

Abstract
Neuroinflammation is associated with glial activation following a variety of brain injuries, including stroke. While activation of perilesional astrocytes and microglia following ischemic brain injury is well documented, the influence of age on these cellular responses after stroke is unclear. This study investigated the influence of advanced age on neuronal degeneration, neuroinflammation, and glial activation in female Sprague-Dawley rats after reversible embolic occlusion of the middle cerebral artery (MCAO). Results indicate that in comparison to young adult rats (3 months), aged rats (18 months) showed enhanced neuronal degeneration, altered microglial response, and a markedly increased expression of proinflammatory cytokines/chemokines following MCAO. In addition, the time-course for activation of STAT3, the signaling mechanism that regulates astrocyte reactivity, was truncated in the aged rats after MCAO. Moreover, the expression of SOCS3, which is associated with termination of astrogliosis, was enhanced as a function of age after MCAO. These findings are suggestive of an enhanced proinflammatory response and a truncated astroglial response as a function of advanced age following MCAO. These data provide further evidence of the prominent role played by age in the molecular and cellular responses to ischemic stroke and suggest that astrocytes may represent targets for future therapies aimed at improving stroke outcome.
Keywords: neurovascular unit, neuropoietic, IL6, SOCS3, astrocyte, microglia
Treatment of acute ischemic stroke injury is hampered by the inability to translate successful animal studies into clinically effective therapies. Despite considerable research interest in specific cardiovascular risk factors for stroke, such as hypertension, hypercholesterolemia, and diabetes, data from the Framingham Heart study demonstrate that age is the single greatest risk factor for stroke (Grossi, 2008). Aging results in enhanced basal expression of proinflammatory cytokines and these same proinflammatory mediators often are associated with neural injury-related activation of microglia and astroglia. Taken together, the aging, inflammation and glial activation phenotypes serve as the basis for the “inflam-aging” hypothesis (Salvioli et al., 2006; Franceschi et al., 2007; Giunta, 2008). According to this hypothesis, increased inflammation during the aging process results from dysregulation of the immune system and a progressive inability to properly handle pathological stimuli (Chung et al., 2001; Giunta, 2008). Studies on aging confirm that proinflammatory cytokines in the IL-6 and TNF-α families are participants in the complex relationship between aging and chronic morbidity (Giunta, 2008). Proinflammatory cytokines in the IL-6 family, also known as neuropoietic cytokines (Bauer et al., 2007), preferentially activate janus kinases (JAK) and the signal transducers and activators of transcription (STAT) pathways (Hirano et al., 2000). Through JAK2/STAT3 signaling, these cytokines activate target genes involved in immune responses, differentiation, survival, apoptosis, and proliferation. Activation of the JAK2/STAT3 pathway is associated with trauma and toxicant-induced astroglial activation (Sriram et al., 2004; Sofroniew, 2009) as well as the acute injury response following stroke (Planas et al., 1996; Yamashita et al., 2005; Satriomoto et al., 2006; Xie et al., 2007; Shyu et al., 2008). Taken together, these observations raise the possibility that aging may affect the proinflammatory and astrocytic response to stroke. Therefore, in the present study, we examined the effects of age on proinflammatory cytokine expression and STAT3 activation during the acute phase of injury following middle cerebral artery occlusion (MCAO) and tissue plasminogen activator (tPA) reperfusion. We found that the aged rat displayed an enhanced proinflammatory and an attenuated astrocytic response to stroke, findings with implications for future therapies.
Materials and Methods
Chemicals and Animals
All chemicals used in this study were of molecular biology grade and purchased from Sigma Chemical (St. Louis, MO), unless otherwise noted. Human recombinant tissue plasminogen activator (tPA) was kindly gifted by Genentech (South San Francisco, CA). Female Sprague-Dawley rats [(3-4 months) and (18-20 months)] were received from Hilltop Animal Laboratory (Scottdale, PA) and housed under 12 hr light/12 hr dark conditions with food and water available ad libitum. All procedures involving rats abided by the West Virginia University Animal Care and Use Committee.
MCAO procedure
Rats from both age groups were randomly divided into MCAO and sham surgery groups. Rats were anesthetized with inhaled isoflurane (4% induction; 2% maintenance) and underwent MCAO for 2 h followed by reperfusion using human recombinant tPA (5 mg/kg; i.v.,femoral artery; 30% bolus and 70% infused over 30 min via syringe drive) as previously described (DiNapoli et al., 2006). Ischemia was defined as a perfusion drop across the MCA region of >80% as determined by laser Doppler and successful reperfusion was denoted as a return to >80% of baseline perfusion rate by 30 min after tPA administration.
Tissue preparation and immunoblot analyses
At 6, 24, and 72 h following MCAO, rats (n=3 rats/group) were euthanized by focused microwave irradiation (3 kW for 1.5 s) to preserve steady-state phosphorylation (O'Callaghan and Sriram, 2004; Scharf et al., 2008), using a microwave applicator (Muromachi Kikai, Inc; Tokyo, Japan). Brains were removed from skulls, cortical hemispheres separated on a cold plate, weighed, homogenized in 10 volumes of hot (85–95 °C) 1% SDS, and stored at -80° C until used. Total protein was determined by bicinchoninic acid method (Smith et al., 1985) using bovine serum albumin as standard. Activation of the STAT3 pathway was assessed by quantifying pSTAT3tyr705 using immunoblot analysis with detection of fluorescent signals using an infrared fluorescence scanner (Licor Biosciences; Lincoln, NE). Primary antibodies used in this study were (rabbit anti-STAT3 (1:500)(Santa Cruz Biotechnology, Santa Cruz, CA) and rabbit anti-phospho-STAT3tyr705 (pSTAT3; 1:500) (Cell Signaling Technology, Inc., Beverly, MA). Following incubation with primary antibodies, blots were washed with phosphate buffered saline with 0.1% Tween 20 (1 × 15 min; 2 × 5 min) and incubated with fluorescent-labeled anti-rabbit and anti-mouse IgG antibodies (1:2500) for 1 h. Antibody specificity was confirmed by immunoblots of the tissue homogenates.
RNA isolation, cDNA synthesis and real-time PCR amplification
Total RNA from the ipsilateral and contralateral hemispheres were isolated from young adult and aged rats (n=4 rats/group) following MCAO using Trizol® reagent (Invitrogen; Carlsbad, CA). Concentration and purity of RNA was determined using a biophotometer and considered for use only if A260/A280 was between 1.8 and 2.1. Total RNA (1μg) was reverse-transcribed to cDNA using SuperScript™ III RNase H- and oligo (dT)12-18 primers (Invitrogen) in a 40μl reaction. Real-time PCR analyses of IL-6 and SOCS3 were performed at 12, 24, and 72 h following MCAO and CNTF, CT-1, OSM, LIF, TNFα and CCL-2 were performed at 12 h following MCAO using an Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems; Foster City, CA) in combination with TaqMan® chemistry. Glyceraldeyde-3-phosphate dehydrogenase (GAPDH) was used as an endogenous control to normalize for differences in amount of cDNA added to reactions. Specific primers and dual-labeled internal fluorogenic (FAM/TAMRA) probe sets (TaqMan® Gene Expression Assays) for these genes were used according to the manufacturer's recommendation (Applied Biosystems). All PCR amplifications (40 cycles) were performed in a total volume of 50μl, containing 1μl cDNA, 2.5μl of the specific Assay on Demand® primer/probe mix, and 25μl of TaqMan® Universal master mix (Applied Biosystems). Relative quantification of gene expression was performed using the comparative threshold (CT) method as described by manufacturer (User Bulletin 2; Applied Biosystems). Changes in mRNA expression level were calculated following normalization to GAPDH (which did not change with age) and expressed as fold change over corresponding age-matched rats (n=3).
Neuropathology
At 6, 24, or 72 h following MCAO, rats (n=3 rats/group) were anesthetized with pentobarbital sodium (65 mg/kg, i.p.) and perfused transcardially with 100 ml wash solution (0.8% NaCl, 0.4% dextrose, 0.8% sucrose, 0.023% CaCl2, 0.025% sodium cacodylate) followed by 150 ml perfusion solution (4.0% sucrose, 4.0% paraformaldehyde, 1.072% sodium cacodylate). Brains were removed from skull, stored in fixative overnight, and incubated serially in 10, 20, and 30% sucrose in Dulbecco's modified phosphate buffered saline (DPBS) for 24 h. Brains were cryosectioned (25 μm) in the horizontal plane and stored in DPBS with 0.1% sodium azide until used. Brain sections were incubated with Fluoro-Jade B, a fluorescent marker for localization of degenerating neurons using a slightly modified technique (Schmued and Hopkins, 2000). Free-floating sections were mounted onto microscope slides, immersed in distilled water for 1 min, 70% ethanol for 2 min, and distilled water for 2 min. Background staining was suppressed by incubation in 0.06% KMnO4 for 10 min with shaking. A rinse in distilled water for 2 min was followed by immersion in staining solution (0.01% stock solution, four ml of stock solution diluted in 96 ml of 0.1% CH3COOH) for 20 min. After staining, slides were washed 3× in distilled water for 1 min and air dried overnight. Slides were placed on a warmer at 55°C for 5 min, cleared in xylene for 5 min and coverslipped.
Immunohistochemistry
To visualize STAT3 immunoreactivity, free floating brain sections (n=3 rats/group) were stained using a modified ABC procedure (Vector Laboratories; Burlingame, CA) (Benkovic et al., 2004). Sections were treated with 10% hydrogen peroxide in DPBS for 15 min to quench endogenous peroxidases. Following 3× rinses in DPBS for 5 min, sections were incubated in a permeabilizing solution (1.8% L-lysine, 4% normal horse serum, 0.2% Triton X-100) for 30 min at room temperature. Sections were transferred directly to primary antibody solution with 4% horse serum (rabbit anti-STAT3; 1:400; Abcam; Cambridge, MA) and incubated overnight at room temperature. The following day, sections were rinsed 3× in DPBS for 5 min and transferred to the secondary antibody for 2 h (anti-rabbit IgG; 1:1000; Invitrogen). Following 3× rinses in DPBS for 5 min, sections were incubated in Avidin D-HRP (1:1000; Vector Laboratories) for 1 h at room temperature; rinsed 3× in DPBS, and incubated with Nova Red (Vector Laboratories) for 5 min. Following a 5 min rinse in distilled water, sections were mounted onto microscope slides, air-dried overnight, dehydrated through a standard ethanol series, and coverslipped.
Double-immunohistochemical detection of STAT3 and GFAP was evaluated using sequential procedures with STAT3 staining performed first using a nickel-enhanced procedure that imparts a blue color to STAT3. The third rinse before and the first rinse after chromagen incubation was in 100 mM tris buffered saline; the chromagen solution contained 50 mg NiCl in 5 ml TBS + 50 mg 3-3′ diaminobenzidine (DAB) in 5 ml TBS + 10 ml 30% H2O2. GFAP was immunolocalized using the following solutions: primary rabbit anti-cow GFAP, 1:10,000, (Dako; Glostrup, Denmark); secondary anti-rabbit IgG, 1:10,000 (Vector Laboratories); and Nova Red chromagen (Vector Laboratories).
Immunohistochemical detection of Iba-1 was performed as described above using the following antibody dilutions for Iba-1: primary rabbit anti-Iba-1 (1:500; Wako, Richmond, VA); secondary anti-rabbit IgG, (1:1000; Vector Laboratories), and chromagen Nova Red.
Microscopic analysis of stained sections was performed on an Olympus BX-50 microscope (Olympus America; Center Valley, PA) interfaced with a Spot II digital camera (Diagnostic Instruments Inc.; Sterling Heights, MI). Images were captured with Spot software (v4.5) and assembled and labeled in Adobe Photoshop CS2 (Adobe Systems Inc.; San Jose, CA).
Statistical analysis
Data are presented as mean ± S.E. Statistical significance in measured and calculated parameters was determined using two-way ANOVA and Tukey's post-hoc analyses. Level of significance was set at p<0.05.
Results
Aged rats displayed an earlier and greater degree of neurodegeneration than young rats following MCAO
In previous studies we used a classical stroke volume measure to show that advanced age leads to enlarged stroke volume following MCAO (DiNapoli et al., 2006). Here, we used a sensitive neuronal degeneration stain to examine the effects of age on neurodegeneration following MCAO. In young rats, at 6h following MCAO, Fluoro-Jade B staining was observed in the striatum of the ipsilateral hemisphere (Fig 1A) while the cortex on the ipsilateral side was devoid of neuronal degeneration (Fig 1B). In aged rats, at 6 h following MCAO, Fluoro-Jade B staining was observed in both the ipsilateral striatum (Fig 1C) and ipsilateral cortex (Fig 1D). In young rats, by 24 h following MCAO, Fluoro-Jade B staining was persistently observed in striatum (Fig 1 E), and appeared in all neuronal layers of the cortex on the ipsilateral side (Fig 1 F) but was restricted to the rostral-caudal extent of the brain supplied by the MCA (data not shown). No change in intensity of Fluoro-Jade B staining was observed in the aged rat at 24 h after MCAO (data now shown) as compared to 6 h following MCAO (Fig 1C and 1D.), however, Fluroro-Jade B staining in aged rats extended beyond the region supplied by the MCA (data not shown). No Fluoro-Jade B staining was observed in the contralateral hemispheres of any rats at 6 or 24 h post MCAO (data not shown).
Figure 1. Aged rats display greater degree of neurodegeneration than young rats following MCAO.

MCAO results in neurodegeneration in both young and aged rats. Arrows show examples of Fluoro-Jade stained (degenerating) neurons. In young adult rats at 6 h post MCAO, neuronal damage was confined to ipsilateral striatum (A) and did not extend into cortex (B). In aged animals at 6 h post MCAO, neuronal damage was observed in both ipsilateral striatum (C) and cortex (D). By 24 h post MCAO in young animals, neurodegeneration was persistently observed in ipsilateral striatum (E) and appeared in ipsilateral cortex (F). Bar = 100 μ.
Aged rats present a different microglial phenotype from the one seen in young adult rats following MCAO
Activated microglia have been associated with proinflammatory responses and neural injury, including stroke. To determine if age affected microglial responses following MCAO, we immunostained brain sections with Iba-1, a calcium-binding protein and microglial marker. We found differences in microglial phenotype between young and aged rats in the ipsilateral cortex at 24 h following MCAO. At 24 h following MCAO and tPA reperfusion, the ipsilateral cortex of young adult rats contained enlarged microglia with thick processes which appeared to contain clumps of engulfed cellular debris (Fig 2B); whereas in the ipsilateral cortex of aged rat, most microglial cells displayed the fully phagocytic phenotype consisting of rounded cell bodies with few extended processes (Fig 2D). In the contralateral hemisphere, both young adult and aged rats displayed microglial cells with typical “resting” or “surveillance” morphology, consisting of a uniform distribution of relatively small cell soma with many highly branched, thin processes at 24 h following MCAO and tPA reperfusion (Fig 2A and 2C).
Figure 2. Aged rats present a different microglial phenotype than young adult rats following MCAO.

At 24 h following MCAO, microglia in the ipsilateral hemisphere of young adult and aged rats exhibited different phenotypes as revealed by Iba-1 immunohistochemistry. In the contralateral cortex of young adult rats, microglia expressed the resting or ramified phenotype with many long, thin, and highly branched processes extending into three-dimensional cortical parenchyma (A). In the ipsilateral cortex of young adult rats, microglial processes appeared swollen and contained clumps of phagocytosed material (B). In the contralateral cortex of aged rats, microglia appeared in the resting phenotype (C). In aged rats, MCAO resulted in the transformation of microglia into the fully phagocytic phenotype: round or oval cell soma with few processes extending from the cell body (D). Arrows denote different microglial phenotypes in young and old rats following MCAO. Bar = 50 μ.
Aged rats have enhanced expression of prioinflammatory cytokines and chemokines following MCAO
Enhanced neurodegeneration and microglial activation have been associated with enhanced proinflammatory responses. To determine if this was the case as a function of age following MCAO, we examined the expression of the IL-6 family of cytokines, TNF-α and the proinflammatory chemokine, CCL2. At 12, 24, and 72 h following MCAO, changes in IL-6 mRNA expression in young adult and aged rats were measured using real-time PCR. Results were denoted as ΔΔCT and signify fold change in mRNA expression as compared to age-matched sham control rats (Fig 3A). At 12 and 24 h following MCAO, a significant (p<0.001) increase in IL-6 mRNA expression was observed in the ipsilateral hemisphere of both young adult and aged rats compared to values obtained for corresponding sham controls. At 12 h following MCAO, a significant increase in IL-6 mRNA expression (p<0.01) was observed for the ipsilateral hemisphere of the aged rats in comparison to values obtained from the ipsilateral hemisphere of young adult rats. At 24 h following MCAO, IL-6 mRNA expression was significantly (p<0.05) decreased in the ipsilateral hemisphere of aged rats compared to young adult rats. At 72 h following MCAO, no difference (p>0.05) in IL-6 mRNA expression was observed between the ipsilateral hemisphere of young adult and aged rats. No difference (p>0.05) was observed in IL-6 mRNA expression between naïve and sham surgery rats. No difference in basal IL-6 mRNA expression was observed between young adult and aged rats for any of the cytokines measured.
Figure 3. Aged rats have enhanced expression of prioinflammatory cytokines/chemokines following MCAO.

(A) Changes in IL-6 mRNA expression were measured using real-time PCR at 6, 24, and 72 h following MCAO and (B-E) changes in other proinflammatory cytokine and chemokine cytokine mRNA were measured at 12 h following MCAO. Results were calculated from ΔΔCT and signify fold change in mRNA expression normalized to age-matched naïve rats. Bars represent mean ± S.E. (n=4 rats/group). No difference (p>0.05) in expression of cytokine/chemokine mRNA were observed among sham rats of either age at the three time points; thus only sham rats at 24 h were displayed (A). Statistical significance was determined using two-way ANOVA with Tukey's post hoc analysis. *, **, *** denote statistical significance (p<0.05, p<0.01 and p<0.001, respectively) as compared to age-matched sham. # ### denote statistical significance (p<0.05 and p<0.001, respectively) as compared to young adult ipsilateral hemisphere at same time point. $ denotes statistical significance (p<0.05) as compared to young adult contralateral hemisphere at same time point.
Based on the magnitude of the increase in IL-6 mRNA at 12 h following MCAO, changes in expression of the IL-6 family of cytokines (CNTF, CT-1, LIF, and OSM) in young adult and aged rats were measured at 12 h following MCAO using real-time PCR. No difference in basal expression of these cytokine mRNAs was observed in the cortex between young adult and aged rats. No effect of age or MCAO on CNTF and CT-1 mRNA expression was observed (p>0.05; data not shown). At 12 h following MCAO, a significant increase in LIF (Fig 3B) and OSM (Fig 3C) mRNA expression was demonstrated in the ipsilateral hemisphere of young adult (29 ± 5 and 6.1 ± 1.2-fold, respectively) and aged (85 ± 18- and 14 ± 4-fold, respectively) rats compared to corresponding mRNA values for the ipsilateral hemispheres of age-matched sham rats. Following MCAO (12 h), a significant (p<0.05) increase in mRNA expression for LIF and OSM was observed in aged rats as compared to corresponding mRNA values for the ipsilateral hemisphere of young adult rats. Following MCAO (12 h), a significant (p<0.05) increase in OSM mRNA expression was measured in the contralateral hemisphere of aged rats (5.6 ± 1.1-fold) as compared to OSM mRNA values in young adult rats (1.9 ± 0.3-fold) (Fig 3B & C). No differences (p>0.05) were observed in CNTF, CT-1, LIF, and OSM mRNA expression values between naïve and sham surgery rats (data not shown).
Real-time PCR of TNFα and CCL2 mRNA was evaluated in young adult and aged rats (Fig. 3D & E). At 12 h following MCAO, both young adult and aged rats showed a significant increase in expression of TNFα (3.1 ± 0.5 and 15.4 ± 2.8-fold, respectively) and CCL2 (508 ± 48 and 1279 ± 343-fold, respectively) mRNA in the ipsilateral hemisphere as compared to values for corresponding age-matched, sham rats (Fig 3D and 3E). Following MCAO, a significant (p<0.05) increase in TNFα mRNA expression was observed in the contralateral hemisphere of aged rats as compared to both the age-matched sham (6.0 ± 0.9-fold) and the contralateral hemisphere of young adult rats. Following MCAO, a significant increase in CCL2 mRNA expression was observed in the contralateral hemisphere of both young adult (10.0 ± 3.2-fold) and aged (59 ± 8-fold) rats as compared to age-matched sham rats. No difference (p>0.05) was observed in TNFα and CCL2 mRNA expression between naïve and sham surgery rats. No difference (p>0.05) in basal TNFα and CCL2 mRNA expression was observed between young adult and aged rats.
MCAO results in astrogliosis and translocation of STAT3 to astrocytic nuclei
Our previous findings demonstrated that neurotoxic damage to nerve terminals and perikarya (Sriram et al., 2004; O'Callaghan et al., 2007) and the traumatic injury from brain slicing (Damiani and O'Callaghan, 2007) results in the elaboration of proinflammatory cytokines and chemokines in the regions of damage. These effects are followed by apparent down-stream activation of JAK2-STAT3 signaling in astrocytes and subsequent astrogliosis (Sriram et al., 2004; Damiani and O'Callaghan, 2007; O'Callaghan et al., 2007). Given the similar proinflammatory response we observed after MCAO, we examined the effects of MCAO and age on astrogliosis and STAT3 translocation using GFAP and STAT3 immunostaining (Fig 4). By 24 h after MCAO, we observed evidence of astrogliosis in young adult (Fig 4B,D,F) and aged (Fig 4H) rats, based on GFAP immunohistochemistry. Astrogliosis appeared in multiple ipsilateral brain regions including striatum, cortex, and hippocampus in young adult and aged rats. In regions supplied by the MCA, obstruction of the artery was associated with poor immunostaining and necrotic loss of tissue proximal to the lesion that was especially evident in aged rats; consequently, the cellular localization of GFAP and STAT3 often was obscured in these areas. In young adult rats, basal GFAP immunoreactivity was observed in contralateral cortices and consisted of somatic staining that extended into a few long, thin processes (Fig 4A). On the ipsilateral side (Fig 4B), enhanced GFAP immunoreactivity was observed in rostral occipital cortex and extended throughout all six layers (Fig 4B); most of the parietal cortex was necrotic and non-staining. Frontal and entorhinal ipsilateral cortices in young adult and aged rats displayed basal immunoreactivity (data not shown). In hippocampus of young adults, basal GFAP immunoreactivity was observed throughout all contralateral hippocampal strata and the dentate gyrus (Fig 4C), and was elevated by MCAO throughout ipsilateral stratum oriens, stratum radiatum proximal to the hippocampal fissure, stratum lacunosum-moleculare, and the molecular layer and hilus of the dentate gyrus (Fig 4D). After MCAO in young adults, astrocytic hypertrophy was notable in the ipsilateral cortex (Fig 4F) viewed at higher magnification and was manifested by thicker GFAP positive astrocytic processes in comparison to astrocytes in the contralateral hemisphere (Fig 4E). In ipsilateral striatum of young adults, enhanced GFAP immunoreactivity was observed compared to the contralateral hemisphere throughout the tissue bordering the necrotic zone (data not shown). In ipsilateral cortex of aged rats (Fig 4H), enhanced GFAP immunoreactivity associated with hypertrophic astrocytes was observed in tissue bordering the necrotic zone compared to GFAP immunoreactivity seen in the contralateral hemisphere (Fig 4G). Overall, there appeared to be less GFAP immunostaining in the ipsilateral cortex of the aged rats (Fig 4H) in comparison to GFAP immunostaining seen in young adults rats (Fig 4F).
Figure 4. MCAO induces reactive gliosis and nuclear translocation of STAT3 in astrocytes.

At 24 h following MCAO in young rats, enhanced immuonstaining of GFAP-positive cells (astrocytes) was observed in ipsilateral cortex (B) compared to the GFAP immunostaining observed in contralateral cortex (A). Basal GFAP immunostaining was observed in contralateral hippocampus (C) that was enhanced in iplateral hippocampus (D). MCAO also resulted in the appearance of STAT3 immunoreactivity in astrocytic nuclei (B, D, F). High magnification microscopy reveals the lack of STAT3 immunoreactivity in astrocytes of the contralateral cortex (E), while the nuclei of astrocytes in ipsilateral cortex appear blue-black from double-immunocytochemical labeling (F). At 24 h following MCAO in aged rats, enhanced GFAP and STAT3 immunostaining also was observed in ipsilateral cortex (H) compared to GFAP and STAT3 immunostaining observed in contralateral cortex. In general GFAP and STAT3 immunostaining in ipsilateral cortex of aged rats (H) was less than that observed in young rats (F). Arrows denote STAT3 immunostaining in GFAP-positive astrocytes. Bar A - B = 100 μ, C - D = 200 μm, E - H = 50 μ.
STAT3 immunostaining 24 h after MCAO revealed a nuclear accumulation of this transcription factor in astrocytic soma ipsilateral to the lesion (Fig 4 B, D, F, H). The greatest STAT3-immunoreactivity was observed in the ipsilateral cortex of young adult rats (Fig 4F) where nuclei of astrocytes appear blue-black from double-immunostaining for GFAP and STAT3 (Fig 4 F). Considerable double-staining also was observed at the rostral extent of parietal cortex and rostral striatum just bordering the zone of necrosis (data not shown). In the ipsilateral cortex of aged rats (Fig 4 H), less double-immunostaining was observed in comparison to double immunostaining observed in young adult rats (Fig 4F).
Age attenuates the duration of STAT3 activation following MCAO
Representative immunoblots for STAT3 and pSTAT3 data are shown in Fig 5A. Western blot analyses confirmed a significant (p<0.001) increase in activated STAT3 (pSTAT3tyr 705) in the ipsilateral hemisphere of young adult and aged rats at 6, 24 and 72 h following MCAO as compared to sham-operated, age-matched controls (Fig 5B). At 6 h, no difference (p>0.05) in pSTAT3 expression was observed in the ipsilateral hemisphere of aged rats as compared to young adult rats. At 6 and 24 h following MCAO, a significant (p<0.05) increase in pSTAT3 expression in both young adult and aged rats was observed in the contralateral hemisphere. At 72 h after MCAO, aged rats showed a decrease in pSTAT3 expression to levels similar to sham-operated, age-matched controls; whereas, young adult rats continued to display expression of pSTAT3 in the ipsilateral hemisphere at significantly (p<0.05) increased levels similar to 6 and 24 h (Fig 5B). Levels of total STAT3 were not affected as a function of age or MCAO (data not shown).
Figure 5. Activation of STAT3 following MCAO is truncated in aged rats.
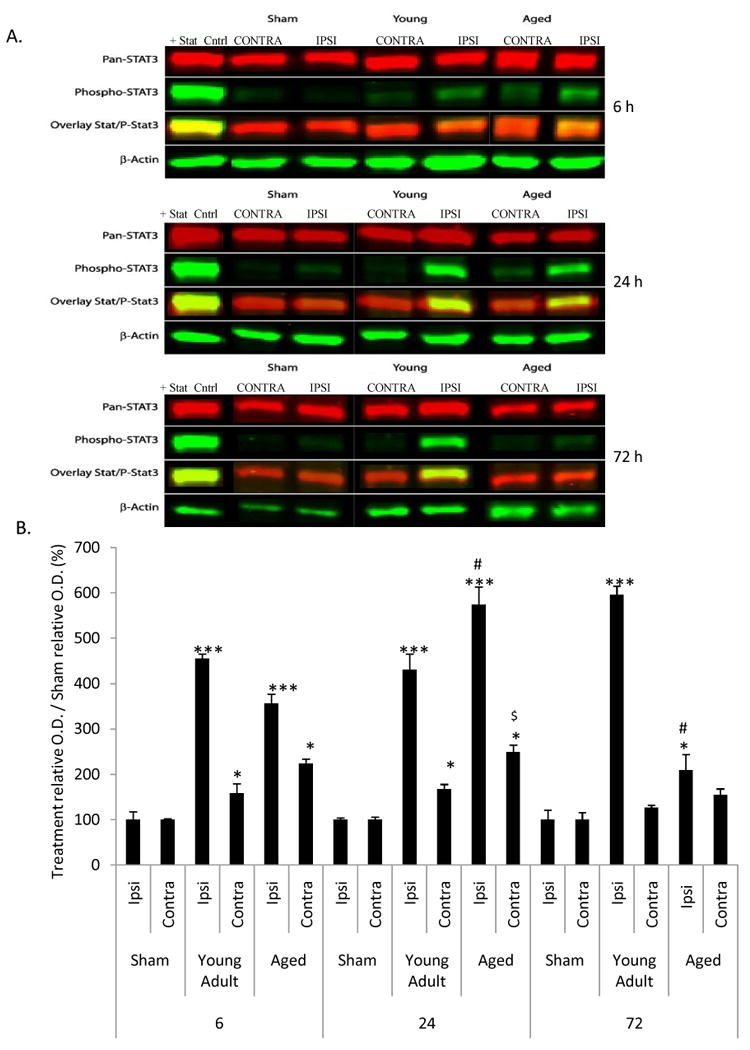
At 6, 24, and 72 h following MCAO, STAT3 and pSTAT3 expression were evaluated quantitatively by immunoblot analysis. Fig 5A shows representative immunoblots for STAT3 and pSTAT3 data. No difference (p>0.05) between basal expression of STAT3 and pSTAT3 expression were noted at any time point. The sham groups on the representative blots were from aged rats. Fig 5B depicts changes in pSTAT3 expression in young adult and aged rats. Bars represent mean ± S.E. (n=3 rats/group). Statistical significance was determined using two-way ANOVA with Tukey's post hoc analysis.*, **, *** denote statistical significance (p<0.05, p<0.01 and p<0.001, respectively) as compared to age-matched sham. # ### denote statistical significance (p<0.05 and p<0.001, respectively) as compared to young adult ipsilateral hemisphere at same time point. $ denotes statistical significance (p<0.05) as compared to young adult contralateral hemisphere at same time point. Total STAT3 remained unchanged as a function of age or MCAO.
Age enhances the expression of SOCS3 mRNA following MCAO
The suppressor of cytokine signaling (SOCS3) is activated through the JAK-STAT pathway and inhibits STAT-mediated signaling. Previously, we showed that rapid induction of SOCS3 mRNA was associated with cessation of STAT3-mediated signaling after nerve terminal damage and associated astrogliosis (Sriram et al., 2004). These prior observations suggest that early termination of STAT3 activation after MCAO in aged rats may be associated with enhanced expression of SOCS3. Real-time PCR of SOCS3 was performed at 12, 24, and 72 h following MCAO. Results were denoted as ΔΔCT and signify fold change in mRNA expression as compared to age-matched sham controls (Fig 6). Both young adult and aged rats showed a significant increase in expression of SOCS3 mRNA (5.6 and 15.3 fold, respectively) as compared to age-matched sham controls (Fig 6). Aged rats displayed a significantly (p<0.001) increased SOCS3 mRNA expression in the ipsilateral hemisphere as compared to young adult rats following MCAO (Fig 6). A significant (p<0.05) increase in SOCS3 mRNA expression was observed in the contralateral hemisphere of aged rats at 12 h following MCAO and in the ipsilateral hemisphere of both young adult and aged rats at 24 h following MCAO. No difference (p>0.05) in SOCS3 mRNA expression was observed in the ipsilateral hemisphere of young adult and aged rats at 72 h following MCAO. No difference (p>0.05) was observed in SOCS3 mRNA expression between naïve and sham surgery rats. No difference in basal SOCS3 mRNA expression was observed between young adult and aged rats.
Figure 6. Age enhances the expression of SOCS3 after MCAO.

At 12, 24, and 72 h following MCAO, changes in SOCS3 mRNA expression in young adult and aged rats were measured using real-time PCR. Results are calculated from ΔΔCT and signify fold change in mRNA expression normalized to age-matched naïve rats. Bars represent mean ± S.E. (n=4 rats/group). No difference (p>0.05) in SOCS3 mRNA expression was observed in sham rats of either age at the three time points; thus, only sham rats at 24 h were displayed. Statistical significance was determined using two-way ANOVA with Tukey's post hoc analysis. *, *** denote statistical significance (p<0.05 and p<0.001, respectively) as compared to age-matched sham. ### denotes statistical significance (p<0.001) as compared to ipsilateral hemisphere of young adult rats following MCAO at same time point.
Discussion
In previous studies, we demonstrated that age worsened ischemic brain damage following MCAO in the rat, as indicated using TTC staining (DiNapoli et al., 2006; DiNapoli et al., 2008, Tan et al., 2009, Kelly et al., 2009). In this study, we show that age increases the proinflammatory response to MCAO in the rat, effects associated with enhanced ipsilateral neuronal damage, based on Fluoro-Jade staining, and a phagocytic microglial phenotype, based on Iba-1 immunostaining. Activation of the JAK2-STAT3 pathway has been linked to neuroinflammation and the induction of astogliosis following neurotoxic insults (Sriram et al., 2004; Damiani and O'Callaghan, 2007; O'Callaghan et al., 2007) as well as to trauma (Okada et al., 2006; Hermann et al., 2008; Sofroniew, 2009) and stroke (Planas et al., 1996; Xie et al., 2007; Shyu et al., 2008). Therefore, we examined whether the enhanced expression of proinflammatory mediators upstream of STAT3 was associated with age-related effects on activation of the STAT3 pathway. While we observed a rapid phosphorylation of STAT3tyr 705 in both young adult and aged rats following ischemic/reperfusion injury, we measured an early attenuation and suppression of this activation in aged animals, despite increased expression of upstream proinflammatory mediators linked to activation of the STAT3 pathway (Sriram et al., 2004; Damiani and O'Callaghan 2007). We also observed that the abbreviated time course of STAT3 phosphorylation in aged animals was associated with a concomitant increase in SOCS3 mRNA in aged rats to levels three times that seen in young adult rats at the equivalent time point. These latter findings provide a mechanism by which STAT3 signaling is terminated early as a function of aging following MCAO. To the extent that the effects observed here relate to neurotoxic and traumatic brain injuries, our findings are in agreement with prior findings showing an enhanced inflammatory response is associated with, but not required for, the induction of astrogliosis (Sriram et al. 2004; Sriram et al. 2006a; O'Callaghan et al., 2008). Our findings also suggest that a truncated astroglial response to injury is associated with and, perhaps, can lead to, increased neuropathology following MCAO in the aged rat.
Proinflammatory mediators, including those examined here (IL-6 family of cytokines, TNF-α, and CCL2) have been implicated in a variety of neurodegenerative disease states and neurotoxic conditions, often in association with the activation of microglia (Liu and Hong, 2003; Sriram et al., 2006b; Block et al., 2007). Both detrimental (Liu and Hong, 2003; Sriram and O'Callaghan, 2007) and beneficial roles (Bauer et al., 2007; Sriram and O'Callaghan, 2007) have been ascribed to this neuroinflammatory condition. While precise roles for brain proinflammatory cytokines or chemokines in health and disease have yet to be established (Dantzer et al., 2008), it has become clear that aging enhances neuroflammation (Dilger and Johnson, 2008). Age-related neuroinflammation is associated with sensitization of the brain to infection, stress, and even injury and disease (Sparkman and Johnson, 2008). “Priming” or sensitization of micoglia appears to be the basis of this age-related enhancement in neuroinflammatory responses (Dilger and Johnson, 2008). In the context of the data presented here, microglia in the aged rats would be “primed” to mount an exaggerated neuroinflammatory response to the triggering stimuli, in this case, MCAO. Much of the literature concerning the effects of age on brain inflammatory responses focuses on adverse effects related to enhanced sickness behavior and depressive episodes that result from an amplified response to systemic infection (Dantzer et al., 2008). If aging also results in an enhanced neuroinflammatory response to stroke, as our results suggest, the consequences may be reflected in enhanced neuropathology with accompanying decrements in function and delays in recovery. Nevertheless, clinical data show both improved (Lampl et al., 2009) and worsened (Abraham et al., 2008; Haag et al., 2008) stroke outcomes due to use of, or treatment with, anti-inflammatory drugs. These latter observations underscore the potential neurotrophic as well as neurodegenerative effects ascribed to proinflammatory mediators, and they suggest that the complex roles for these pleiotrophic mediators in stroke need to be more clearly defined before effective therapeutic manipulations can be implemented.
Beyond its role as a mediator of acute phase responses in a variety of target organs, including brain, STAT3 signaling has been shown to be a critical regulator of astrogliosis (Herrmann et al., 2008; Sofroniew, 2009). Conditional ablation of STAT3 inhibits astrocytic hypertrophy and scar formation and promotes the spread of inflammation at the lesion site following spinal cord injury (Okada, et al., 2006; Hermann et al., 2008). These effects are associated with an enhanced lesion volume and attenuated motor recovery (Herrmann et al., 2008). In contrast, conditional ablation of the protein suppressor of STAT3, SOCS3, promotes repair and recovery from spinal cord injury (Okada et al., 2006). Our data for pSTAT3tyr705 and expression of SOCS3 after MCAO in aged rats are remarkably consistent with these previous findings. Thus, the truncated pSTAT3tyr705 signaling in aged rats following MCAO was associated with enhanced expression of SOCS3 consistent with early termination of astrogliosis. Indeed, the translocation of STAT3 to the nucleus of astrocytes and astrogliosis observed in young rats following MCAO was less evident in the aged rats. As in the STAT3 ablated mice (Herrmann et al., 2008), enhanced inflammatory responses and a phagocytic microglial phenotype were observed in aged rats following MCAO. What emerges from these combined observations is a role for an age-related truncation of astrocytic response to injury that enhances the detrimental effects of MCAO. While evidence exists to implicate a role for STAT3 in neurons as well as in astrocytes with respect to adverse stroke outcome (Planas et al. 1996), our data here and prior data with genetic deletions of STAT3 point to a role for this transcription factor in astrocytes. The STAT3 genetic ablation studies after spinal cord injury show that eliminating the astrocyte response to injury alone can result in enhanced neuronal damage and neuroinflammation (Okada et al., 2006; Herrmann et al., 2008). Thus, while aging may prime microglia for enhanced responsiveness to brain injury, our data for aging and MCAO is suggestive of a desensitized astrocytic response that results in enhanced neuroinflammatory responses to injury.
Our data do not allow for delineation of the instigating factors that account for the worsened stroke outcome associated with aging. Cross-talk among astrocytes, microglia, damaged neurons and infiltrating blood elements all have been implicated in neural injury and disease. Multiple beneficial as well as detrimental roles for microglia and astroglia have been proposed to be associated with different types and phases of neural injury. Likewise, multiple signaling pathways underlying these important cellular responses likely will be involved. Elucidation of signaling mechanisms underlying stroke outcomes in aged animals seems likely to point the way for identification of novel therapeutic targets (Pekny and Nilsson, 2005; Correa-Cerro and Mandell, 2007; Buffo et al., 2009; Sofroniew 2009). Recent evidence shows a number of remarkable age-related differences in pathophysiological responses to ischemic brain injury. Our findings suggest that altered responsiveness of astrocytes to ischemia should be included as an additional brain cellular phenotype associated with aging. Viewing astrocytes as viable therapeutic targets for neuroprotection against stroke and other neurological diseases becomes increasingly more likely as molecular mechanisms of astrogliosis are discovered and characterized.
Acknowledgments
The authors would like to thank Genentech for their kind gift of tPA (OR-208707) to perform these studies and to the National Institutes of Health for support of this project through the National Institute of Neurological Diseases and Stroke (RO1 NS061954 to J.D.H.). The excellent technical support of Brenda K. Billig and Christopher M. Felton is gratefully acknowledged.
Footnotes
Disclosure/Conflict of Interest: None of the authors have a conflict of interest related to the studies and results presented.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham NS, Castillo DL, Hartman C. National mortality following upper gastrointestinal or cardiovascular events in older veterans with recent nonsteroidal anti-inflammatory drug use. Aliment Pharmacol Ther. 2008;28:97–106. doi: 10.1111/j.1365-2036.2008.03706.x. [DOI] [PubMed] [Google Scholar]
- Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci. 2007;8:221–32. doi: 10.1038/nrn2054. [DOI] [PubMed] [Google Scholar]
- Benkovic SA, O'Callaghan JP, Miller DB. Sensitive indicators of injury reveal hippocampal damage in C57BL/6J mice treated with kainic acid in the absence of tonic-clonic seizures. Brain Res. 2004;1024:59–76. doi: 10.1016/j.brainres.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Buffo A, Rolando C, Ceruti S. Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol. 2010;79:77–89. doi: 10.1016/j.bcp.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Chung HY, Kim HJ, Kim JW, Yu BP. The inflammation hypothesis of aging: molecular modulation by calorie restriction. Ann N Y Acad Sci. 2001;928:327–35. [PubMed] [Google Scholar]
- Correa-Cerro LS, Mandell JW. Molecular mechanisms of astrogliosis: new approaches with mouse genetics. J Neuropathol Exp Neurol. 2007;66:169–76. doi: 10.1097/01.jnen.0000248555.53079.d5. [DOI] [PubMed] [Google Scholar]
- Damiani CL, O'Callaghan JP. Recapitulation of cell signaling events associated with astrogliosis using the brain slice preparation. J Neurochem. 2007;100:720–6. doi: 10.1111/j.1471-4159.2006.04321.x. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilger RN, Johnson RW. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J Leukoc Biol. 2008;84:932–9. doi: 10.1189/jlb.0208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinapoli VA, Rosen CL, Nagamine T, Crocco T. Selective MCA occlusion: a precise embolic stroke model. J Neurosci Methods. 2006;154:233–8. doi: 10.1016/j.jneumeth.2005.12.026. [DOI] [PubMed] [Google Scholar]
- DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. 2008;29:753–64. doi: 10.1016/j.neurobiolaging.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Giunta S. Exploring the complex relations between inflammation and aging (inflamm-aging): anti-inflamm-aging remodelling of inflamm- aging, from robustness to frailty. Inflamm Res. 2008;57:558–63. doi: 10.1007/s00011-008-7243-2. [DOI] [PubMed] [Google Scholar]
- Grossi E. The framingham study and treatment guidelines for stroke prevention. Curr Treat Options Cardiovasc Med. 2008;10:207–15. doi: 10.1007/s11936-008-0022-0. [DOI] [PubMed] [Google Scholar]
- Haag MD, Bos MJ, Hofman A, Koudstaal PJ, Breteler MM, Stricker BH. Cyclooxygenase selectivity of nonsteroidal anti-inflammatory drugs and risk of stroke. Arch Intern Med. 2008;168:1219–24. doi: 10.1001/archinte.168.11.1219. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–43. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–56. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- Kelly KA, Li X, Tan Z, VanGilder RL, Rosen CL, Huber JD. NOX2 inhibition with apocynin worsens stroke outcome in aged rats. Brain Res. 2009;1292:165–72. doi: 10.1016/j.brainres.2009.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampl Y, Boaz M, Gilad R, Lorberboym M, Dabby R, Rapoport A, Anca-Hershkowitz M, Sadeh M. Minocycline treatment in acute stroke: an open-label evaluator-blinded study. Neurology. 2007;69:1404–1410. doi: 10.1212/01.wnl.0000277487.04281.db. [DOI] [PubMed] [Google Scholar]
- Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Sriram K. Focused microwave irradiation of the brain preserves in vivo protein phosphorylation: comparison with other methods of sacrifice and analysis of multiple phosphoproteins. J Neurosci Methods. 2004;135:159–68. doi: 10.1016/j.jneumeth.2003.12.006. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Sriram K, Benkovic SA, Miller DB. Activation of the JAK-STAT3 pathway is associated with the induction of astrogliosis in multiple models of neurotoxicity. Soc Neurosci Abstracts 2007 [Google Scholar]
- O'Callaghan JP, Sriram K, Miller DB. Defining “neuroinflammation”. Ann N Y Acad Sci. 2008;1139:318–30. doi: 10.1196/annals.1432.032. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–34. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–34. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Planas AM, Soriano MA, Berruezo M, Justicia C, Estrada A, Pitarch S, Ferrer I. Induction of Stat3, a signal transducer and transcription factor, in reactive microglia following transient focal cerebral ischaemia. Eur J Neurosci. 1996;8:2612–8. doi: 10.1111/j.1460-9568.1996.tb01556.x. [DOI] [PubMed] [Google Scholar]
- Salvioli S, Capri M, Valensin S, Tieri P, Monti D, Ottaviani E, Franceschi C. Inflamm-aging, cytokines and aging: state of the art, new hypotheses on the role of mitochondria and new perspectives from systems biology. Curr Pharm Des. 2006;12:3161–71. doi: 10.2174/138161206777947470. [DOI] [PubMed] [Google Scholar]
- Satriotomo I, Bowen KK, Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J Neurochem. 2006;98:1353–68. doi: 10.1111/j.1471-4159.2006.04051.x. [DOI] [PubMed] [Google Scholar]
- Scharf MT, Mackiewicz M, Naidoo N, O'Callaghan JP, Pack AI. AMP-activated protein kinase phosphorylation in brain is dependent on method of killing and tissue preparation. J Neurochem. 2008;105:833–41. doi: 10.1111/j.1471-4159.2007.05182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–30. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Shyu WC, Lin SZ, Chiang MF, Chen DC, Su CY, Wang HJ, Liu RS, Tsai CH, Li H. Secretoneurin promotes neuroprotection and neuronal plasticity via the Jak2/Stat3 pathway in murine models of stroke. J Clin Invest. 2008;118:133–48. doi: 10.1172/JCI32723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–47. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkman NL, Johnson RW. Neuroinflammation associated with aging sensitizes the brain to the effects of infection or stress. Neuroimmunomodulation. 2008;15:323–30. doi: 10.1159/000156474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, Benkovic SA, Hebert MA, Miller DB, O'Callaghan JP. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J Biol Chem. 2004;279:19936–47. doi: 10.1074/jbc.M309304200. [DOI] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-alpha. FASEB J. 2006;20:670–82. doi: 10.1096/fj.05-5106com. [DOI] [PubMed] [Google Scholar]
- Sriram K, Miller DB, O'Callaghan JP. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J Neurochem. 2006;96:706–18. doi: 10.1111/j.1471-4159.2005.03566.x. [DOI] [PubMed] [Google Scholar]
- Sriram K, O'Callaghan JP. Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol. 2007;2:140–53. doi: 10.1007/s11481-007-9070-6. [DOI] [PubMed] [Google Scholar]
- Tan Z, Li X, Kelly KA, Rosen CL, Huber JD. Plasminogen activator inhibitor type 1 derived peptide, EEIIMD, diminishes cortical infarct but fails to improve neurological function in aged rats following middle cerebral artery occlusion. Brain Res. 2009;1281:84–90. doi: 10.1016/j.brainres.2009.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie HF, Xu RX, Wei JP, Jiang XD, Liu ZH. P-JAK2 and P-STAT3 protein expression and cell apoptosis following focal cerebral ischemia-reperfusion injury in rats. Nan Fang Yi Ke Da Xue Xue Bao. 2007;27:208–11. [PubMed] [Google Scholar]
- Yamashita T, Sawamoto K, Suzuk S, Suzuki N, Adachi K, Kawase T, Mihara M, Ohsugi Y, Abe K, Okano H. Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J Neurochem. 2005;94:459–68. doi: 10.1111/j.1471-4159.2005.03227.x. [DOI] [PubMed] [Google Scholar]