was being measured. For example, electrical stimulation of digit 2 was homotopic for FDI and heterotopic for ADM. Electrical stimulation of digit 5 was homotopic for ADM and heterotopic for FDI.
F-waves
In addition to analysis of MEP amplitudes associated with TMS, supramaximal electrical stimulation of the ulnar nerve at the wrist was performed in a separate session in order to measure F-waves from ADM. F-waves are a measure of spinal cord excitability and can be reliably elicited from the small hand muscles, in contrast to other measures of spinal cord excitability such as the H-reflex. The F-waves were recorded to provide data on changes in spinal cord excitability with movement and peripheral stimulation that could be compared with the MEP data—which is a result of both cortical, subcortical and spinal influences. The resulting F-wave area averaged from 20 trials of ADM was determined in both rest and movement sessions. ADM compound muscle action potential (CMAP) was also determined during both movement and rest sessions. F-wave ulnar stimulation at the wrist was delivered 174 ms after peripheral stimulation at the digit to evaluate the spinal cord excitability at the time when the descending corticospinal volley from the TMS stimulation would be reaching the C7-C8 level of the spinal cord.
Data analysis
To compare MEP amplitudes in ADM and FDI between control and stimulated trials we calculated a repeated measures analysis of variance (rmANOVA) with the factors STIMULATION (non-stimulated vs. stimulated) and GROUP (dystonia patients vs. healthy volunteers). Results for MEPs were expressed as mean ± SE. Subsequently, MEP amplitudes of stimulated trials were normalized to non-stimulated trials to demonstrate the amount of LAI. The amount of unintentional coactivation in ADM during index finger flexion was defined as the percentage of entire motor neuron pool activated (background ADM EMG amplitude divided by ADM CMAP amplitude).
For group comparisons of changes in LAI in ADM and FDI due to movement and peripheral stimulation, we used rmANOVA with the factors MOVE (movement vs. rest) and GROUP (dystonia patients vs. healthy volunteers). Here, trials with homo- and heterotopic stimulation were treated separately for each muscle, as what was considered homotopic for ADM was heterotopic for FDI. Further, the same statistical analysis (rmANOVA MOVE x GROUP) was performed to test for group differences in F-wave area. Paired samples t tests were performed as post hoc comparisons given a significant F statistic. P-values were Bonferroni-corrected for multiple comparisons.
We analyzed ADM and FDI separately as the hotspot for the TMS stimulation was over ADM’s cortical representation and optimized the stimulation intensity to see changes in ADM. Although we evoked potentials in FDI with this stimulation area, it may not have been optimal for this muscle and therefore inhibition may be subject to floor or ceiling effects.
Results
The mean RMT value for healthy volunteers was 49.96% (±8.51% SD) and for dystonia patients 44.90% (±5.86% SD) of stimulator output. There was a tendency towards lower RMT in dystonia patients (univariate ANOVA F1,29=3.555, p=0.069).
In order to match the MEP size during active movement with the rest condition, the mean TMS intensity was reduced by 14.23% (±5.25% SD) or from 140% to 110% of the RMT. ADM MEP amplitudes before and after stimulator adjustment did not significantly differ (rmANOVA F1,30=0.007, p=0.933). The averages of ADM MEP amplitudes in all conditions (rest, movement, homo- and heterotopic stimulation) are shown in Table 1.
Table 1.
Mean MEP amplitudes (mV ± SE) of the non-stimulated (without peripheral electrical stimulation) and stimulated (with peripheral electrical stimulation) trials
| Rest – control trials |
Rest – conditioned trials |
Movement – control trials |
Movement – conditioned trials |
||
|---|---|---|---|---|---|
| Homotopic | 1.71 ± 0.44 | 1.74 ± 0.54 | 1.47 ± 0.36 | 1.70 ± 0.34 | |
| Patients | |||||
| Heterotopic | 1.59 ± 0.36 | 1.41 ± 0.36 | 1.46 ± 0.48 | 1.47 ± 0.48 | |
|
| |||||
| Homotopic | 1.60 ± 0.30 | 1.28 ± 0.32 | 1.45 ± 0.22 | 1.71 ± 0.31 | |
| Controls | |||||
| Heterotopic | 1.58 ± 0.28 | 1.37 ± 0.30 | 1.87 ± 0.47 | 1.74 ± 0.37 | |
At rest we found a significant decrease in MEP amplitudes in stimulated compared to the corresponding non-stimulated trials in both ADM (F1,59=8.931, p=0.004) and FDI (F1,59=57.52, p<0.0001). There was no significant main effect for the factor GROUP (ADM F1,59=0.2, p=0.656, FDI F1,59=0.622, p=0.434). However, we observed a trend towards an interaction of the factors STIMULATION and GROUP for ADM (F1,59=2.973, p=0.09) and FDI (F1,59=3.158, p=0.081) suggesting that dystonia patients had less LAI at rest. In ADM in healthy volunteers MEP amplitudes were significantly decreased from 1.589 ± 0.225 to 1.327 ± 0.253 mV in stimulated compared to non-stimulated trials (paired samples t test p=0.001), whereas in patients MEP size remained more or less constant (1.652 ± 0.253 mV in non-stimulated trials, 1.582 ± 0.283 mV in stimulated trials, p=0.424). At rest, FDI amplitudes were decreased in healthy volunteers (from 2.358 ± 0.311 mV to 1.804 ± 0.272 mV, p<0.001) and in FHD patients (from 2.866 ± 0.349 mV to 1.972 ± 0.305 mV, p<0.001).
In the rmANOVA (MOVE x GROUP) there was a significant effect of the factor MOVE during homotopic and heterotopic stimulation in ADM (homotopic: F1,29=34.717, p<0.001, heterotopic: F1,28=12.647, p<0.01, Figure 2) and FDI (homotopic: F1,27=17.654, p<0.001, heterotopic: F1,28=26.346, p<0.001, Figure 3). LAI was significantly decreased during movement compared to rest. We observed no significant main effect of the factor GROUP (ADM homotopic: p=0.212, heterotopic: p=0.557, FDI homotopic: p=0.820, heterotopic: p=0.695). There was no significant interaction of the factors MOVE x GROUP (ADM homotopic: p=0.677, heterotopic: p=0.93, FDI homotopic: p=0.711, heterotopic: p=0.703).
FIG. 2.
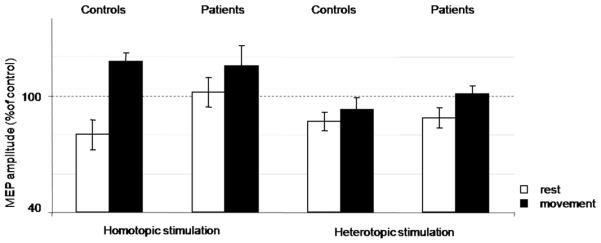
LAI ADM data. Means and standard error bars are shown for the primary outcome (effect of voluntary movement on LAI) in a muscle in the surround of the index finger flexion. At rest, there was a trend towards less LAI in patients compared with controls (p=0.09). No group differences with movement were found.
FIG. 3.

LAI FDI data. Means and standard error bars are shown for the effect of voluntary movement on LAI in a muscle stabilizing the joint during index finger flexion. At rest, there was a trend towards less LAI in patients compared with controls (p=0.081). No significant differences between groups with movement were found.
F-wave area was greater during movement in both homo- (F1,25=11.639, p<0.01) and heterotopic stimulation (F1,24=10.69, p<0.01). We found no difference in F-wave area between patients and healthy volunteers (main GROUP effect F1,25=0.044, p=0.836) (Figure 4).
FIG. 4.

LAI ADM F waves. Means and standard error bars are shown for F wave area during homo- and heterotopic stimulation both at rest and with movement. No significant differences were seen between groups.
The level of coactivation was 2.23% (±2.12% SD) in healthy volunteers and 2.29% (±2.70% SD) in dystonia patients. Although in some patients the level of coactivation was relatively high (ranging up to 29.06%), the two groups did not significantly differ (univariate ANOVA (F1,54=0.011, p=0.917). The degree of LAI, during homotopic and heterotopic stimulation, was not correlated with the level of coactivation (Pearson correlation homotopic: r=−0.075, p=0.704, heterotopic: r=−0.179, p=0.363).
Discussion
We confirmed the finding that LAI occurs at rest in healthy volunteers (Chen, Corwell et al. 1999; Abbruzzese, Marchese et al. 2001). Although we did not replicate the finding of absent LAI in FHD patients, we did see a trend toward less LAI in FHD patients compared to our controls (Abbruzzese, Marchese et al. 2001). In addition, it appears that there was more motor facilitation during movement and stimulation in our patient group than our controls, suggesting impairment in surround inhibition; however, this finding was not statistically significant. We then have to conclude that LAI decreased similarly with voluntary movement in both FHD patients and controls. This implies that LAI does not contribute to the generation of normal surround inhibition. In addition, we can conclude that a disruption in the circuitry responsible for LAI does not account for the abnormal surround inhibition in FHD patients as they behaved similarly to controls in this experiment. We, however, cannot state for certain whether the decrease in LAI with movement is purely a cortical phenomenon given the same direction of change seen with movement in F-wave area, representing an increase in spinal excitability.
There are several possible explanations of these results and some methodological issues to touch on as well. First, the presence of LAI in our patients at rest (even with a trend towards a decrease) is in contrast to previous findings (Abbruzzese, Marchese et al. 2001). It is possible that this is due to methodological differences as the previous investigators used higher electrical stimulation intensities (e.g. three times sensory perceptual threshold). It is also possible that LAI in this paradigm is not a robust measure of the sensorimotor abnormality present in FHD.
Our main result showed that LAI decreased with voluntary movement in both FHD patients and in controls. Interestingly, this decrease in LAI occurred with both homotopic and heterotopic stimulation. Our statistical power calculations were focused on assessing a difference between LAI during rest and movement and not on differential modulation of LAI during movement with homotopic vs. heterotopic stimulation. It is possible if sufficiently powered, we might have detected differential modulation. We were not able to replicate the previous findings from Voller et al. that in healthy volunteers, LAI increased in a muscle in the surround of a voluntary movement (Voller, St Clair Gibson et al. 2005). The earlier study used a randomized block design (stimulation and no-stimulation in different blocks) whereas we used a randomization based on individual trials, possibly indicating that attention to electrical stimulation may play a role in the amount of inhibition detected. We also did not find a correlation with co-activation in the amount of LAI seen in either the controls or the patients. As the authors discuss in the earlier paper, there also could be an effect of training and attention on the results of this study. In order to ensure in each trial that the peripheral electrical stimulation, the voluntary index finger flexion and the TMS administration all occurred at fixed times of 0 ms, 160 ms and 180 ms, respectively, all subjects were trained in the task for 20-30 minutes prior to the start of data collection. All subjects were able to flex their index finger exactly at the fourth auditory tone (−40 ms to +20 ms) and were able to sustain this accuracy for at least 25 trials in a row. It is possible that a training effect resulted in a general increase in M1 excitability. A general increase in excitability is also supported by the fact that with FDS movement, ADM MEP amplitudes increased. To account for this, the TMS stimulator output was decreased to match the MEP amplitude in both the rest and movement conditions. Furthermore, the increase in F-wave area with movement despite electrical stimulation suggests an increase in spinal cord excitability, which could be masking any cortical inhibitory changes—especially if these cortical changes were small in magnitude.
The inhibitory mechanisms underlying LAI are unclear, despite investigations into the pharmacology of LAI and interactions of LAI with other inhibitory circuits. There is evidence that at rest LAI is deficient in FHD patients and in patients with Parkinson disease (PD) (Abbruzzese, Marchese et al. 2001; Sailer, Molnar et al. 2003). Deficient LAI in PD patients was not improved with dopaminergic medication alone, but there was partial restoration of LAI in PD patients with deep brain stimulation and dopaminergic medication administration (Sailer, Molnar et al. 2003; Sailer, Cunic et al. 2007). The authors suggest that LAI may play a role in non-dopaminergic manifestations of PD and in sensorimotor integration. It is not known whether the same pharmacological effects would apply to FHD. In addition, LAI has been shown to inhibit other cortical inhibitory circuits such as long intracortical inhibition (LICI) and intrahemispheric inhibition (IHI) (Sailer, Molnar et al. 2002; Kukaswadia, Wagle-Shukla et al. 2005). Finally, the anatomical networks involved in LAI are not well described, but they are thought to be cortical in origin (Chen, Corwell et al. 1999; Abbruzzese, Marchese et al. 2001). The networks activated in LAI are also likely to be widespread given that with electrical stimulation of the median nerve activity is seen in bilateral primary and secondary somatosensory cortices (Hari, Reinikainen et al. 1984; Allison, McCarthy et al. 1992) and contralateral posterior parietal cortex (Forss, Hari et al. 1994). With the widely distributed anatomical network and complex intracortical inhibitory circuitry, it is unclear where precisely the reduced LAI seen in FHD patients at rest is abnormally generated.
The pathophysiology of FHD includes both abnormal sensorimotor integration and a loss of inhibitory mechanisms (e.g. surround inhibition). It is likely that afferent information whether processed abnormally or generated abnormally influences the degree of motor output through the magnitude of surround inhibition generated. This could lead to varying degrees of movement selectivity and overflow into irrelevant muscles. Based on our current study, however, it does not appear that LAI contributes to the generation of normal surround inhibition nor does it explain the abnormal surround inhibition in FHD patients. In their first description of surround inhibition in the motor system, Sohn et al. (2004) observed suppression of ADM MEP amplitudes at intervals of 3 and 15 ms after movement onset. At a later time point (200 ms after movement onset), they even found facilitation (Sohn and Hallett, 2004b). They also showed during index finger movement MEP amplitudes of ADM increased less than those that were found in the movement-related muscles FDS, FDI, and EIP. This indicates that surround inhibition may be a relative rather than an absolute phenomenon. Some of the increased MEP amplitude is due to a generalized increase in spinal excitability that masks what occurs cortically (Sohn and Hallett, 2004b). Therefore, the timing appears to be crucial when investigating surround inhibition. Our present study did not account for differential changes in LAI based on the latency to movement onset.
Even if a contribution of LAI to surround inhibition remains to be clarified, the trend towards less LAI in FHD patients at rest as well as the greater – albeit non-significant facilitation seen with movement in patients indicates a disruption of this pathway. The restoration of LAI as seen in PD patients after DBS, suggests that this abnormality is potentially modifiable (Sailer, Cunic et al. 2007). The effect that this “normalization” of LAI could potentially have on motor behavior in FHD patients is unclear and deserves further study.
Acknowledgement
This research was supported by the Intramural Research Program of the NIH, NINDS, by the International Graduate School of Neuroscience, Ruhr University, Bochum (B.B.) and a travel grant of German Exchange Service (B.B.).
References
- Abbruzzese G, Berardelli A. Sensorimotor integration in movement disorders. Mov Disord. 2003;18(3):231–40. doi: 10.1002/mds.10327. [DOI] [PubMed] [Google Scholar]
- Abbruzzese G, Marchese R, et al. Abnormalities of sensorimotor integration in focal dystonia: a transcranial magnetic stimulation study. Brain. 2001;124(Pt 3):537–45. doi: 10.1093/brain/124.3.537. [DOI] [PubMed] [Google Scholar]
- Allison T, McCarthy G, et al. The relationship between human long-latency somatosensory evoked potentials recorded from the cortical surface and from the scalp. Electroencephalogr Clin Neurophysiol. 1992;84(4):301–14. doi: 10.1016/0168-5597(92)90082-m. [DOI] [PubMed] [Google Scholar]
- Chen R, Corwell B, et al. Modulation of motor cortex excitability by median nerve and digit stimulation. Exp Brain Res. 1999;129(1):77–86. doi: 10.1007/s002210050938. [DOI] [PubMed] [Google Scholar]
- Classen J, Steinfelder B, et al. Cutaneomotor integration in humans is somatotopically organized at various levels of the nervous system and is task dependent. Exp Brain Res. 2000;130(1):48–59. doi: 10.1007/s002210050005. [DOI] [PubMed] [Google Scholar]
- Forss N, Hari R, et al. Activation of the human posterior parietal cortex by median nerve stimulation. Exp Brain Res. 1994;99(2):309–15. doi: 10.1007/BF00239597. [DOI] [PubMed] [Google Scholar]
- Hallett M. Dystonia: abnormal movements result from loss of inhibition. Adv Neurol. 2004;94:1–9. [PubMed] [Google Scholar]
- Hari R, Reinikainen K, et al. Somatosensory evoked cerebral magnetic fields from SI and SII in man. Electroencephalogr Clin Neurophysiol. 1984;57(3):254–63. doi: 10.1016/0013-4694(84)90126-3. [DOI] [PubMed] [Google Scholar]
- Kukaswadia S, Wagle-Shukla A, et al. Interactions between long latency afferent inhibition and interhemispheric inhibitions in the human motor cortex. J Physiol. 2005;563(Pt 3):915–24. doi: 10.1113/jphysiol.2004.080010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt JG, Muenter MD, et al. Epidemiology of focal and generalized dystonia in Rochester, Minnesota. Mov Disord. 1988;3(3):188–94. doi: 10.1002/mds.870030302. [DOI] [PubMed] [Google Scholar]
- Quartarone A, Rizzo V, et al. Homeostatic-like plasticity of the primary motor hand area is impaired in focal hand dystonia. Brain. 2005;128(Pt 8):1943–50. doi: 10.1093/brain/awh527. [DOI] [PubMed] [Google Scholar]
- Rosenkranz K, Altenmuller E, et al. Alteration of sensorimotor integration in musician’s cramp: impaired focusing of proprioception. Clin Neurophysiol. 2000;111(11):2040–5. doi: 10.1016/s1388-2457(00)00460-0. [DOI] [PubMed] [Google Scholar]
- Sailer A, Cunic DI, et al. Subthalamic nucleus stimulation modulates afferent inhibition in Parkinson disease. Neurology. 2007;68(5):356–63. doi: 10.1212/01.wnl.0000252812.95774.aa. [DOI] [PubMed] [Google Scholar]
- Sailer A, Molnar GF, et al. Effects of peripheral sensory input on cortical inhibition in humans. J Physiol. 2002;544(Pt 2):617–29. doi: 10.1113/jphysiol.2002.028670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailer A, Molnar GF, et al. Short and long latency afferent inhibition in Parkinson’s disease. Brain. 2003;126(Pt 8):1883–94. doi: 10.1093/brain/awg183. [DOI] [PubMed] [Google Scholar]
- Sohn YH, Hallett M. Disturbed surround inhibition in focal hand dystonia. Ann Neurol. 2004;56(4):595–9. doi: 10.1002/ana.20270. [DOI] [PubMed] [Google Scholar]
- Stinear CM, Byblow WD. Task-dependent modulation of silent period duration in focal hand dystonia. Mov Disord. 2005;20(9):1143–51. doi: 10.1002/mds.20514. [DOI] [PubMed] [Google Scholar]
- Tarsy D, Simon DK. Dystonia. N Engl J Med. 2006;355(8):818–29. doi: 10.1056/NEJMra055549. [DOI] [PubMed] [Google Scholar]
- Tinazzi M, Farina S, et al. Task-specific impairment of motor cortical excitation and inhibition in patients with writer’s cramp. Neurosci Lett. 2005;378(1):55–8. doi: 10.1016/j.neulet.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Tinazzi M, Rosso T, et al. Role of the somatosensory system in primary dystonia. Mov Disord. 2003;18(6):605–22. doi: 10.1002/mds.10398. [DOI] [PubMed] [Google Scholar]
- Voller B, St Clair Gibson A, et al. Long-latency afferent inhibition during selective finger movement. J Neurophysiol. 2005;94(2):1115–9. doi: 10.1152/jn.00333.2005. [DOI] [PubMed] [Google Scholar]
- Weise D, Schramm A, et al. The two sides of associative plasticity in writer’s cramp. Brain. 2006;129(Pt 10):2709–21. doi: 10.1093/brain/awl221. [DOI] [PubMed] [Google Scholar]