

Abstract
Oncogenic mutations of the receptor tyrosine kinase c-KIT play an important role in the pathogenesis of gastrointestinal stromal tumors (GIST), systemic mastocytosis, and some acute myeloid leukemias (AML). Whilst juxtamembrane mutations commonly detected in GIST are sensitive to tyrosine kinase inhibitors, the kinase domain mutations frequently encountered in systemic mastocytosis and AML confer resistance and are largely unresponsive to targeted inhibition by the existing agent imatinib. In this study we show that myeloid cells expressing activated c-KIT mutants that are imatinib-sensitive (V560G) or –resistant (D816V) can inhibit the tumor suppressor activity of protein phosphatase 2A (PP2A). This effect was associated with reduced expression of PP2A structural (A) and regulatory subunits (B55α; B56α; B56γ and B56δ). Overexpression of PP2A-Aα in D816V c-KIT cells induced apoptosis and inhibited proliferation. In addition, pharmacological activation of PP2A by FTY720 reduced proliferation, inhibited clonogenic potential and induced apoptosis of mutant c-KIT+ cells, whilst having no effect on WT c-KIT cells or empty vector controls. FTY720 treatment caused dephosphorylation of the D816V c-KIT receptor and its downstream signaling targets pAkt, pSTAT5 and pERK1/2. Additionally, in vivo administration of FTY720 delayed the growth of V560G and D816V c-KIT tumors, inhibited splenic and bone marrow infiltration, and prolonged survival. Our findings show that PP2A inhibition is essential for c-KIT-mediated tumorigenesis, and that reactivating PP2A may offer an attractive strategy to treat drug-resistant c-KIT+ cancers.
Keywords: protein phosphatase 2A, c-KIT, imatinib resistance, FTY720, acute myeloid leukemia
INTRODUCTION
c-KIT is a type 3 receptor tyrosine kinase that is characterised by 5 extracellular immunogloblin–like domains and an intracellular split tyrosine kinase domain (1). Binding of stem cell factor (SCF) to c-KIT activates multiple signaling pathways important for cellular proliferation, differentiation and survival (2). Gain-of-function c-KIT mutations have been documented in core-binding factor acute myeloid leukemia (CBF-AML) (3), systemic mastocytosis (SM) (4), gastrointestinal stromal tumors (GIST) (5), testicular seminoma (6) and melanoma (7). In GIST samples ~85% of c-KIT aberrations are located in the juxtamembrane domain (e.g V560G) (5), whereas the majority of CBF-AML and SM patients express kinase domain mutations (e.g. D816V). In most cases, the presence of an activating c-KIT mutation is associated with a higher relapse rate and reduced survival compared to patients expressing the wild-type (WT) receptor (8).
The tyrosine kinase inhibitor (TKI), imatinib, has shown remarkable success in treating metastatic GIST patients that harbor juxtamembrane c-KIT mutations (9). In contrast, mutations involving the kinase domain are resistant to imatinib inhibition, and as such, c-KIT+ CBF-AML and SM patients are unresponsive to imatinib therapy (10, 11). Recent clinical trials with the second generation TKI, dasatinib (SPRYCEL, Bristol-Myers Squib), have also reported disappointing results (12). To improve treatment outcomes a greater understanding of the signaling pathways activated downstream of c-KIT is required.
Protein phosphatase 2A (PP2A) is a major serine/threonine phosphatase which negatively regulates numerous signal transduction pathways that are involved in cell cycle progression, DNA replication and apoptosis (13). PP2A is emerging as an important tumor suppressor that is mutated or down-regulated in a number of cancers including breast, lung and colon (14–16). It is also inactivated by the oncogenic tyrosine kinase, BCR/ABL, in chronic myeloid leukemia (CML) (17). As BCR/ABL and c-KIT activate similar oncogenic pathways, we hypothesized that PP2A may also be regulated by c-KIT. In this study we report that PP2A activity is inhibited in myeloid precursors expressing imatinib-sensitive (V560G) and -resistant (D816V) mutant c-KIT. Using in vitro and in vivo models of c-KIT tumorigenesis we demonstrate that over-expression of PP2A-Aα, or reactivation of PP2A with FTY720 (fingolimod; Novartis) (18, 19), inhibits c-KIT-mediated growth and survival. Furthermore, FTY720 exhibits selectivity for activating c-KIT mutations over the WT c-KIT receptor. As mutant c-KIT is the driving force behind multiple cancer types, this data suggests that PP2A activation may be a powerful strategy for the treatment of c-KIT+ malignancies.
MATERIALS AND METHODS
Cell lines, retroviral infection and patient samples
The FDC-P1 mouse growth factor-dependent myeloid cell line (20) expressing empty vector (EV), WT, and the oncogenic V560G and D816V mutant forms of human c-KIT have been described previously (21). The expression of c-KIT was routinely monitored by flow cytometry (21). The Kasumi-1 myeloid leukemia cell line (22) and HMC-1 human mast cell lines (23) expressing c-KIT mutations have been previously described. The human AML cell lines MV4-11 (24) and THP-1 (25) were a kind gift from Dr Kyu-Tae Kim (University of Newcastle, Australia). Retroviral expression vectors pMIG EV and pMIG PP2A-Aα (Addgene plasmid 9044 and 10884; deposited byProf. William C. Hahn, Dana Faber Cancer Institute, Boston, MA) have been previously reported (26). FDC-P1 D816V c-KIT cells expressing pMIG EV or PP2A-Aα were generated by retroviral transduction followed by FACS-mediated sorting of GFP+ cells (27).
A bone marrow sample was obtained from a 71 y/o male CBF-AML patient according to institutional guidelines. Studies were approved by the Royal Adelaide Hospital Human Ethics Committee. Mononuclear cells were isolated by Ficoll-Hypaque density-gradient centrifugation, washed, and resuspended at 2 × 105/ml in IMDM/0.5% FBS. c-KIT mutation analysis was performed using high resolution melt analysis and direct sequencing as described (28).
Drug treatments
Imatinib was provided by Prof. Bruce Lyons (University of Tasmania, Hobart, Australia) and resuspended in milli-Q water at 10mM for cell culture or 10 mg/ml for in vivo administration. For cell culture, FTY720 (Cayman Chemicals) and FTY720-P (Echelon Biosciences Inc.) were dissolved in DMSO at 50 mM stock. For in vivo administration, FTY720 was prepared daily in normal saline (0.9% w/v NaCl) at 1 mg/ml. Okadaic acid (Sigma) was dissolved in DMSO to 1 mM.
Phosphatase activity assay
PP2A activity was determined using the PP2Ac immunoprecipitation phosphatase assay kit, as per manufacturer’s instructions (Millipore). The percentage of phosphatase activity was determined by dividing the free phosphate (PO4) of the test cells by that of the untreated EV cells.
Immunoblotting and immunoprecipitation
Cells were lysed in 1% Triton X-100, 50 mM HEPES pH 7.4, 0.5% sodium deoxycholate, 0.1% SDS and 5 mM EDTA supplemented with 1 mM PMSF, 50 mM NaF, 1 mM Na2VO4, 20 μg/ml leupeptin, 20 μg/ml aprotinin and 25 μl/ml complete protease inhibitor cocktail (Sigma). Total protein (50 μg) was separated by SDS-PAGE and transferred onto nitrocellulose membranes (GE Healthcare). For immunoblotting, the following antibodies were used: anti-PP2A-A (Calbiochem); anti-PP2A-B56α, -B56γ3, -B56δ and -B56ε (Novus Biochemicals); anti-PP2AcY307 (Epitomics), anti-I2PP2A (SET; clone H-120) (Santa Cruz Biotechnology), anti-pAktT308, -pBADS112, -pERK1/2T202/Y204, -pMEKS221, -pp38MAPKT180/Y182 and -pSTAT5Y694 (Cell Signaling Technology Inc.); anti-actin (Sigma). Affinity-purified rabbit polyclonal antibodies were raised against a PP2A-B55α peptide (CFSQVKGAVDDDV) (29) and a PP2Ac peptide (PHVTRRTPDYFL) (30). Membranes were incubated with HRP-conjugated secondary antibodies and protein-antibody complexes visualised by enhanced chemiluminescence (GE Healthcare). Images were captured with a FUJIFILM LAS-3000 detection system and analysed using Multi Gauge V3.0. Immunoprecipitation of c-KIT and detection of tyrosine phosphorylation was performed as previously described (31).
Immunofluorescent staining
Cells were cytocentrifuged onto a glass slide, fixed in 3.7% formaldehyde/PBS for 10 min at room temperature and permeabilised in 0.1% Triton/PBS for 3 min. The slides were blocked in 10% FCS/PBS for 20 min, incubated with either 1:500 anti-PP2A-Aα (clone 6F9; Covance) or –cleaved caspase-3 (clone Asp175; Cell Signaling Technology Inc.) for 1 h, then with 1:1000 dilution of the appropriate Alexa Fluor-555 conjugated secondary antibody (Invitrogen), followed by DAPI mounting (Invitrogen). Images were obtained using a FlouView FV1000 confocal microscope (Olympus) and FV10-ASW V2.0 software (Olympus).
Cell proliferation, apoptosis and cell cycle
FDC-P1 and human AML cell lines (2 × 104/well) were seeded in a 96 well plate with appropriate factors and the indicated concentrations of drug for 48 h. Proliferation was assessed using the CellTiter-Blue™ Cell Viability Assay (Promega). The concentration of drug that reduces cell viability by 50% (ID50) was analysed using cubic spline regression (32). Assays were plated in quadruplicate and repeated at least three times. Apoptosis was measured using the annexin-V FITC apoptosis detection kit (BD Biosciences). Samples were run on a FACSCalibur flow cytometer, and data analysed using CellQuest software (BD Biosciences). For cell cycle analysis, cells (5 × 105) were resuspended in 0.5 ml 0.1% glucose/PBS with 2.5 ml 70% EtOH for 1 h at 4°C. Cells were washed in PBS then incubated with 50 μg/mL propidium iodide (PI) (BD Biosciences) and 0.2 mg/ml RNAse (Fermentas) for 30 minutes at 37°C. Samples were analysed on a FACSAria (BD Biosciences), and histograms fitted for cell-cycle ratios using ModFit LT v3.2 (Verity Software House). Four independent experiments were performed for each assay. For the AML patient sample, cells were treated with 0, 0.1, 0.3, 1 and 10μM FTY720, and AML blast survival was measured at 24 hours by co-staining with annexin-V FITC (Roche). The ID50 of apoptosis induction by FTY720 was calculated as above.
Clonogenic Assay
Cells (2 × 102) were plated into 1% methylcellulose medium (MethoCult; Stem Cell Technologies Inc., Vancouver, BC, Canada) in the presence of appropriate factors and indicated drug concentrations. Colonies (>50 cells) were scored 7 days later. Assays were performed in triplicate and repeated three times.
Syngeneic mouse model
Eight- to 10-week-old female DBA/2J mice (Animal Resources Centre, Canning Vale, WA, Australia) were s.c. injected on both flanks with 5 × 106 FDC-P1 V560G c-KIT or 3.5 × 106 FDC-P1 D816V c-KIT cells in 200 μl 1:1 PBS/Matrigel (Trevigen). When tumors reached a volume of ~200 mm3 (day 5), mice were randomised into two groups that received daily i.p. injections of either saline or 10 mg/Kg FTY720. Tumor volume was determined using the formula: 0.5 × length (mm) × width2 (mm). On day 14, three mice from FDC-P1 V560G c-KIT and six mice from FDC-P1 D816V c-KIT injected groups were sacrificed. The remaining mice were used for survival studies and culled when tumor volumes reached ~2100 mm3. The tumors and organs were fixed in formalin and paraffin-embedded. Immunohistochemical staining of anti-human c-KIT (CD117; Dako, Glostrup, Denmark) was detected with the Vectastain Elite ABC kit and diaminobenzidine (DAB; Vector Laboratories, Inc), and imaged with a ColorView II camera and analySIS software (Olympus Soft Imaging Systems GmbH, Munster, Germany). Apoptosis was assessed by TUNEL staining using an In Situ Cell Death Detection kit (Roche). Images were acquired with an Axioplan 2 imaging system and merged with Axio Vision v4.7 (Carl Zeiss). All animal studies were performed with approval of The University of Newcastle Animal Care and Ethics Committee.
Statistical Analysis
Statistical significance between samples was assessed using an unpaired two-tailed Student’s t-test. Survival probabilities between groups were determined by the Kaplan-Meier method and differences in survival distributions were evaluated by the log-rank test. Values shown are the mean ± SEM. All analyses were performed using GraphPad Prism software.
RESULTS
c-KIT inhibits PP2A activity and alters the expression of PP2A subunits
To evaluate whether PP2A is regulated by c-KIT, we measured the activity of PP2Ac immunoprecipitates extracted from FDC-P1 mouse myeloid cells expressing EV, WT c-KIT, the oncogenic imatinib-sensitive juxtamembrane V560G c-KIT mutant or the imatinib-resistant kinase domain D816V c-KIT mutant (21). The phosphatase activity of PP2A was significantly reduced to 72% and 65% in cells expressing V560G and D816V c-KIT, respectively, compared to EV controls (Supplementary Table 1). Thus, activating mutations of c-KIT reduce PP2A activity.
The cellular localisation and substrate specificity of PP2A is regulated by post-translational modification of the catalytic subunit and binding of regulatory B subunits (33, 34). To investigate the mechanism by which constitutive c-KIT activation inhibits PP2A, expression of PP2A subunits were examined by immunoblotting. There was no change in the total expression or phosphorylation of PP2Ac in cells expressing WT or mutant c-KIT (Fig. 1; Supplementary Fig. S1). In contrast, protein levels of PP2A-A, B55α, B56α and B56δ were significantly decreased in mutant c-KIT+ cells. Furthermore, B56γ was barely detectable in mutant c-KIT+ cells, whilst WT c-KIT cells expressed a lower molecular weight isoform (Fig. 1; Supplementary Fig. S1). No change was observed in B56ε. B55β and B55γ are neuronal specific isoforms and are not expressed in the FDC-P1 cells (data not shown). Quantitative real-time PCR (qRT-PCR) showed no significant difference in PP2A subunit mRNA levels between EV controls and mutant c-KIT+ cells (Supplementary Fig. S2). Interestingly, total expression of the PP2A inhibitory protein, SET, was elevated in the mutant c-KIT+ cells, together with the presence of a lower molecular weight isoform (Fig. 1).
Figure 1. Expression of PP2A subunits in FDC-P1 c-KIT+ cell lines.

Immunoblot of FDC-P1 cell lysates showing expression of PP2A-A, PP2Ac, PP2Ac pY307, PP2A-B55α, -B56α, -B56γ, -B56δ, -B56ε, and SET. Actin was used as a loading control. Blots are a representative of three independent experiments.
Over-expression of PP2A-Aα reverses the tumorigenic phenotype
To investigate the functional importance of reduced PP2A expression in oncogenic c-KIT+ cells, the pMIG vector encoding the PP2A-Aα subunit or corresponding EV control was introduced into the FDC-P1 D816V c-KIT cells by retroviral transduction. After 24 h (day 0), the top 10% of GFP+ cells were selected from a heterogeneous population, and the presence of Aα was confirmed by immunofluorescent staining on cytospins (Fig. 2A). On day 0 the viability of the D816V c-KIT PP2A-Aα cells, as determined by trypan blue exclusion, was 55%, markedly lower than 85% observed for the EV control population. It should be noted that for both cell lines, only the viable population was sorted for subsequent experiments. Notably, the percentage of annexin-V+ cells at day 0 was elevated twofold in the PP2A-Aα population compared to the EV controls (Fig. 2B). Furthermore, the percentage of cleaved caspase-3 positive cells increased from 4% in the EV controls to 16% in the PP2A-Aα cells (Fig. 2B). Accordingly, over-expression of PP2A-Aα inhibited the proliferation of D816V c-KIT cells (Fig. 2C), supporting the hypothesis that functional loss of PP2A contributes to c-KIT-mediated tumorigenesis.
Figure 2. Over-expression of PP2A-Aα induces apoptosis and inhibits proliferation of FDC-P1 D816V c-KIT cells.

The pMIG EV and PP2A-Aα expression vectors were retrovirally transduced into FDC-P1 D816V c-KIT cells. A, Immunofluorescent staining of cytopsins detecting PP2A-Aα 24 h post-infection (day 0). B, The induction of apoptosis at day 0 was assessed by annexin-V binding (left) and cleaved caspase-3 immunofluorescent staining of cytopsins (right). C, Total cell numbers were determined by trypan blue exclusion starting at day 0. Scale bars, 50 μm or 20 μm (600× magnification). UTF, untransfected.
Reactivation of PP2A inhibits the proliferation of mutant c-KIT+ cells
To determine the biological relevance and potential therapeutic implications of PP2A loss-of-function in c-KIT+ malignancies, we utilised the pharmacological activator, FTY720 (18, 19). PP2A activity was enhanced 1.7 fold by FTY720 (2.5 μM; 6 h) in cells expressing V560G c-KIT, and 2.4 fold in FTY720-treated D816V c-KIT cells compared to untreated controls (Fig. 3A). In contrast, minimal effect on PP2A activity was seen in WT c-KIT cells or EV controls (Fig. 3A).
Figure 3. Reactivation of PP2A induces apoptosis and suppresses the clonogenic potential of mutant c-KIT cells.

A, FDC-P1 cells were treated with FTY720 or FTY720-P (2.5 μM; 6 h). PP2A activity was determined by incubating the isolated PP2Ac complex with a PP2A-specific phosphopeptide and measuring free phosphate release using a colorimetric assay. PP2A activity is normalised to untreated EV controls. B, FDC-P1 cells were treated with FTY720 (2.5 μM, 24 h) and assessed for annexin-V by flow cytometry. C, FDC-P1 cells were treated with FTY720 (2.5 μM; 36 h) and stained with PI to analyse DNA content. Plots are a representative of four independent experiments. D, E, FDC-P1 cells were grown in methylcellulose for 7days in the presence of indicated drugs. Columns, mean colony number (n=4); bars, SEM. *, p<0.05; **, p<0.01, Student’s t-test compared to untreated.
FTY720 is phosphorylated in vivo by sphingosine kinase 2 to yield FTY720-P, which binds to sphingosine-1-phosphate-specific G protein-coupled receptors (S1PR) and induces signaling that regulates a variety of cellular processes (35). To determine whether reactivation of PP2A is dependent on S1PR-induced signaling, we incubated the cells with FTY720-P (2.5 μM; 6 h). In all cell lines PP2A activity did not increase (Fig. 3A), suggesting that non-phosphorylated FTY720 is responsible for PP2A reactivation.
The effect of FTY720 on viable cell number was determined using a colorimetric assay. Importantly, the ID50 values for V560G and D816V c-KIT cells were significantly reduced compared to WT c-KIT and EV cell lines (Table 1a; Supplementary Fig. S3A). Treatment with a chemically distinct PP2A activator, forskolin (17), showed similar results, whereby mutant c-KIT cells were more sensitive to reactivation of PP2A than EV controls (Table 1a). Incubation with FTY720-P up to 20 μM had no effect on the growth of FDC-P1 cell lines (Table 1a). That FTY720 and forskolin have distinct mechanisms of action, yet both activate PP2A, strongly suggests that the cytotoxicity observed in mutant c-KIT+ cells is due to enhanced activation of PP2A. Furthermore, this effect is not mediated via S1PR signaling.
Table 1.
Cytotoxicity of PP2A activators in mutant c-KIT+ myeloid cells
| FTY720 (μM) | Forskolin (μM) | FTY720-P (μM) | ||||
|---|---|---|---|---|---|---|
| a) FDC-P1 Cell Line | ID501 | Sensitivity2 | ID50 | Sensitivity | ID50 | Sensitivity |
| EV | 5.5 ± 0.4 | 1 | 40.8 ± 7.1 | 1 | > 20 | N/A |
| WT c-KIT | 4.4 ± 0.4 | 1.25 | 37.5 ± 12.9 | 1.09 | > 20 | N/A |
| V560G c-KIT | 2.8 ± 0.3 | 1.96** | 19.4 ± 6.7 | 2.10** | > 20 | N/A |
| D816V c-KIT | 2.4 ± 0.2 | 2.20** | 10.6 ± 6.2 | 3.85** | > 20 | N/A |
| b) Human Cell Lines | FTY720 ID50 (μM)1 | |||||
| HMC 1.1 (V560G c-KIT) | 3.8 ± 0.2 | |||||
| HMC 1.2 (V560G/D816V c-KIT | 3.4 ± 0.1 | |||||
| Kasumi-1 (N822K c-KIT) | 4.9 ± 0.4 | |||||
| MV4-11 (WT c-KIT) | 7.5 ± 0.3 | |||||
| THP-1 (WT c-KIT) | 5.5 ± 0.1 | |||||
| c) Patient Sample | Age/Sex | % Blasts | Cytogenetics | c-KIT | FTY720 ID50 (μM)3 | |
| CBF-AML-1 | 71/M | 69 | Inv(16) | D816Y | 0.62 ± 0.2 | |
ID50 is the concentration (μM) of drug required to reduce cell viability by 50% at 48 h and was calculated using fit-spline lowess regression. Data is presented as the mean of at least three independent experiments performed in quadruplicate ± SEM.
Sensitivity was determined by dividing the ID50 of the FDC-P1 EV cells by the ID50 of the FDC-P1 c-KIT cell lines.
p<0.01 compared to EV, Student’s t-test.
Primary AML cells were treated with 0, 0.1, 0.3, 1 and 10 μM FTY720. Induction of apoptosis was measured at 24 h by annexin-V binding and ID50 was calculated as above. N/A, not available.
We then tested the efficacy of FTY720 on a panel of human AML cell lines. The human mast cell line HMC-1.1, expresses a V560G c-KIT mutation, and HMC-1.2 expresses an additional D816V c-KIT mutation (23). The Kasumi-1 CBF-AML cell line harbors a t(8;21) translocation together with a N822K c-KIT mutation (22). Each of these cell lines displayed increased sensitivity to FTY720 than human AML cell lines expressing WT c-KIT (MV4-11 (24) and THP-1 (25)) (Table 1b; Supplementary Fig. S3B). Furthermore, blasts isolated from a CBF-AML patient with inv(16) and D816V c-KIT displayed remarkable sensitivity to apoptosis induction by FTY720 with an ID50 of 0.6 μM (Table 1c). This data shows that myeloid malignancies characterised by mutant c-KIT are sensitive to FTY720.
FTY720 induces apoptosis and inhibits the clonogenic potential of mutant c-KIT cells
The ability of FTY720 to induce apoptosis in c-KIT-expressing myeloid precursors was investigated by staining with annexin-V (Fig. 3B). In both the mutant c-KIT cell lines, FTY720 (2.5 μM) markedly increased the percentage of annexin-V+ cells at 24 h compared to untreated controls (Fig 3B; Supplementary Fig. S4), and induced a shift into the sub-G0 phase at 36 h (Fig. 3C). Importantly, FDC-P1 EV or WT c-KIT cells remained unaffected by the presence of 2.5 μM FTY720 (Fig. 3B and 3C) up to 48 h (data not shown). Accordingly, the clonogenic potential of FDC-P1 cells expressing V560G c-KIT or D816V c-KIT was significantly inhibited by 2.5 μM FTY720 (Fig. 3D). In contrast, no difference was observed between untreated and FTY720-treated WT c-KIT cells or EV controls (Fig. 3D).
To confirm the observed cytotoxic effects were a consequence of PP2A reactivation by FTY720, FDC-P1 D816V c-KIT cells were co-treated with the serine-threonine phosphatase inhibitor okadaic acid (0.25 nM), at a concentration that specifically inhibits the activity of PP2A (36). Importantly, the addition of okadaic acid rescued the colony formation of FTY720-treated cells (Fig. 3D). Furthermore, the clonogenic potential of V560G and D816V c-KIT cells treated with 2.5 μM FTY720-P resembled that of untreated controls (Fig. 3D). Together this data indicates that FTY720 mediates its effects on FDC-P1 mutant c-KIT cells by specifically reactivating PP2A.
Reactivation of PP2A dephosphorylates the c-KIT receptor and its downstream signaling targets
c-KIT activity is regulated by tyrosine phosphorylation (37). To determine if PP2A reactivation affects c-KIT phosphorylation, the receptor was immunoprecipitated from FDC-P1 cells treated with FTY720 (2.5μM; 6, 12 and 24 h). FTY720 caused marked inhibition of V560G and D816V c-KIT phosphorylation at 6 h (Fig. 4A). In contrast, phosphorylation of the WT c-KIT receptor remained stable, consistent with these cells being less responsive to FTY720 (Fig. 4A).
Figure 4. Reactivation of PP2A dephosphorylates the c-KIT receptor and its downstream signaling targets.
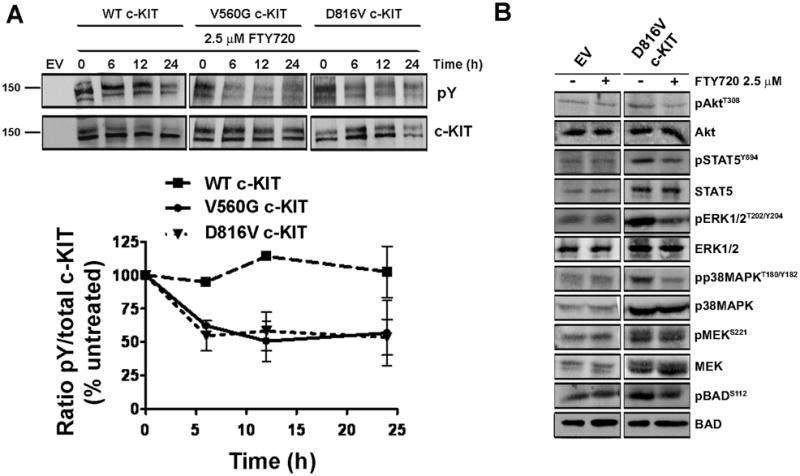
A, c-KIT was immunoprecipitated from FDC-P1 cells treated with 2.5 μM FTY720 (0, 6, 12, 24 h). Levels of tyrosine phosphorylated (pY) and total c-KIT were determined by immunoblotting (top). Quantitation of three independent experiments (bottom). B, Immunoblots detecting phosphorylated or total Akt, STAT5, ERK1/2, p38MAPK, MEK and BAD after FTY720 treatment (2.5μM; 12h).
We next analysed the effect of FTY720 on known PP2A and c-KIT signaling targets. Phosphorylation of Akt, STAT5, ERK1/2, p38MAPK and BAD in WT and V560G c-KIT expressing cells was undetectable (data not shown), which is consistent with previous reports (21, 38, 39). As such, our investigations focussed on the D816V c-KIT cells, which showed reduced phosphorylation of pAkt, pSTAT5, pERK1/2, pp38MAPK and pBAD with FTY720 treatment (2.5 μM; 12 h) (Fig. 4B; Supplementary S5). No change in pMEK was observed. While the D816V cells expressed increased total levels of a number of proteins, the expression did not alter with FTY20 (Fig 4B; Supplementary Fig. S6). Importantly, FTY720 did not affect signaling through the PI3K, STAT or MAPK pathways in the EV controls (Fig. 4B). These findings suggest that PP2A inhibition is required for sustained c-KIT activation, and that enhancing the activity of PP2A results in the dephosphorylation of D816V c-KIT and its downstream targets.
FTY720 delays mutant c-KIT tumor growth in vivo
The efficacy of FTY720 against established tumors expressing either the V560G or D816V mutant c-KIT was evaluated in a syngeneic mouse model. For mice harbouring V560G c-KIT tumors, FTY720 treatment prolonged their survival compared to untreated controls (21 days vs 24 days; p<0.05) (Fig. 5A). Similarly, for tumors expressing D816V c-KIT, the median survival for FTY720-treated mice was 22 days, a significant increase from 18 days for saline-treated mice (p<0.01) (Fig. 5A). As expected, imatinib was effective against V560G c-KIT tumors only (Fig. 5A; Supplementary Fig. S7).
Figure 5. FTY720 delays mutant c-KIT tumor growth in vivo.

FDC-P1 cells expressing either V560G (left) or D816V c-KIT (right) were s.c. inoculated into a separate group of mice. Treatment started day 5 post-tumor injection with daily i.p. injections of saline, 100 mg/Kg imatinib or 10 mg/Kg FTY720. A, The estimated probabilities for survival were calculated using the Kaplan-Meier method, and log-rank test was used to determine the survival difference compared to saline-treated mice. B, On day 14, mice were sacrificed and tumors weighed. Columns, mean of individual tumor weights; bars, SEM. *, p<0.05, n=6. **, p<0.01, n=12, Student’s t-test compared to saline. C, Apoptosis within the V560G and D816V c-KIT tumors assessed by TUNEL staining. Images were acquired at 400x on DAPI (blue) and FITC (green) channels with an Axioplan 2 imaging system. Scale bars, 20 μm. D, Splenic weight (mg) recorded from mice harboring D816V c-KIT tumors on day 14 (top). Columns, mean; bars, SEM. *, p<0.05; **, p<0.01. Immunohistochemical staining of human c-KIT on spleen and bone marrow harvested at day 14 (bottom). Representative pictures were taken with a ColorView II camera and analysis software at 100× and 400× magnification. Scale bars, 100 μm (100×) and 20 μm (400×). The % TUNEL (C) or human c-KIT+ (D) cells were determined by counting the number of positive cells divided by total cells seen in 10 individual fields from 3 different mice. Columns, mean percentage of positive cells; bars, SEM. **, p<0.01, n=3, Student’s t-test compared to saline.
At day 14, a subset of mice from each group were sacrificed. Compared to saline-treated mice, FTY720 significantly reduced the tumor mass in both the V560G c-KIT and D816V c-KIT groups (Fig. 5B). Consistent with these observations, a significant increase in TUNEL-positive cells was observed in tumors treated with FTY720 compared to saline controls (Fig. 5C, Supplementary Fig. S8 and S9).
The D816V c-KIT tumors grew at a faster rate than those expressing V560G c-KIT (Supplementary Fig. S7). Consistent with this observation, saline-treated mice from the D816V c-KIT group developed splenomegaly by day 14, as indicated by an increase in splenic weight compared to age-matched controls (Fig. 5D). Immunohistochemical analysis revealed a disruption of splenic architecture and intense c-KIT-positive staining in saline-treated mice only (Fig. 5D). Importantly, the spleen size of FTY720-treated mice was significantly lower than saline-treated mice, the histopathology resembled that of age-matched controls, and the presence of c-KIT+ cells was almost non-detectable. Similar results were observed in the bone marrow (Fig. 5D). It should be noted that splenomegaly was not observed at day 14 in mice injected with V560G c-KIT cells (Supplementary Fig. S10). No signs of acute or delayed toxicity were observed with FTY720 treatment (data not shown).
DISCUSSION
This study has shown that inhibition of PP2A is a crucial mediator of c-KIT tumorigenesis. Constitutive activation of c-KIT via the juxtamembrane domain mutation, V560G, or kinase domain mutation, D816V, impairs the activity of the tumor suppressor PP2A. The mechanism of PP2A inhibition is associated with decreased expression of PP2A structural and regulatory subunits. Furthermore, pharmacological reactivation of PP2A specifically reduces the growth and survival of both imatinib-sensitive and -resistant c-KIT+ cells. Thus specific activation of PP2A provides a unique target for therapeutic intervention in cancers expressing mutant c-KIT.
Regulation of PP2A activity and specificity is complex. Translation of the catalytic subunit is tightly controlled (13), and indeed, no changes in the total protein expression or phosphorylation of PP2Ac in cells with or without c-KIT was found (Fig. 1). However, inhibition of PP2A in the mutant c-KIT+ cells was associated with reduced expression of the structural and a number of regulatory PP2A subunits (Fig. 1). Down-regulation of PP2A subunits has been reported in human gliomas, breast cancers and B-cell chronic lymphocytic leukemia (B-CLL) primary samples (15, 40, 41), and in melanoma and lung cancer cell lines (29, 42). Furthermore, suppression of PP2A-Aα or B56γ using shRNA induces tumorigenicity in transformed HEK293T cells (26, 29). That overexpression of the PP2A-Aα subunit induces apoptosis and inhibits the proliferation of FDC-P1 D816V c-KIT cells (Fig. 2), suggests that functional loss of PP2A is required for c-KIT-mediated tumorigenesis, and further implicates PP2A as a tumor suppressor.
We demonstrate that impaired PP2A activity can be restored in mutant c-KIT+ myeloid cells using the pharmacological agent, FTY720 (Fig. 3A). Importantly, treatment with FTY720 induces c-KIT tyrosine dephosphorylation on both imatinib-sensitive and -resistant activating mutants (Fig. 4A). The mechanisms underlying this are unclear. As PP2A is a serine/threonine phosphatase, dephosphorylation of c-KIT most likely involves the recruitment of additional proteins that act directly on the receptor, such as the c-KIT targeting tyrosine phosphatase SHP-1 (17, 43). Furthermore, FTY720 specifically inhibited components of the PI3K, MAPK and JAK/STAT pathways in the D816V c-KIT cells (Fig 4B). Thus the cytotoxicity in these cells can be explained by the fact that FTY720 inhibits both the D816V c-KIT receptor and its downstream signals.
Consistent with the dephosphorylation of oncogenic c-KIT and its downstream targets, reactivation of PP2A by FTY720 reduced the proliferation (Table 1), induced apoptosis (Fig. 3B) and suppressed the clonogenic potential of cells expressing V560G or D816V c-KIT (Fig. 3D). Importantly, at the same concentration of FTY720, these effects were not observed in GM-CSF dependent EV cells or WT c-KIT cells grown in SCF. This highlights that the growth inhibition with FTY720 treatment is dependent on the functional status of PP2A, whereby cells expressing activating c-KIT mutations are more sensitive due to impaired PP2A activity. The increased sensitivity of c-KIT mutants compared to WT c-KIT suggests that FTY720 could target malignant cells at low doses without disrupting normal SCF/c-KIT signaling.
We demonstrate that daily administration of FTY720 markedly delays the in vivo growth of tumors expressing activating c-KIT mutations via induction of apoptosis at the cellular level (Fig 5C). Importantly, the reduction in tumor mass resulted in prolonged survival of FTY720-treated mice compared to saline-treated controls (Fig. 5A). Moreover, FTY720 inhibited the engraftment of FDC-P1 D816Y c-KIT tumor cells into secondary lymphoid organs including the spleen and bone marrow (Fig. 5D). FTY720 also inhibited the proliferation of mutant c-KIT human cell lines, and induced apoptosis of primary blasts from a CBF-AML patient with a D816V c-KIT mutation (Table 1). Taken together, these results demonstrate that cancers harboring human c-KIT mutations are sensitive to FTY720 treatment.
FTY720 is a water-soluble, non-toxic drug with high oral bioavailability that is currently being evaluated as an immunomodulator in Phase III trials for multiple sclerosis patients (35, 44). FTY720 becomes phosphorylated in vivo by sphingosine kinase 2 and FTY720-P binds to S1PR, which induces its internalization (45). While the mechanism of action is not currently known, FTY720 has also been shown to activate purified PP2A in vitro (18), and its ability to activate PP2A and induce cytotoxicity has been demonstrated in a variety of leukemic models (19, 46). In agreement with the mechanism whereby FTY720 induces apoptosis of BCR/ABL+ myeloid and lymphoid progenitors (19), we have shown that PP2A reactivation does not require FTY720 phosphorylation (Fig. 3A). Accordingly, incubation with FTY720-P has no effect on cell viability or the clonogenic potential of mutant c-KIT+ cell lines (Table 1; Fig. 3). Furthermore, the induction of apoptosis by FTY720 specifically requires activation of PP2A, as inhibition of FTY720-induced PP2A activation by okadaic acid restores the clonogenic potential of mutant c-KIT+ cells (Fig. 3D). We also demonstrate that a chemically distinct PP2A activator, forskolin, inhibits the growth of c-KIT+ cells (Table 1). The hypersensitivity of oncogenic c-KIT+ cells to two distinct PP2A activators strongly indicates that reactivation of PP2A is the essential mechanism of action.
In summary, our data demonstrates for the first time that activating c-KIT mutations inhibit the tumor suppressor, PP2A, and that reactivating PP2A effectively suppresses the in vitro and in vivo growth of imatinib-sensitive and –resistant c-KIT+ cells. As kinase domain mutations, such as D816V, are most commonly detected in non-responsive patients, and there is currently no effective inhibitor against these mutants, PP2A reactivation represents a unique and powerful strategy for treating drug-resistant patients. Furthermore, our study together with those related to the interplay between PP2A and BCR/ABL (17) indicate that functional inactivation of PP2A tumor suppressor activity represents a key step in the induction and maintenance of leukemias and, perhaps, other solid tumors characterized by the aberrant activation of oncogenic tyrosine kinases.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the Cancer Council NSW, Anthony Rothe Memorial Trust, Cure Cancer Australia Foundation, and Hunter Medical Research Institute (NMV); the National Cancer Institute CA095512, NIH, Bethesda MD, and the US Army, CML Research Program, W81XWH-07-1-0270 (DP); and the American-Italian Cancer Foundation (PN). KGR is supported by an Australian Postgraduate Award and a Cancer Institute NSW Research Scholar Award; AMS by an Australian Postgraduate Award and an Arrow Bone Marrow Transplant Foundation Scholarship; DP is a Scholar of The Leukemia and Lymphoma Society; and NMV is a National Health and Medical Research Council Peter Doherty Fellow. We thank Prof. William Hahn for providing PP2A antibodies, Ms Lauren Watt for assistance with confocal microscopy (University of Newcastle, Australia), and Angela Tan and David Westerman for the c-KIT mutation screening of AML samples (Peter MacCallum Cancer Centre, Melbourne, Australia). A provisional patent has been filed for the use of PP2A activators to treat c-KIT cancers based on these data.
Abbreviations
- AML
acute myeloid leukemia
- CBF
core binding factor
- CML
chronic myeloid leukemia
- GIST
gastrointestinal stromal tumor
- PP2A
protein phosphatase 2A
- WT
wild-type
Footnotes
The authors declare no additional competing financial interests.
References
- 1.Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells. 2005;23:16–43. doi: 10.1634/stemcells.2004-0117. [DOI] [PubMed] [Google Scholar]
- 2.Linnekin D. Early signaling pathways activated by c-Kit in hematopoietic cells. Int J Biochem Cell Biol. 1999;31:1053–74. doi: 10.1016/s1357-2725(99)00078-3. [DOI] [PubMed] [Google Scholar]
- 3.Paschka P, Marcucci G, Ruppert AS, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;24:3904–11. doi: 10.1200/JCO.2006.06.9500. [DOI] [PubMed] [Google Scholar]
- 4.Worobec AS, Semere T, Nagata H, Metcalfe DD. Clinical correlates of the presence of the Asp816Val c-kit mutation in the peripheral blood mononuclear cells of patients with mastocytosis. Cancer. 1998;83:2120–9. doi: 10.1002/(sici)1097-0142(19981115)83:10<2120::aid-cncr10>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 5.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 6.Kemmer K, Corless CL, Fletcher JA, et al. KIT mutations are common in testicular seminomas. The American journal of pathology. 2004;164:305–13. doi: 10.1016/S0002-9440(10)63120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smalley KSM, Nathanson KL, Flaherty KT. Genetic Subgrouping of Melanoma Reveals New Opportunities for Targeted Therapy. Cancer research. 2009;69:3241–4. doi: 10.1158/0008-5472.CAN-08-4305. [DOI] [PubMed] [Google Scholar]
- 8.Mrozek K, Marcucci G, Paschka P, Bloomfield CD. Advances in molecular genetics and treatment of core-binding factor acute myeloid leukemia. Curr Opin Oncol. 2008;20:711–8. doi: 10.1097/CCO.0b013e32831369df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 10.Pardanani A, Elliott M, Reeder T, et al. Imatinib for systemic mast-cell disease. Lancet. 2003;362:535–6. doi: 10.1016/s0140-6736(03)14115-3. [DOI] [PubMed] [Google Scholar]
- 11.Piccaluga PP, Malagola M, Rondoni M, et al. Imatinib mesylate in the treatment of newly diagnosed or refractory/resistant c-KIT positive acute myeloid leukemia. Results of an Italian Multicentric Phase II Study. Haematologica. 2007;92:1721–2. doi: 10.3324/haematol.11345. [DOI] [PubMed] [Google Scholar]
- 12.Verstovsek S, Tefferi A, Cortes J, et al. Phase II Study of Dasatinib in Philadelphia Chromosome-Negative Acute and Chronic Myeloid Diseases, Including Systemic Mastocytosis. Clin Cancer Res. 2008;14:3906–15. doi: 10.1158/1078-0432.CCR-08-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–39. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calin GA, di Iasio MG, Caprini E, et al. Low frequency of alterations of the alpha (PPP2R1A) and beta (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene. 2000;19:1191–5. doi: 10.1038/sj.onc.1203389. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki K, Takahashi K. Reduced expression of the regulatory A subunit of serine/threonine protein phosphatase 2A in human breast cancer MCF-7 cells. International journal of oncology. 2003;23:1263–8. [PubMed] [Google Scholar]
- 16.Takagi Y, Futamura M, Yamaguchi K, Aoki S, Takahashi T, Saji S. Alterations of the PPP2R1B gene located at 11q23 in human colorectal cancers. Gut. 2000;47:268–71. doi: 10.1136/gut.47.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neviani P, Santhanam R, Trotta R, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355–68. doi: 10.1016/j.ccr.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 18.Matsuoka Y, Nagahara Y, Ikekita M, Shinomiya T. A novel immunosuppressive agent FTY720 induced Akt dephosphorylation in leukemia cells. Br J Pharmacol. 2003;138:1303–12. doi: 10.1038/sj.bjp.0705182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neviani P, Santhanam R, Oaks JJ, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest. 2007;117:2408–21. doi: 10.1172/JCI31095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dexter TM, Garland J, Scott D, Scolnick E, Metcalf D. Growth of factor-dependent hemopoietic precursor cell lines. J Exp Med. 1980;152:1036–47. doi: 10.1084/jem.152.4.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frost MJ, Ferrao PT, Hughes TP, Ashman LK. Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild-type c-kit whereas the kinase domain mutant D816VKit is resistant. Mol Cancer Ther. 2002;1:1115–24. [PubMed] [Google Scholar]
- 22.Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K, Kamada N. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood. 1991;77:2031–6. [PubMed] [Google Scholar]
- 23.Furitsu T, Tsujimura T, Tono T, et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest. 1993;92:1736–44. doi: 10.1172/JCI116761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lange B, Valtieri M, Santoli D, et al. Growth factor requirements of childhood acute leukemia: establishment of GM-CSF-dependent cell lines. Blood. 1987;70:192–9. [PubMed] [Google Scholar]
- 25.Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1) Int J Cancer. 1980;26:171–6. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- 26.Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer research. 2005;65:8183–92. doi: 10.1158/0008-5472.CAN-05-1103. [DOI] [PubMed] [Google Scholar]
- 27.Perrotti D, Cesi V, Trotta R, et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet. 2002;30:48–58. doi: 10.1038/ng791. [DOI] [PubMed] [Google Scholar]
- 28.Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment cell & melanoma research. 23:210–5. doi: 10.1111/j.1755-148X.2010.00671.x. [DOI] [PubMed] [Google Scholar]
- 29.Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell. 2004;5:127–36. doi: 10.1016/s1535-6108(04)00026-1. [DOI] [PubMed] [Google Scholar]
- 30.Sim AT, Collins E, Mudge LM, Rostas JA. Developmental regulation of protein phosphatase types 1 and 2A in post-hatch chicken brain. Neurochem Res. 1998;23:487–91. doi: 10.1023/a:1022422332404. [DOI] [PubMed] [Google Scholar]
- 31.Roberts KG, Odell AF, Byrnes EM, et al. Resistance to c-KIT kinase inhibitors conferred by V654A mutation. Mol Cancer Ther. 2007;6:1159–66. doi: 10.1158/1535-7163.MCT-06-0641. [DOI] [PubMed] [Google Scholar]
- 32.Verrills NM, Po’uha ST, Liu ML, et al. Alterations in gamma-actin and tubulin-targeted drug resistance in childhood leukemia. J Natl Cancer Inst. 2006;98:1363–74. doi: 10.1093/jnci/djj372. [DOI] [PubMed] [Google Scholar]
- 33.Sim AT, Ludowyke RI, Verrills NM. Mast cell function: Regulation of degranulation by serine/threonine phosphatases. Pharmacol Ther. 2006 doi: 10.1016/j.pharmthera.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 34.Janssens V, Longin S, Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail) Trends in Biochemical Sciences. 2008;33:113–21. doi: 10.1016/j.tibs.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-Out” Signaling of Sphingosine-1-Phosphate: Therapeutic Targets. Pharmacol Rev. 2008;60:181–95. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen P, Klumpp S, Schelling DL. An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 1989;250:596–600. doi: 10.1016/0014-5793(89)80803-8. [DOI] [PubMed] [Google Scholar]
- 37.Ashman LK. The biology of stem cell factor and its receptor C-kit. Int J Biochem Cell Biol. 1999;31:1037–51. doi: 10.1016/s1357-2725(99)00076-x. [DOI] [PubMed] [Google Scholar]
- 38.Casteran N, De Sepulveda P, Beslu N, et al. Signal transduction by several KIT juxtamembrane domain mutations. Oncogene. 2003;22:4710–22. doi: 10.1038/sj.onc.1206587. [DOI] [PubMed] [Google Scholar]
- 39.Chen H, Isozaki K, Kinoshita K, et al. Imatinib inhibits various types of activating mutant kit found in gastrointestinal stromal tumors. Int J Cancer. 2003;105:130–5. doi: 10.1002/ijc.11025. [DOI] [PubMed] [Google Scholar]
- 40.Colella S, Ohgaki H, Ruediger R, et al. Reduced expression of the Aalpha subunit of protein phosphatase 2A in human gliomas in the absence of mutations in the Aalpha and Abeta subunit genes. Int J Cancer. 2001;93:798–804. doi: 10.1002/ijc.1423. [DOI] [PubMed] [Google Scholar]
- 41.Kalla C, Scheuermann MO, Kube I, et al. Analysis of 11q22-q23 deletion target genes in B-cell chronic lymphocytic leukaemia: evidence for a pathogenic role of NPAT, CUL5, and PPP2R1B. Eur J Cancer. 2007;43:1328–35. doi: 10.1016/j.ejca.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Ito A, Koma Y, Watabe K, et al. A truncated isoform of the protein phosphatase 2A B56gamma regulatory subunit may promote genetic instability and cause tumor progression. Am J Pathol. 2003;162:81–91. doi: 10.1016/s0002-9440(10)63800-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozlowski M, Larose L, Lee F, Le DM, Rottapel R, Siminovitch KA. SHP-1 binds and negatively modulates the c-Kit receptor by interaction with tyrosine 569 in the c-Kit juxtamembrane domain. Molecular and cellular biology. 1998;18:2089–99. doi: 10.1128/mcb.18.4.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown BA, Kantesaria PP, McDevitt LM. Fingolimod: a novel immunosuppressant for multiple sclerosis. Ann Pharmacother. 2007;41:1660–8. doi: 10.1345/aph.1G424. [DOI] [PubMed] [Google Scholar]
- 45.Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–60. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 46.Liu Q, Zhao X, Frissora F, et al. FTY720 demonstrates promising preclinical activity for chronic lymphocytic leukemia and lymphoblastic leukemia/lymphoma. Blood. 2008;111:275–84. doi: 10.1182/blood-2006-10-053884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.