

Abstract
Post traumatic seizures (PTS) are a significant complication from traumatic brain injury (TBI). Adenosine, a major neuroprotective and neuroinhibitory molecule, is important in experimental epilepsy models. Thus, we investigated the adenosine A1 receptor (A1AR) gene and linked it with clinical data extracted for 206 subjects with severe TBI. Tagging SNPs rs3766553, rs903361, rs10920573, rs6701725, and rs17511192 were genotyped, and variant and haplotype associations with PTS were explored. We investigated further genotype, grouped genotype, and allelic associations with PTS for rs3766553 and rs10920573. Multivariate analysis of rs3766553 demonstrated an association between the AA genotype and increased early PTS incidence. In contrast, the GG genotype was associated with increased late and delayed onset PTS rates. Multivariate analysis of rs10920573 revealed an association between the CT genotype and increased late PTS. Multiple risk genotype analysis shows subjects with both risk genotypes have a 46.7% chance of late PTS. To our knowledge, this is the first report implicating genetic variability in the A1AR with PTS, or any type of seizure disorder. These results provide a rationale for further studies investigating how adenosine neurotransmission impacts PTS, evaluating anticonvulsant in preventing and treating PTS, and developing and testing targeted adenosinergic therapies aimed at reducing PTS.
Keywords: A1AR, adenosine, ADORA1, brain injury, genetic susceptibility, post-traumatic seizure
1.INTRODUCTION
5.3 million American civilians have disabilities after traumatic brain injury (TBI), and the national cost is estimated at $48.3 billion per year (Thurman et al., 1999). Post-traumatic seizures (PTS) are a significant complication after TBI. PTS accounts for 20% of symptomatic seizures and 5% of all seizures in the general population (Englander et al., 2003a). Up to 86% of patients with one late seizure after TBI will have a second within 2 years (Haltiner et al., 1997). Despite recommendations supporting limited treatment for seizure prophylaxis (Temkin, 2002), people with TBI often receive PTS prophylaxis on a long term basis with anticonvulsant medications that require regular monitoring and are associated with unwanted side effects, including sedation. Patients who go on to develop PTS also experience similar issues with long term treatment (Dikmen et al., 1991; Smith Jr. et al., 1994). Importantly, the impact of these drugs on neurorecovery is largely unknown. Moreover, PTS is associated with increased disability and limitations with activities of daily living (Armstrong et al., 1990). As such, identifying mechanisms and risk factors for PTS as well as preventing and treating PTS are major public health issues.
Although its occurrence is commonly related to injury severity (Temkin et al., 1990), several other injury related risk factors are also associated with PTS. These include biparietal contusions, dural penetration with bone and metal fragments, multiple intracranial operations, cortical contusions, subdural hematoma, significant midline shift, early PTS, and skull fractures (Englander et al., 2003b; Annegers et al., 1998). Intrinsic or individual factors are less well studied. Evidence indicates that progesterone has anticonvulsant properties (Rhodes and Frye, 2005) and that catemenial seizures are prevalent among women (Herzog, 2008), but studies have reported gender as a non-significant factor with PTS (Lee et al., 1995). However, age has been shown to have some relationship with PTS (Frey, 2003; Annegers et al., 1998). Although studies suggest genetic susceptibility to seizure disorders (Cavalleri et al., 2007) and treatment response (Hung et al., 2005), little has been studied about genetic susceptibility to PTS.
Adenosine A1 receptors (A1AR) are located on neurons in regions susceptible to both seizures and TBI (e.g. hippocampus, cortex) and are spatially associated with NMDA receptors (Deckert and Jorgensen, 1988). Adenosine binding to A1AR modulates ionic conductances in the neuronal membrane, limiting excitotoxicity (Rudolphi et al., 1992). Adenosine is an important acute neuroprotectant in the setting of TBI. Previous work by our group and others demonstrates elevated glutamate levels in the cerebrospinal fluid (CSF) of adults with severe TBI (Bell et al., 2001) and a significant relationship between adenosine and glutamate levels in pediatric TBI (Robertson et al., 2001). Experimental models suggest a relationship between excitatory amino acids and PTS after cortical impact (Nilsson et al., 1994). A1AR activation is a key local regulator of excitatory amino acid (EAA) release during ischemia (Andine et al., 1990). Thus, A1AR activation is of particular importance to the neuroprotective effects of adenosine.
Adenosine and its target A1AR play an important role in excitoxicity (Rudolphi et al, 1992) and with non-traumatic seizure development. In fact, A1AR activation influences the extent of status epilepticus in both ex vivo and in vivo seizure models (Fedele et al., 2006; Avsar and Empson, 2004). Additionally, glial scar, common to TBI, has been implicated in seizure development by compromising adenosine synthesis (Fedele et al., 2005). Importantly, profound PTS, leading to lethal status epilepticus, occurs in A1AR knockout (KO) mice when subjected to the controlled cortical impact (CCI) model of experimental TBI (Kochanek et al., 2006).
Given the role of adenosine and the A1AR in neuroprotection and non-traumatic seizures and PTS in experimental models, the goal of this study was to determine if genetic variability within the A1AR gene and the flanking region was associated with the development of PTS in a human population with severe TBI. This study explored several tagging single nucleotide polymorphisms (tSNPs) in the A1AR gene for associations with PTS. After accounting for mortality, univariate and multivariate analyses demonstrate a significant association between rs3766553 and rs10920573 and PTS. These findings have implications for PTS risk stratification and treatment strategies after TBI. Further work is required to replicate findings and to determine how rs3766553 and rs10920573 tSNPs influence PTS susceptibility.
2.METHODS
2.1 Study Design and Subjects
This study was approved by the Institutional Review Board at the University of Pittsburgh. We genotyped tSNPs selected for the A1AR gene and retrospectively extracted patient information regarding PTS for 206 persons with severe TBI (Glasgow coma scale (GCS) score≤8) who were prospectively enrolled in a larger study assessing genetic relationships with TBI outcome at our level 1 trauma center between 2000 and 2006. Patients were enrolled if they were between the ages of 18-75, had a severe TBI based on a GCS ≤8 with positive findings on head CT, and required an extraventricular drainage catheter (EVD) for intracranial pressure (ICP) monitoring and management. Patients with penetrating head injury as well as cardiac or respiratory arrest at injury were excluded from the study.
2.2 Critical Care Management of Severe TBI
Subjects enrolled in this study were admitted to the neurotrauma intensive care unit to receive treatment consistent with The Guidelines for the Management of Severe Head Injury (Bullock et al., 1996). This included initial placement of an EVD, central venous catheter, and arterial catheter. When clinically necessary, surgical intervention for decompression of mass lesions was provided. Elevated ICP was treated in a stepwise fashion to regain control and maintain the pressure within normal parameters (<20 mmHg), and cerebral perfusion pressure (CPP) was maintained at >60mmHg. If CPP remained low, then mean arterial pressure (MAP) was supported with pressors or inotropes to keep MAP>90mmHg. Although routine continuous EEG is not our standard of care, standard EEG was ordered intermittently for patients for clinical suspicion of PTS activity. In general, patients with severe injuries received PTS prophylaxis for 1 week based on previously published studies (Temkin et al., 1990). Temperature was monitored regularly, and a subset of subjects received moderate hypothermia as a part of clinical care or if they were enrolled in a randomized controlled clinical trial evaluating moderate hypothermia after severe TBI. Subjects not receiving hypothermia were treated to maintain a normothermic state. In total, 43 patients in the final analysis received hypothermia and 142 remained normothermic.
2.3.Demographic and Injury Data
Demographic and premorbid variables including age, gender, racial background, and reference to premorbid seizure disorder were recorded. These variables were assessed for their relationship to both PTS and genotype. Additionally, injury and treatment variables known to influence the occurrence of PTS were also recorded (Clifton et al., 1993; Englander et al., 2003a; Annegers et al., 1998; Temkin, 2003). Hypothermia status and mechanism of injury were recorded for each subject. Initial hospital GCS scores were recorded for each patient after resuscitation and without the influence of paralytics. Acute care hospital length of stay (LOS), depressed skull fracture, and receipt of surgical intervention to treat a TBI lesion was also recorded.
Subject medical records were reviewed for antiepileptics drug (AED) use during acute care. Indications for seizure prophylaxis or treatment were recorded whenever possible. Common AEDs used at our center for seizure prophylaxis or treatment were included in the analysis. Drugs with anticonvulsant properties that were explicitly designated in the medical record for a condition other than seizure treatment/prophylaxis, were excluded from analysis.
2.4 Single Nucleotide Polymorphism Selection
tSNPs with a minor allele frequency of at least 20% were selected for the A1AR gene. tSNPs evaluated include rs3766553, rs903361, rs10920573, rs6701725, and rs17511192. Selection was based on data from the HapMap database (build 35 accessed Feb 2008; hapmap.org) and SNP database (www.ncbi.nlm.nih.gov/projects/SNP). At the time of selection, these tSNPs captured the variability of the gene including 1,000 bases 5’ upstream into the promoter region (see figure 1 which is based on the updated data from build 36). We evaluated each tSNP for allelic, genotype and grouped genotype as independent variables for associations with PTS and outcome. The two groupings used were presence of variant (homozygous variant and heterozygotes combined) versus homozygous wild type and presence of wild type (homozygous wild type and heterozygotes combined) versus homozygous variant (see table 1). For some analyses with rs10920573, the heterozygote (CT) was compared to the homozygote groups (CC+TT). One SNP, rs17852405, was also evaluated because it was the only non-synonymous SNP located within the block of linkage disequilibrium tagged by rs3766553. Selection was based on data from HapMap Build 35 and locations within the UCSC genome browser (genome.ucsc.edu).
Figure 1.
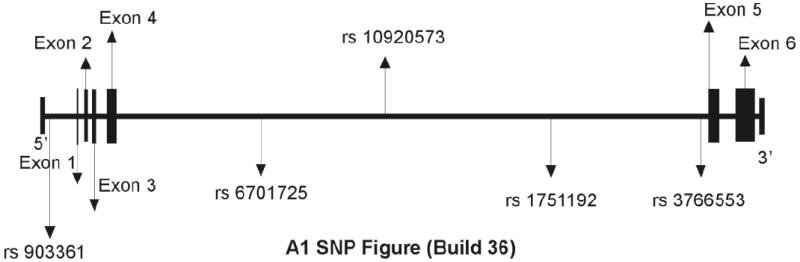
Gene map depiction of the 5 tSNPs explored in the study along with the 6 exons in the A1 gene (Information generated from the HapMap Database). The two tSNPs found to be significant are rs3766553 and rs10920573.
Table 1.
Univariate Tagging SNP Results.
| SNP | HapMap positionc | HapMap positiond | Allele Freq | Early PTS: Within One Week | Late PTS: Beyond One Week | Delayed Onset PTS: Beyond Six Months | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ˆ overall | WTa | Varb | Geno | Allele Freq | WT | Varb | Geno | Allele Freq | WTa | Varb | Geno | Allele Freq | |||
| rs903361 | chr1:199,822,931 | Chr1:201,357,897 | A=.68 (wt) | 1 | 0.058 | 0.188 | A=.70 | 1 | 0.502 | 0.718 | A=.66 | 1 | 0.577 | 0.658 | A=.67 |
| G=.32 (var) | G=.30 | G=.34 | G=.33 | ||||||||||||
| rs6701725 | chr1:199,834,385 | Chr1:201,369,351 | G=.55 (wt) | 0.663 | 1 | 0.888 | G=.55 | 0.36 | 0.621 | 0.496 | G=.56 | 1 | 1 | 1 | G=.55 |
| A=.45 (var) | A=.45 | A=.44 | A=.45 | ||||||||||||
| rs10920573 | chr1:199,840,165 | Chr1:201,375,131 | T=.34 (wt) | 0.115 | 1 | 0.212 | T=.30 | *0.037 | 0.735 | *0.039 | T=.36 | 0.147 | 1 | 0.142 | T=.35 |
| C=.66 (var) | C=.70 | C=.64 | C=.65 | ||||||||||||
| rs17511192 | chr1:199,851,148 | Chr1:201,386,114 | C=.59 (wt) | 0.173 | 1 | 0.336 | C=.54 | 0.761 | 0.33 | 0.557 | C=.57 | 1 | 0.761 | 0.93 | C=.56 |
| T=.41 (var) | T=.46 | T=.43 | T=.44 | ||||||||||||
| rs3766553 | chr1:199,864,699 | Chr1:201,399,665 | G=.57 (wt) | *0.032 | 0.68 | *0.052 | G=.53 | 0.764 | *0.044 | 0.112 | G=.55 | 0.459 | *0.005 | *0.019 | G=.56 |
| A=.43 (var) | A=.47 | A=.45 | A=.44 | ||||||||||||
= presence of wild type (wt) versus homozygous variant
= presence of variant (var) versus homozygous wild type
= based on build 35, accessed February 2008
= based on build 36, accessed March 2010
= p-values for chi-square analysis less than 0.005
= overall allele frequency for total population
2.5 DNA Collection and Genotyping Methods
DNA was extracted from one of two sources for each subject, whole blood or cerebrospinal fluid (CSF). Whole blood was collected into EDTA vacutainer tubes, processed to retrieve the buffy coat and DNA extracted using a simple salting out procedure (Miller et al., 1988). CSF was collected by passive drainage as part of clinical care, and DNA was extracted using the Qiaamp DNA extraction protocol for extraction from body fluids (Qiagen Corporation, Valencia, CA).
tSNPs (rs3766553, rs903361, rs10920573, rs6701725, and rs17511192) were genotyped using TaqMan allele discrimination technology and commercially available 5’ exonuclease Assay-on-Demand TaqMan assays (Applied Biosystems Incorporated, Foster City, CA). Briefly, this approach uses dual-labeled hybridization probes, each containing one reporter dye (FAM, or carboxyfluorescein), whose fluorescence is cancelled by the quencher dye (TAMRA, or 6-carboxy-tetramethylrhodamine) while in close proximity prior to amplification. During DNA polymerization, the reporter is separated from the quencher, resulting in a quantifiable, allele-specific release of fluorescence. The sequence detector measures the fluorescence spectra of all 96 wells continuously during PCR thermal cycling, which in turn determines the genotype.
Amplification and genotype assignments were conducted using ABI7000 and SDS 2.0 software (Applied Biosystems Incorporated, Foster City, CA). Double-masked genotype assignments were made for each SNP. They were compared, and each discrepancy was addressed using raw data or re-genotyping.
rs3766553 is an intronic tSNP for A1AR that tags a block of DNA that is approximately 9,263 basepairs based on the haplotype block data from HapMap Build 35 and locations within the UCSC genome browser (genome.ucsc.edu). This block of DNA was evaluated for potential functional SNPs and only one non-synonymous SNP, rs17852405 within exon 3, was noted in the SNP database. This SNP codes for a proline to glutamine at amino acid 261, however no allele frequencies were provided. Rs17852405 was genotyped using PCR-RFLP. Amplification primers were 5’-GCCCTCATCCTCTTCCTCTT-3’ for the forward primer and 5’-CGGAAATGGTCATTCCAAAT-3’ for the reverse primer. This produced a 215bp product using an annealing temperature of 54°C. BsrI (New England Biolabs, Ipswich, MA) recognized the C→A transversion. Thirty subjects (10 homozygous wild type, 10 homozygous variant and 10 heterozygotes for the rs3766553 tSNP) were then selected for DNA sequence verification. Sequencing was conducted by first cleaning the PCR products using exoSAP reagents (USBiochemicals, Cleveland, Ohio) and sequenced using the reverse primer and Big Dye Cycle Sequencing reagents (Applied Biosystems Incorporated, Foster City, CA). The sequencing products were electrophoresed using an ABI377 automated sequencer (Applied Biosystems Incorporated, Foster City, CA), and sequence data was viewed using Sequencer software (Gene Codes Corporation, Ann Arbor, MI).
2.6 Haplotyping
We used PHASE software (Stephens et al., 2001) to generate the most likely haplotypes (and haplotype pairs, or diplotypes) for each individual in this unrelated cohort using the 5 tSNPs. A total of 29 haplotypes were generated, and haplotype pairings were assigned to each subject using only haplotypes with a high enough probability of occurrence such that at least one individual could be assigned that haplotype. As such, 18 possible haplotypes were assigned. Both the presence of a specific haplotype and haplotype pairings, that is, diplotypes, were analyzed (see statistical analysis).
2.7 Outcome Measures
The time period of first documented PTS (i.e. time to first post-traumatic seizure) was the primary measure of outcome for this study. Time to first seizure was divided into three different time based categories: early, late and delayed-onset late PTS. In order to meet standard early PTS criteria (Temkin et al., 1990) subjects must have had a documented seizure within the first week post injury. Individuals categorized as having late PTS (Temkin et al., 1990) must have had first documented seizure beyond the first week post-injury. Subjects were designated as having delayed-onset of PTS if the time to first documented seizure occurred beyond 6 months post injury. Subjects characterized as having delayed onset PTS were also included in the late PTS analyses because their first PTS was documented beyond one week post TBI to follow published definitions of late PTS (Temkin et al., 1990). All electronic inpatient and outpatient subject medical records available from our center were reviewed to determine first documented PTS. Inpatient notes used to determine time to first seizure included ambulance emergency room reports, progress notes, nursing notes, EEG reports, patient history and physical reports, and discharge or transfer summaries. Determining late PTS and delayed-onset late PTS required at least one discharge/death summary or inpatient/outpatient note related to the TBI and referencing the appropriate PTS period. Medical record availability for late PTS and delayed-onset PTS ranged from at least 6 months post-injury to 6 years post-injury. Subjects were included for analysis if there was at least one clinical note on record related to their injury. Notation in medical records referring to convulsions, seizures, status epilepticus or seizure disorder was taken as PTS occurrence for an individual.
Patients were administered Glasgow Outcome Scale (GOS) scores at 6 months post-injury. The GOS is a widely utilized measure that classifies outcome into five categories: 5=good recovery, 4=moderate disability, 3=severe disability, 2=persistent vegetative state, and 1=death (Jennett and Bond, 1975). Mortality during acute care hospitalization and mortality after acute care discharge was recorded by reviewing medical records and the Social Security Death Index (http://ssdi.rootsweb.ancestry.com/). From this information, subjects who died were coded as having expired during the first week of injury, beyond the first week from injury, or beyond six months from injury.
2.8 Statistical analysis
As ancestral background can significantly influence the allele frequency of the tSNPs selected for the A1AR, our analyses were limited to Caucasian subjects due to the small number of African Americans (n=11) available. Additionally, subjects with documented premorbid seizure disorder (n=8) were also removed from the final sample set. There were a small number of subjects with missing data. Individuals with missing information were included where possible in univariate analysis and descriptive analyses, however they were removed for multivariate analysis.
Descriptive statistics were performed using SPSS (version 17). Summary statistics, including means, standard error of the mean (SEM), and medians, for all continuous variables were computed. Frequencies and percentages were determined for categorical variables. Chi-square analysis was conducted using the Fisher Exact test to determine if statistically significant differences were present among subjects with no PTS, early PTS (within a week of injury), late PTS (beyond a week), and delayed-onset PTS (beyond six months) of injury with respect to categorical descriptive variables. Independent t-tests were used to assess differences among PTS groups for continuous variables. Chi-square analysis using the Fisher Exact test was also used to determine associations between SNP genotypes and other descriptive variables. Additionally, chi-square analysis was used with the Fisher exact test to determine differences in early, late, and delayed onset late PTS with respect to allelic, grouped genotype, and genotype distribution for each tSNP. Chi-square analysis was also used to test differences in PTS among all haplotype pairs and with regard to the presence vs. absence of the four most common haplotypes in the population. All PTS groups were compared to subjects who had no documented PTS at any time point (No PTS group). Further, subjects with an early PTS designation were not included in analyses of the late and delayed onset PTS cohorts. Conversely, subjects with late or delayed onset PTS were not included in early PTS analyses.
tSNPs with at least one significant univariate association (p≤0.05) within a PTS time interval were then followed by multivariate logistic regression analyses across each time interval to determine adjusted relationships between the tSNP of interest and PTS using Statistical Analysis System (SAS) (9.2). Covariates tested were those that were associated in univariate analysis with PTS (p<0.10). Clinically relevant covariates (e.g. age, GCS, gender) were also included in the multivariate model, regardless of univariate results. Multiple risk genotypes associated with PTS were then assessed in the same multivariate model (i.e. multivariant analysis) using the same covariate structure. Due to small numbers in some categories, covariate*genotype interactions with PTS were not explored in regression analyses. To avoid the confounding factor of death on seizure incidence, we removed subjects from analysis who died within a week of injury and did not seize when comparing tSNPs and early PTS. All documented deceased subjects, except one subject with late PTS, were removed to assess tSNP associations with late and delayed-onset late PTS.
3. RESULTS
3.1 Description of the Population for Tagging SNP Analysis
Descriptive analysis first was done for the entire population with tSNP data after non-Caucasian subjects and subjects with premorbid seizures were removed. Of the 187 subjects included in the entire population for all tSNP analysis, 19.8% were female, and 80.2% were male. The median GCS score was 6, and the mean age was 34.39 ± 1.05 years. Approximately 17.2% of the population had documented evidence in the medical record of PTS. Of those that seized, 24.2% developed early PTS during their acute hospital admission, and 75.7% developed late or delayed-onset PTS. The length of acute hospital stay ranged from less than 1 day to 61 days post-injury, and the mean length of stay (LOS) for each cohort was within one day of each other. (Early=23.38, Late=24.34, Delayed-Onset=24.01). During the acute care stay, 94.1% of the population received AED’s for seizure prophylaxis/treatment, and 14.6% percent received an EEG as a part of their acute care management. The primary mechanisms of injury were motor vehicle collisions (52.4%), falls (16.6%), and motorcycle collisions (16.0%). 23.2% of the population received hypothermia. 15.5% of the population died within the first week of injury, while an additional 12.3% of cohort expired after a week. Covariates were also explored for both rs3766553 and rs10920573, and only the heterozygote for rs3766553 was found to be significantly less likely to have a cranial surgery (p=0.01).
3.2 Evaluation of Tagging SNPs and Haplotypes
Each tSNP was independently evaluated for associations with PTS, and allelic/genotype frequencies for each cohort are presented in Table 1. Hap map position based on build 35 (used at the time of tSNP selection) and build 36 (currently published) is provided. Allelic frequencies are provided for each tSNP based on the entire population of 187 subjects described above and also by PTS cohort. In these cohorts, subjects with PTS were grouped according to definitions for early, late, and delayed onset as described in the methods, and subjects who died were removed as previously described. In each cohort, subjects with PTS were compared to subjects who did not have PTS at any time point. Both rs3766553 and rs10920573 were significantly associated with PTS (table 1). The AA genotype for rs3766553 was associated with higher early PTS rates, while the GG genotype was associated with higher late and delayed-onset PTS. For the tSNP rs10920573, the heterozygote genotype (CT) was associated with late PTS. Chi-square analysis showed no differences between haplotype pairs and PTS at any time period. Subjects with one of the four most common haplotypes were each compared to the rest of the population, and there were no significant associations between groups at any time period (data not shown). All tSNPs were in Hardy-Weinberg equilibrium, indicating that the expected proportion of genotypes in this population was obtained. Additional analyses focused on rs3766553 and rs10920573. 12.4% of subjects had both risk genotypes, while 42.2% had one risk genotype for late or delayed-onset PTS.
3.3 Description of Population by Post Traumatic Seizure Status
Table 2 further describes general demographic and characteristics of those with early, late, and delayed-onset PTS for rs3766553 or rs10920573. Ninety-four percent of the early PTS cohort, 95% of the late PTS cohort, and 95% of the delayed onset cohort were treated with AED’s during acute hospitalization. There were no significant differences between PTS groups with regard to acute care length of stay. Neurosurgical intervention to treat a TBI specific lesion was significantly associated with late and delayed-onset PTS. Factors previously reported in other studies to affect PTS, including depressed skull fracture, did not significantly affect PTS.
Table 2.
Descriptive Comparisons for No PTS vs. Early, Late, and Delayed Onset PTS
| Variable | a No PTS vs. Early PTS (N=133) | c p-value | b No PTS vs. Late PTS (N=127) | c p-value | b No PTS vs. Delayed Onset PTS (N=117) | c p-value | |||
|---|---|---|---|---|---|---|---|---|---|
| Gender | p=1.000 | p=0.525 | p=0.719 | ||||||
| Male | 103(93.6%) | 7 (6.4%) | 85 (82.5%) | 18 (17.5%) | 85 (87.6%) | 12 (12.4%) | |||
| Female | 22 (95.6%) | 1 (4.35%) | 17 (70.8%) | 7 (29.2%) | 17 (85.0%) | 3 (15.0%) | |||
| AED treatment | p=1.000 | p=0.597 | p=1.000 | ||||||
| Yes | 117 (93.6%) | 8 (6.4%) | 96 (79.3%) | 25 (20.7%) | 96 (86.5%) | 15 (13.5%) | |||
| No | 8 (100.0%) | 0 (0.0%) | 6 (100.0%) | 0 (0.0%) | 6 (100.0%) | 0 (0.0%) | |||
| GCS Score | p=0.681 | p=0.591 | p=0.519 | ||||||
| 3-4 | 34 (97.1%) | 1 (2.7%) | 23 (85.2%) | 4 (14.8%) | 23 (92.0%) | 2 (8.0%) | |||
| 5-8 | 90 (92.8%) | 7 (7.2%) | 78 (78.8%) | 21 (21.2%) | 78 (85.7%) | 13 (14.3%) | |||
| Hypothermia Status | p=0.198 | p=0.427 | p=0.321 | ||||||
| Cooled | 30 (100.0%) | 0 (0.0%) | 23 (76.7%) | 7 (23.3%) | 23 (82.1%) | 5 (17.9%) | |||
| Normothermic | 95 (92.2%) | 8 (7.8%) | 79 (83.2%) | 16 (16.8%) | 79 (89.8%) | 9 (10.2%) | |||
| Depressed Skull Fracture | p=1.000 | p=0.623 | p=0.220 | ||||||
| Yes | 7 (100.0%) | 0 (0.0%) | 5 (71.4%) | 2 (28.6%) | 5 (71.4%) | 2 (28.6%) | |||
| No | 118 (93.6%) | 8 (6.3%) | 97 (80.8%) | 23 (19.2%) | 97 (88.2%) | 13 (11.8%) | |||
| Injury Mechanism | p=0.211 | p=0.327 | p=.595 | ||||||
| Motor Vehicle | 74 (94.9%) | 4 (5.1%) | 63 (85.1%) | 11 (14.9%) | 63 (90.0%) | 7 (10.0%) | |||
| Fall | 15 (93.7%) | 1 (6.2%) | 10 (76.9%) | 3 (23.1%) | 10 (83.3%) | 2 (16.7%) | |||
| Motorcycle | 17(85.0%) | 3 (15.0%) | 13 (68.4%) | 6 (31.6%) | 13 (81.2%) | 3 (18.7%) | |||
| Other | 19 (100.0%) | 0 (0.0%) | 16 (76.2%) | 5 (23.8%) | 16 (84.2%) | 3 (15.8%) | |||
| Cranial Surgery | p=0.119 | p=0.003 | p=0.009 | ||||||
| Yes | 40 (88.9%) | 5 (11.1%) | 31 (66.0%) | 16 (34.0%) | 31 (75.6%) | 10 (24.4%) | |||
| No | 85 (96.6%) | 3 (3.4%) | 71 (88.7%) | 9 (11.2%) | 71 (93.4%) | 5 (6.6%) | |||
| Age | 33.68± 1.26 | 35.15± 4.39 | p=0.770 | 31.63±1.29 | 31.42±2.15 | p=0.46 | 31.63± 1.29 | 31.65±2.82 | p=1.00 |
| Acute Care LOS | 23.58± 3.34 | 20.25± 4.93 | p=0.400 | 24.31± 1.03 | 24.44± 2.21 | p=0.71 | 24.31±1.04 | 21.93±2.43 | p=0.36 |
Analysis does not include subjects who died in during the first week after TBI and did not have PTS
Analysis does not include subjects who died in acute care or had documented death after one week post inury and did not have PTS
p-values based on chi-square analysis and independent t-test as appropriate
3.4 rs3766553 Associations with Early Post-traumatic Seizures
When assessing early PTS frequency within each genotype group, we compared those who had PTS within the first week after injury to those who survived at least one week and did not seize. 17.4% of the population with the AA genotype had PTS, while only 3.9% of the AG and 3.2% of the GG subjects developed early PTS (figure 2a) (p=0.052). Further grouped genotype analysis shows that subjects with the AA genotype (risk group) had a 17.4% PTS rate compared to those with the AG+GG genotype (3.7%) (p=0.032). No significant associations were noted with PTS when GG subjects were assigned as the risk group (see table 1). Allelic associations show that 8.9% of those with the A allele have early PTS, while 3.6% with the G allele develop early PTS (p=0.118).
Figure 2.
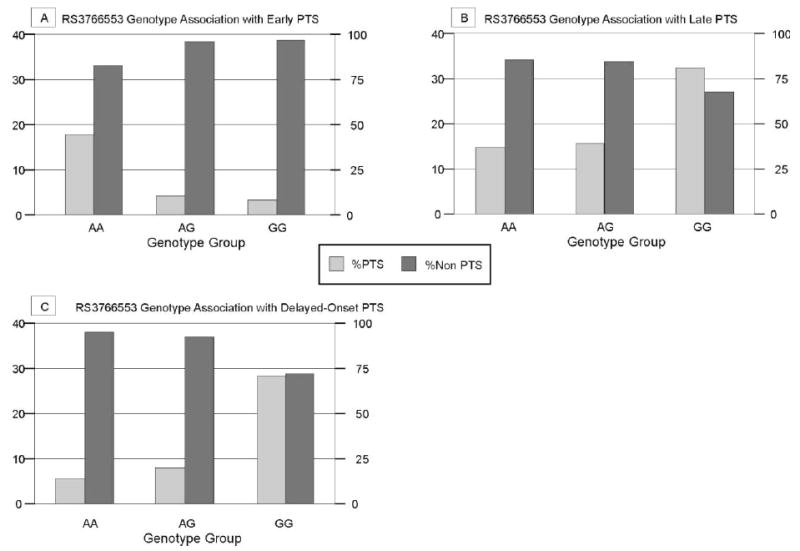
PTS frequency by genotype for rs3766553 A) associated with early post traumatic seizures within one week of initial injury (p=0.052) (Population sizes by genotype: AA=23, AG= 77, GG=31) B) associated with late post traumatic seizures beyond one week after injury, (p=0.112) (Population sizes by genotype: AA=21, AG= 71, GG=34), C) associated with delayed onset post traumatic seizures beyond six months after injury (p=0.019) (Population sizes by genotype: AA=19, AG= 65, GG=32)
3.5 rs3766553 Associations with Late and Delayed-Onset Post-traumatic Seizures
When assessing late and delayed PTS frequency within each genotype group for rs3766553 we compared those who had PTS at these time points only to those who survived and did not seize. 32.4% of GG subjects had late PTS, while 15.5% of those with the AG and 14.3% of those with the AA genotype had late PTS (figure 2b). There was a significant difference with regard to grouped genotype and late PTS incidence (p=0.044) where the GG genotype (risk group) had a 32.2% late PTS rate compared to those with the AG+AA genotype (15.2%). compared to those with the AG+AA genotype. No significant associations were noted with PTS when AA subjects were assigned as the risk group (table 1). Allelic associations with late PTS show that 23.7% of those carrying the G allele have late PTS while 15.0% of those with the A allele develop PTS (p=0.112).
5.3% of those with an AA genotype had delayed-onset PTS, while 7.7% of the AG group and 28.1% of the GG group had delayed onset PTS (p=0.019) (figure 2c). Similar to late PTS, there was an even stronger grouped genotype difference with regard to PTS occurring beyond six months where subjects with the GG genotype (risk group) had a 28.1% delayed-onset PTS rate compared to those with the AG+AA groups (7.1%) (p=0.005). No significant associations were noted with PTS when subjects with AA genotype were assigned as the risk group (table 1). Allelic associations show that 17.8% of those carrying the G allele have delayed-onset PTS and only 6.8% of those with the A allele develop delayed-onset PTS (p=0.0171).
3.6 rs3766553 Multivariate Analysis with Post-traumatic Seizures
Multivariate analysis was performed to determine the adjusted relationships between risk genotype AA and early PTS and between risk genotype GG in the development of late and delayed onset PTS (table 3). When assessing genotype relationships with early PTS, AA subjects had a higher odds of developing early PTS compared to the AG group (OR=5.360; p=0.045) (table 3). Grouped genotype analysis also showed that after adjusting for age, gender, GCS, and neurosurgical intervention, AA subjects had a higher odds (OR=5.482; p=0.028) of developing early PTS compared to the AG+GG group.
Table 3.
Multivariate Analysis—rs3766553 Associations with PTS
| Variable | Odds Ratio | 95% Confidence Interval | P-Value | |
|---|---|---|---|---|
| Genotype Associations with PTS in Early PTS Cohort (N=129) | Age | 0.996 | 0.945-1.050 | 0.890 |
| Gender | 0.682 | 0.074-6.252 | 0.735 | |
| GCS Score | 0.352 | .040-3.111 | 0.348 | |
| Brain Surgery | 0.947 | 0.211-4.252 | 0.944 | |
| rs3766553 AA Genotype versus AG | 5.36 | 1.047-27.479 | 0.045 | |
| rs3766553 AA Genotype versus GG | 5.849 | 0.566-60.401 | 0.138 | |
| Genotype Associations with PTS in Late PTS Cohort (N=124) | Age | 0.995 | 0.957-1.035 | 0.810 |
| Gender | 2.024 | 0.683-5.998 | 0.203 | |
| GCS Score | 0.667 | 0.201-2.217 | 0.509 | |
| Brain Surgery | 0.965 | .374-2.488 | 0.941 | |
| rs3766553 GG Genotype versus AG | 3.02 | 1.106-8.251 | 0.031 | |
| rs3766553 GG Genotype versus AA | 2.899 | 0.663-12.687 | 0.158 | |
| Genotype Associations with PTS in Delayed Onset PTS Cohort (N=115) | Age | 1.004 | 0.959-1.052 | 0.853 |
| Gender | 1.174 | 0.265-5.205 | 0.833 | |
| GCS Score | 0.535 | 0.105-2.721 | 0.451 | |
| Brain Surgery | 1.431 | 0.417-4.905 | 0.569 | |
| rs3766553 GG Genotype versus AG | 4.624 | 1.378-15.524 | 0.013 | |
| rs3766553 GG Genotype versus AA | 7.309 | 0.799-66.850 | 0.078 | |
Multivariate analysis assessing genotype associations with late PTS while adjusting for age, gender, GCS, and neurosurgical intervention showed that the GG group had a greater odds of developing late PTS compared to those with the AG genotype (OR=3.02; p=0.031) (table 3). Grouped genotype analysis, adjusting for the same covariables, showed that the GG group had a greater odds of developing late PTS, compared to those with the AA+AG genotypes (OR=2.993; p=0.024). Similar multivariate results were found for delayed-onset PTS. Here, genotype analyses showed that the GG group had a significantly higher proportion of subjects with delayed-onset PTS than the AG group (OR=4.624; p=0.013), and there was a trend for the GG group to have a higher PTS incidence compared to those with the AA genotype (OR: 7.309; p=0.078) (table 3). Grouped genotype analysis adjusting for the same covariates showed that the GG group had significantly higher odds of delayed-onset PTS when compared to the AG+AA group. (OR=5.049; p=0.006).
3.7 rs10920573 Associations with Early Late and Delayed-Onset Post-traumatic Seizures
When assessing PTS frequency within each genotype group for rs10920573, we removed subjects who died and did not seize as described for rs3766553. For rs10920573, there were no significant differences in early PTS incidence (figure 3a). However, 28.8% of the population with the CT genotype had late PTS, while 10.2% with the CC genotype and 14.3% with the TT genotype developed late PTS (figure 3b) (p=0.039). Further analysis shows significantly higher late PTS incidence in subjects with the CT genotype (risk genotype) compared to those with the CC+TT genotype (19.2% versus 6.7%; p=0.022). Significant associations also occurred when comparing seizure rate for the CC subjects (26%) to the CT+TT genotypes (26.0% versus 10.2%) (p=0.037) (table 1). However, no significant associations were noted with PTS when TT subjects were compared to the rest of the population. Allelic associations with late PTS for this SNP showed a PTS rate of 13.9% for those with the C allele and 18.5% for the T allele (p=0.350).
Figure 3.
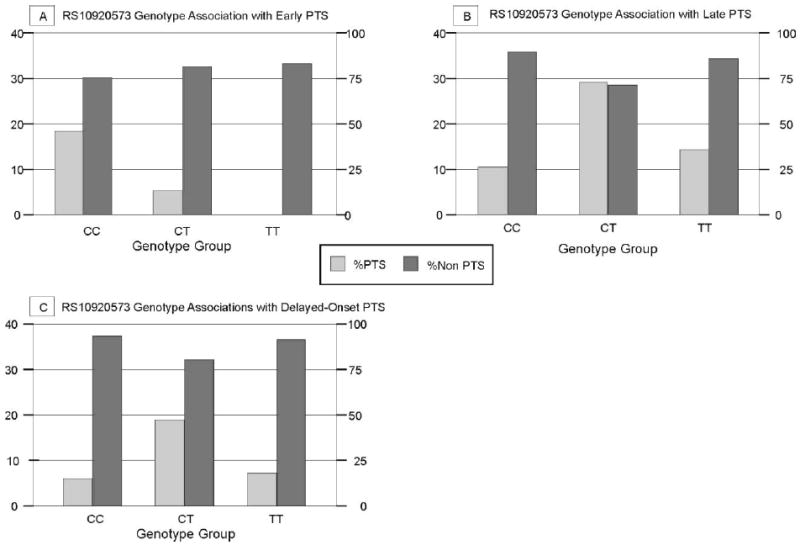
PTS frequency by genotype for rs10920573 A) associated with early post traumatic seizures occurring in the first week post injury (p=0.212) (Population sizes by genotype: CC=64 CT=50 TT=14) B) late post traumatic seizures occurring beyond one week after injury (p=0.039) (Population sizes by genotype: CC=49 CT=59 TT=14) C) delayed onset post traumatic seizures occurring beyond six months after injury (p=0.142) (Population sizes by genotype: CC=47 CT=52 TT=13)
For delayed-onset PTS, individuals with the CT genotype had a 19.2% PTS rate, while the CC group had a 6.4% and the TT group had a 7.7% PTS rate (figure 3c). Although not significant when CC or TT was designated the risk group (see table 1), delayed onset PTS was higher with CT as the risk genotype and trended towards significance (p=0.083). Allelic frequencies for rs10920573 and delayed-onset PTS were 11% for the C allele and 15.4% for the T allele (p=0.397).
3.8 rs10920573 Multivariate Analysis with Late and Delayed-Onset Post-traumatic Seizures
Multivariate analysis was performed to determine the adjusted relationships between risk genotype CT in the development of late PTS (table 4). When adjusting for age, gender, GCS score, and neurosurgical intervention, genotype analyses showed that the CT group had higher odds of developing PTS when compared to those with the CC group (OR=3.547; p=0.024). Grouped genotype analysis showed that CT subjects were more likely to have late PTS than those with the CC+TT genotype (OR=3.665; p=0.013). Multivariate analysis, adjusting for the same covariates showed a trend for individuals with the CT genotype to have a higher odds of delayed onset PTS compared to the CC group (OR=3.549; p=0.072) (Table 4). Grouped genotype analyses also showed a trend for higher delayed-onset PTS rates in subjects with the CT genotype compared to the CC+TT genotypes (OR=3.307; p=0.063).
Table 4.
Multivariate Analysis—rs10920573 Associations with PTS
| Variable | Odds Ratio | 95% Confidence Interval | P-Value | |
|---|---|---|---|---|
| Genotype Associations with PTS in Late PTS Cohort (N=120) | Age | 0.999 | 0.959-1.040 | 0.082 |
| Gender | 1.831 | 0.607-5.528 | 0.283 | |
| GCS Score | 0.634 | 0.183-2.194 | 0.471 | |
| Brain Surgery | 0.91 | 0.350-2.364 | 0.846 | |
| rs10920573 CC versus CT | 3.547 | 1.182-10.642 | 0.024 | |
| rs10920573 TT Genotype versus CT | 4.266 | 0.490-37.114 | 0.189 | |
| Genotype Associations with PTS in Delayed Onset PTS Cohort (N=111) | Age | 1.003 | 0.957-1.051 | 0.901 |
| Gender | 1.061 | 0.244-4.613 | 0.937 | |
| GCS Score | 0.55 | 0.108-2.797 | 0.471 | |
| Brain Surgery | 1.4223 | 0.430-4.705 | 0.564 | |
| rs10920573 CC versus CT | 3.549 | 0.891-14.136 | 0.072 | |
| rs10920573 TT Genotype versus CT | 2.541 | 0.271-23.795 | 0.414 | |
3.9 Analysis of Multiple Risk Genotypes with Late and Delayed-Onset Post-Traumatic Seizures
Both rs3766553 and rs10920573 were associated with or had trends with late PTS and delayed-onset PTS. As such, we explored the combined effects of the risk genotypes rs3766553 (GG) and rs10920573 (CT) on late and delayed-onset PTS. For late PTS, 8.7% of subjects with no risk genotypes seized, while 21.6% subjects with one risk genotypes seized, and 46.7% of subjects with both risk genotypes seized (p=0.007) (figure 4a). Similar findings were noted for delayed-onset PTS, where 2.3% of subjects with no risk genotypes seized, 14.5% of subjects with one risk genotype seized, and 38.5% of subjects with both risk genotypes seized (p=0.003) (figure 4b).
Figure 4.
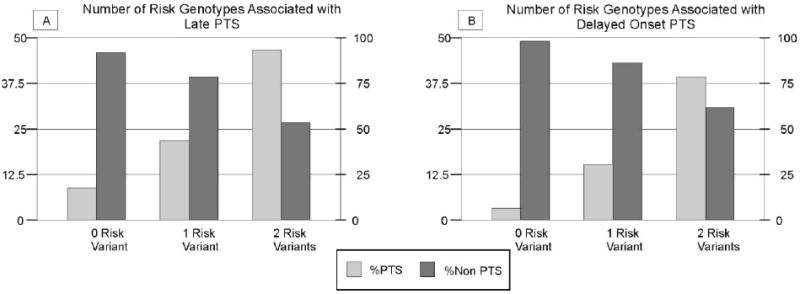
PTS frequency by number of risk genotypes A) Number of risk genotypes associated with late post traumatic seizures beyond one week after injury (p=0.007) (Population sizes by number of risk genotypes: Zero=46 One=60 Two=15) and B) Number of risk genotypes associated with delayed onset post traumatic seizures beyond six months after injury (p=0.003) (Population sizes by number of risk genotypes: Zero=43 One=55 Two=13). For both late and delayed onset PTS, the risk genotype for rs10920573 was CT and the risk genotype for rs3766553 was GG.
Multivariate analysis shows that if individuals carried either the GG genotype for rs3766553 or the CT genotype for rs10920573, they were more likely have late PTS (OR=3.812; 95%; p=0.052) and trended towards a higher odds for delayed onset PTS (OR=7.612; p=0.065) (Table 5). Furthermore, individuals who carried both risk genotypes had much higher odds of late PTS (OR=13.124; p=0.001) and delayed-onset PTS (OR=28.869; p=0.005). In comparing those who had one risk genotype to those who carried both, subjects with both genotypes had higher odds of late PTS (OR=3.443; 95% p=0.049) and trended towards higher odds for delayed-onset PTS (OR=3.792; p=0.061).
Table 5.
Multivariate Analysis---Multi-genotype Associations with Late and Delayed Onset PTS
| Variable | Odds Ratio | 95% Confidence Interval | P-Value | |
|---|---|---|---|---|
| Multivariate Associations with PTS in Late PTS (N=119) | Age | 0.998 | 0.958-1.040 | 0.939 |
| Gender | 1.713 | 0.547-5.365 | 0.355 | |
| GCS Score | 0.652 | 0.187-2.278 | 0.503 | |
| Brain Surgery | 0.827 | 0.308-2.223 | 0.707 | |
| 1 versus 0 risk variants | 3.812 | 0.989-14.692 | 0.052 | |
| 2 versus 0 risk variants | 13.124 | 2.672-64.458 | 0.001 | |
| 1 versus 2 risk variants | 3.443 | 1.004-11.813 | 0.049 | |
| Multivariate Associations with PTS in Delayed Onset PTS Cohort (N=110) | Age | 1.007 | 0.959-1.058 | 0.773 |
| Gender | 0.862 | 0.189-3.928 | 0.848 | |
| GCS Score | 0.614 | 0.118-3.204 | 0.563 | |
| Brain Surgery | 1.15 | 0.328-4.030 | 0.827 | |
| 1 versus 0 risk variants | 7.612 | 0.883-65.633 | 0.065 | |
| 2 versus 0 risk variants | 28.869 | 2.815-296.059 | 0.005 | |
| 1 versus 2 risk variants | 3.792 | 0.938-15.329 | 0.061 | |
3.10 rs3766553 and rs10920573 Associations with Mortality and 6-month GOS Score
Chi-square analysis showed that neither rs3766553 genotype (p=0.476) nor grouped genotype [(AA versus AG+GG) (p=0.346) or (GG versus AG+AA) (p=0.461)] was significantly associated with mortality. Additionally, neither rs10920573 genotype (p=0.722) nor risk-related variant (CT versus CC+TT) (p=0.504)) was significantly associated with mortality. Similarly, neither rs3766553 genotype (p=0.091) nor risk-related variant (AA versus AG+GG; p=1.000) and (GG versus AG+AA; p=0.693) was significantly associated with 6 month GOS score among survivors when GOS was dichotomized as 2-3 (vegetative/severe disability) versus 4-5 (moderate/disability-good outcome). In contrast, rs10920573 genotype was associated with the dichotomized GOS (p=0.040) among survivors. However the risk variant (CT vs. TT+CC) was not significantly associated with GOS (p=0.511).
3.11 Evaluation of rs17852405
The functional SNP identified as part of the DNA block representing rs3766553 was analyzed to determine if it was responsible for the apparent association between rs3766553 and PTS. This SNP was evaluated via PCR-RFLP and sequencing, and it was monomorphic for the C allele. Since the SNP was monomorphic, DNA for 10 subjects from each genotype for rs3766553 then underwent sequence verification for rs1785205 to verify genotyping results.
4. DISCUSSION
Post-traumatic seizures are a significant complication after TBI that often leads to post-traumatic epilepsy, long term AED treatment, and increased disability. The mechanisms underlying secondary injury and PTS development are poorly understood. However, the neurobiology of secondary TBI may be tightly linked to the mechanisms underlying PTS. Not every patient with clinical risk factors for PTS goes on to develop PTS, and this variability in occurrence may be attributable to genetic variation that patients bring to post-TBI recovery. Adenosine and its highest affinity target receptor, A1AR, are important in TBI neuroprotection and in traumatic/non-traumatic seizure development in experimental models (D’Ambrosio et al. 2004; Fedele et al., 2005; Fedele et al., 2006; Kochanek et al., 2006). Thus, we hypothesized that genetic variation in A1AR is important for the clinical development of PTS.
We explored several tSNPs in the A1AR gene and demonstrated that rs3766553 was significantly associated with early, late, and delayed onset PTS. For rs3766553, we noted an association between the AA genotype and early PTS. In contrast, the GG genotype was associated with both late/delayed-onset PTS. The CT heterozygote genotype for tSNP rs10920573 also was associated with late/delayed-onset PTS. Further, we found that risk variants for both rs3766553 and rs10920573 were independently associated with increased late and delayed onset PTS in multi-genotype multivariate analysis and had a cumulative effect on seizure susceptibility. The incidence of PTS in our cohort is similar to that reported elsewhere (Frey, 2003). To our knowledge, this is the first clinical research report implicating genetic variability within the A1AR gene in the development of either non-traumatic seizures or PTS.
For rs3766553 in the early PTS cohort, the A allele is the susceptibility allele. In contrast, it is the G allele that is important to susceptibility with late/delayed-onset PTS. In early, late, and delayed-onset PTS, rs3766553 heterozygotes are relatively protected from PTS compared to subjects homozygote for the risk allele. This indicates that in fact having a least one copy of the non-risk allele is protective. Further, it suggests that heterozygosity for rs3766553 may provide the best protection against PTS over the recovery continuum. The opposite is true for rs10920573, where heterozygotes are at greater risk for PTS beyond one week post injury.
rs3766533 is a tagging SNP. Although our haplotype analysis for the entire gene does not indicate significant haplotype differences in PTS rates, without fully characterizing all variation within the haplotype block tagged by this SNP, it is still possible that haplotype differences confined to this block are at the core of this finding. DNA sequencing and in vitro assessments with the A1AR cloned from the A allele vs. G allele will be important to determine functional differences in the receptor associated with each genotype. Additionally, these findings may indicate the importance of the receptor, but through different mechanisms for early versus late PTS.
Changing levels of adenosine availability as glial scar forms and matures over time may be why different A1AR rs3766553 alleles are associated with the development of early vs. late PTS. For example, glial scar formation is implicated in non-traumatic seizure development by compromising adenosine synthesis via increased astrocytic production of adenosine kinase (Fedele et al., 2005). Glial scarring commonly occurs after TBI (Silver and Miller, 2004), and may play role in adenosine receptor mediated seizure susceptibility via adenosine availability. The A1AR can undergo dimerization and heteromerization to modulate neurotransmission (Ciruela et al., 2006; Franco et al., 2000), and genetic variation in the A1AR gene represented by rs3766553 and rs10920573 may influence PTS susceptibility via these mechanisms.
rs3766553 is intronic and is a tSNP for A1AR that tags a block of DNA that is approximately 9,263 basepairs. This area covers the last exon and the 3’ UTR of the gene and corresponds to amino acids 113 through 326 of the receptor (Rosen et. al., 2003). This corresponds to the middle of the second intracellular domain, the fourth through seventh transmembrane regions, as well as the cytoplasmic portion of the receptor that interacts with the G-protein (Olah and Stiles, 2000). Thus, genetic variation associated with rs3766553 may be associated with both ligand binding and signal transduction (Ji et al., 1998) to affect PTS susceptibility. The fact that rs3766553 is intronic does not necessarily mean it is not the susceptibility SNP, especially since it tags a portion of the coding region for the gene. Only one known coding SNP is documented within this region in the NCBI’s SNP database, rs17852405. However, this SNP had no variation, and does not account for the association with the tSNP rs3766553. The rs3766553 also tags a portion of the 3’ region of the myosin binding protein H (MYBPH) gene. Because MYBPH is expressed and functions as a structural constituent of muscle (Vaughan et al., 1993) it is unlikely to be involved with PTS.
rs10920573 is also intronic, located within intron 4, and flanked by two additional intron 4 SNPs that are not significantly associated with PTS, therefore the region of the gene tagged by rs10920573 is limited to a portion of intron 4 that does not include splice site recognition sequences. One explanation for an association limited to intronic variability is the potential impact that intronic sequences can have on gene expression (Mackenzie et al., 1999; Hernandez-Saavedra et al., 2009). Future studies assessing the functional implications of this SNP on gene transcription, mRNA stability, and/or associated variability within this haplotype block may be informative (Mackenzie et al., 1999). Approximately 12.4% of the population has both risk genotypes for late and delayed-onset PTS. As indicated in figure 1, rs3766553 and rs10920573 are separated by rs1751192. This finding, along with the multivariate analysis showing increased PTS risk with 0 vs. 1 vs. 2 risk genotypes, supports the concept that these two tSNPs represent independent risk factors for late and delayed-onset PTS.
A1AR influences status epilepticus in both ex vivo and in vivo seizure models (Fedele et al., 2006; Avsar and Empson, 2004). Fedele et al (2006) reported that kainic acid injection causes non-convulsive status epilepticus in wild-type mice but induces rapid convulsive status epilepticus and death A1R-KO mice. Similarly, we reported that profound PTS leading to lethal status epilepticus occurs in A1AR KO mice in the CCI model of experimental TBI (Kochanek et al., 2006), a model that typically does not result in PTS activity in wild type. Our clinical findings implicating A1AR variability in PTS susceptibility, the spatial proximity of A1ARs to glutamatergic receptors, (Deckert and Jorgensen, 1988) and the relationship between adenosine and glutamate in human TBI (Robertson et al., 2001) shows that adenosine and A1AR are important in countering the contribution of excitotoxic injury to immediate/early PTS.
To minimize the confounding factor of death in artificially reducing incidence of PTS and masking gene associations with PTS, we adjusted for mortality in our analyses. Since early PTS was defined as occurring within the first week after TBI, we removed all subjects for this analysis who died during that period and did not seize. Similarly, we removed all subjects who died (except one subject with late PTS) when analyzing gene associations with late and delayed-onset PTS. Additionally, analyses suggest no associations between rs3766553, rs10920573 and mortality, indicating that genotype did not significantly impact PTS incidence by affecting mortality.
Gender, age, and GCS score were not associated with PTS. Gender is not a risk factor in other studies, however, age and injury severity have been cited as risk factors. Studies typically assess injury severity over the range of mild to severe (Annegers et al., 1998, Frey et al., 2003). Since our cohort only includes subjects with severe injuries, differentiating PTS risk by age may not informative. Also, other large epidemiological studies include both pediatric and adult populations in their analyses, and results show younger patients at more risk for early PTS and older patients at more risk for late PTS (Annegers et al., 1998, Frey et al., 2003). The majority of subjects were treated acutely with some type of AED. All subjects with PTS were given an AED, precluding AED use in multivariate analysis. However, it was difficult to determine if AEDs were given as a prophylactic or as treatment in response to a specific event. Cranial surgery was a significant univariate predictor of late and delayed-onset PTS. This variable was also significantly related to genotype for rs3766553. As such, cranial surgery was a confounding variable for this SNP which may be, in part, why cranial surgery was not significant in multivariate analyses.
While this study suggests an association between the portions of the A1AR gene tagged by rs3766553 and rs10920573 with regard to PTS, additional work needs to focus on identifying the specific portion of DNA underlying these tSNP associations with PTS and how differences in the biological functioning of portions of the receptor represented by rs3766553 and rs10920573 could contribute to the development of early, late and delayed onset PTS. The role of rs10920573 tagged haploblock with mRNA stability also should be explored. Further analysis with DNA sequencing and prospective validation studies will be required to confirm our findings. Further, targeted multivariate analysis of genes for other proteins and receptors involved in adenosine synthesis and neurotransmission will be needed to fully understand how genetic variability affects the adenosine neurotransmitter system with regard to PTS susceptibility and treatment. Future studies should also consider EEG monitoring for subclinical seizures (Vespa et al., 2007) in the acute phase post-TBI as a function of genotype.
Targeted genomic exploration of the neurobiological substrates thought to underlie the development of PTS may lead to new treatments aimed at disrupting the mechanisms that contribute to PTS and pharmacogenetic management and risk stratification for PTS therapy. The result may lead to prevention and more effective management of this complication, rather than simply treating disease associated symptoms. Also, future work with in vitro models incorporating genetically modified receptors known to have clinical associations with PTS may be useful in further understanding PTS.
The work is novel in that it implicates A1AR in a clinical population with PTS, yet there are several caveats to consider when interpreting these results. We systematically coded PTS using standard definitions for early (<7 days) and late (>7 days) categories used in the literature (Temkin et al., 1990). However, a reliable classification for immediate PTS (<24 hours) was not possible. We also explored genetic relationships with delayed onset PTS as a means to further characterize how genetic variability influences PTS onset over a broad time course. The Hapmap database available at the onset of this study was build 35. Since then build 36 of the Hapmap database became available. The selection of relevant tSNPs may have changed had we initiated the study using this build, and therefore, gene coverage may not be complete based on this newer information. However, this does not negate the PTS associations with rs3766553 and rs10920573 identified in this study. The fact that medical records available for review vary based on the time since injury make identifying the true incidence of later PTS less accurate. Due to limitations with clinical chart abstraction about seizure recurrence, time to first seizure was the primary end point evaluated, and the findings are not necessarily informative regarding whether A1AR gene variability can determine who will develop recurrent seizures and post traumatic epilepsy.
5. CONCLUSIONS
Here we provide evidence that genetic variability within the rs3766553 and the rs10920573 regions of the A1AR gene are associated with increased susceptibility for PTS. Understanding the role of A1AR in TBI and PTS, along with the mechanisms by which these tSNPs influence PTS, may lead to a better understanding of this common complication and targeted effective therapies for prevention and treatment.
Acknowledgments
The authors wish to thank Sandra Deslouches for her genotyping efforts, Candace Kammerer, PhD for haplotype consultation, and the subjects and their families for their generous participation. This work was supported by NIH R01HD048162 and DOD W81XWH-07-1-0701 to AKW, NIH R01NR008424 and DOD W81XWH-07-1-0701 to YPC, and NIH 5P01NS030318-16 and NIHR01NS38087 to PMK
Footnotes
AUTHOR DISCLOSURE STATEMENT: No competing financial interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andine, Rudolphi KA, Fredholm BB, Hagberg H. Effect of propentofylline (HWA 285) on extracellular purines and excitatory amino acids in CA1 of rat hippocampus during transient ischaemia. Br J Pharmacol. 1990;100:814–8. doi: 10.1111/j.1476-5381.1990.tb14097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;33:20–4. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- Armstrong KK, Sahgal V, Bloch R, Armstrong KJ, Heinemann A. Rehabilitation outcomes in patients with posttraumatic epilepsy. Arch Phys Med Rehabil. 1990;71:156–60. [PubMed] [Google Scholar]
- Avsar E, Empson RM. Adenosine acting via A1 receptors, controls the transition to status epilepticus-like behaviour in an in vitro model of epilepsy. Neuropharmacology. 2004;47:427–37. doi: 10.1016/j.neuropharm.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Bell MJ, Robertson CS, Kochanek PM, Goodman JC, Gopinath SP, Carcillo JA, Clark RS, Marion DW, Mi Z, Jackson EK. Interstitial brain adenosine and xanthine increase during jugular venous oxygen desaturations in humans after traumatic brain injury. Crit Care Med. 2001;29(2):399–404. doi: 10.1097/00003246-200102000-00033. [DOI] [PubMed] [Google Scholar]
- Bullock R, Chesnut RM, Clifton G, Ghajar J, Marion DW, Narayan RK, Newell DW, Pitts LH, Rosner MJ, Wilberger JW. Guidelines for the management of severe head injury. Eur J Emerg Med. 1996;3:109–27. doi: 10.1097/00063110-199606000-00010. [DOI] [PubMed] [Google Scholar]
- Cavalleri GL, Walley NM, Soranzo N, Mulley J, Doherty CP, Kapoor A, Depondt C, Lynch JM, Scheffer IE, Heils A, Gehrmann A, Kinirons P, Gandhi S, Satishchandra P, Wood NW, Anand A, Sander T, Berkovic SF, Delanty N, Goldstein DB, Sisodiya SM. A multicenter study of BRD2 as a risk factor for juvenile myoclonic epilepsy. Epilepsia. 2007;48:706–12. doi: 10.1111/j.1528-1167.2007.00977.x. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Luján R, Burgueño J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, Cortés A, Canela EI, López-Giménez JF, Milligan G, Lluis C, Cunha RA, Ferré S, Franco R. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci. 2006;26(7):2080–7. doi: 10.1523/JNEUROSCI.3574-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton GL, Allen S, Barrodale P, Plenger P, Berry J, Koch S, Fletcher J, Hayes RL, Choi SC. A phase II study of moderate hypothermia in severe brain injury. J Neurotrauma. 1993;10:263–71. doi: 10.1089/neu.1993.10.263. [DOI] [PubMed] [Google Scholar]
- Cox DR. Regression Models and Life-Tables (with Discussion) J Royal Statist Soc, Series B. 1972;34:187–220. [Google Scholar]
- Deckert J, Jorgensen MB. Evidence for pre- and postsynaptic localization of adenosine A1 receptors in the CA1 region of rat hippocampus: a quantitative autoradiographic study. Brain Res. 1988;446:161–4. doi: 10.1016/0006-8993(88)91308-x. [DOI] [PubMed] [Google Scholar]
- Dikmen SS, Temkin NR, Miller B, Machamer J, Winn HR. Neurobehavioral effects of phenytoin prophylaxis of posttraumatic seizures. JAMA. 1991;265:1271–7. [PubMed] [Google Scholar]
- Englander J, Bushnik T, Duong TT, Cifu DX, Zafonte R, Wright J, Hughes R, Bergman W. Analyzing risk factors for late posttraumatic seizures: a prospective, multicenter investigation. Arch Phys Med Rehabil. 2003;84:365–73. doi: 10.1053/apmr.2003.50022. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Gouder N, Güttinger M, Gabernet L, Scheurer L, Rülicke T, Crestani F, Boison D. Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain. 2005;128:2383–95. doi: 10.1093/brain/awh555. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Li T, Lan JQ, Fredholm BB, Boison D. Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Exp Neurol. 2006;200(1):184–90. doi: 10.1016/j.expneurol.2006.02.133. [DOI] [PubMed] [Google Scholar]
- Franco R, Ferré S, Agnati L, Torvinen M, Ginés S, Hillion J, Casadó V, Lledó P, Zoli M, Lluis C, Fuxe K. Evidence for adenosine/dopamine receptor interactions: indications for heteromerization. Neuropsychopharmacology. 2000;23(4 Suppl.):S50–9. doi: 10.1016/S0893-133X(00)00144-5. Review. [DOI] [PubMed] [Google Scholar]
- Frey LC. Epidemiology of posttraumatic epilepsy: a critical review. Epilepsia. 200;44(Suppl 10):11–7. doi: 10.1046/j.1528-1157.44.s10.4.x. [DOI] [PubMed] [Google Scholar]
- Haltiner AM, Temkin NR, Dikmen SS. Risk of seizure recurrence after the first late posttraumatic seizure. Arch Phys Med Rehabil. 1997;78:835–40. doi: 10.1016/s0003-9993(97)90196-9. [DOI] [PubMed] [Google Scholar]
- Herzog AG. Catamenial epilepsy: Definition, prevalence pathophysiology and treatment. Seizure. 2008;17(2):151–9. doi: 10.1016/j.seizure.2007.11.014. Review. [DOI] [PubMed] [Google Scholar]
- Hernandez-Saavedra D, McCord JM. Association of a new intronic polymorphism of the SOD2 gene (G1677T) with cancer. Cell Biochem Funct. 2009;27:223–227. doi: 10.1002/cbf.1560. [DOI] [PubMed] [Google Scholar]
- Hung CC, Tai JJ, Lin CJ, Lee MJ, Liou HH. Complex haplotypic effects of the ABCB1 gene on epilepsy treatment response. Pharmacogenomics. 2005;6:411–7. doi: 10.1517/14622416.6.4.411. [DOI] [PubMed] [Google Scholar]
- Jennett B, Bond M. Assessment of outcome after severe brain damage. Lancet. 1975;1:480–4. doi: 10.1016/s0140-6736(75)92830-5. [DOI] [PubMed] [Google Scholar]
- Ji TH, Grossmann M, Ji I. G Protein-coupled Receptors. J Biol Chemistry. 1998;273:17299–17302. doi: 10.1074/jbc.273.28.17299. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Vagni VA, Janesko KL, Washington CB, Crumrine PK, Garman RH, Jenkins LW, Clark RS, Homanics GE, Dixon CE, Schnermann J, Jackson EK. Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2006;26:565–75. doi: 10.1038/sj.jcbfm.9600218. [DOI] [PubMed] [Google Scholar]
- Lee ST, Lui TN, Wong CW, Yeh YS, Tzaan WC. Early seizures after moderate closed head injury. Acta Neurochir (Wien.) 1995;137:151–4. doi: 10.1007/BF02187187. [DOI] [PubMed] [Google Scholar]
- MacKenzie A, Quinn J. A serotonin transporter gene intron 2 polymorphic region, correlated with affective disorders, has allele-dependent differential enhancer-like properties in the mouse embryo. Proc Nat Acad Sci. 1999;96:15251–15255. doi: 10.1073/pnas.96.26.15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson P, Ronne-Engstrom E, Flink R, Ungerstedt U, Carlson H, Hillered L. Epileptic seizure activity in the acute phase following cortical impact trauma in rat. Brain Res. 1994;637:227–32. doi: 10.1016/0006-8993(94)91237-8. [DOI] [PubMed] [Google Scholar]
- Olah ME, Stiles GL. The role of receptor structure in determining adenosine receptor activity. Pharmacol Ther. 2000;85(2):55–75. doi: 10.1016/s0163-7258(99)00051-0. [DOI] [PubMed] [Google Scholar]
- Rhodes ME, Frye CA. Actions at GABA(A) receptors in the hippocampus may mediate some antiseizure effects of progestins. Epilepsy Behav. 2005;6:320–7. doi: 10.1016/j.yebeh.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Robertson CL, Bell MJ, Kochanek PM, Adelson PD, Ruppel RA, Carcillo JA, Wisniewski SR, Mi Z, Janesko KL, Clark RS, Marion DW, Graham SH, Jackson EK. Increased adenosine in cerebrospinal fluid after severe traumatic brain injury in infants and children: association with severity of injury and excitotoxicity. Crit Care Med. 2001;29:2287–93. doi: 10.1097/00003246-200112000-00009. [DOI] [PubMed] [Google Scholar]
- Rosen N, Chalifa-Caspi V, Shmueli O, Adato A, Lapidot M, Stampnitzky J, Safran M, Lancet D. GeneLoc: Exon-based integration of human genome maps. Bioinformatics. 2003;19(S1):i222–4. doi: 10.1093/bioinformatics/btg1030. [DOI] [PubMed] [Google Scholar]
- Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Adenosine and brain ischemia. Cerebrovasc Brain Metab Rev. 1992;4:346–69. Review. [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–56. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Smith KR, Jr, Goulding PM, Wilderman D, Goldfader PR, Holterman-Hommes P, Wei F. Neurobehavioral effects of phenytoin and carbamazepine in patients recovering from brain trauma: a comparative study. Arch Neurol. 1994;51:653–60. doi: 10.1001/archneur.1994.00540190029012. [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith N, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Human Genetics. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin NR, Dikmen SS, Wilensky AJ, Keihm J, Chabal S, Winn HR. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med. 1990;323:497–502. doi: 10.1056/NEJM199008233230801. [DOI] [PubMed] [Google Scholar]
- Temkin NR. Prophylactic Anticonvulsants after Neurosurgery. Epilepsy Curr. 2002;2:105–7. doi: 10.1046/j.1535-7597.2002.00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin NR. Risk factors for posttraumatic seizures in adults. Epilepsia. 2003;44(Suppl 10):18–20. doi: 10.1046/j.1528-1157.44.s10.6.x. [DOI] [PubMed] [Google Scholar]
- Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: A public health perspective. J Head Trauma Rehabil. 1999;14:602–15. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- Vaughan KT, Weber FE, Ried T, Ward DC, Reinach FC, Fischman DA. Human myosin-binding protein H (MyBP-H): complete primary sequence, genomic organization, and chromosomal localization. Genomics. 1993;16(1):34–40. doi: 10.1006/geno.1993.1136. [DOI] [PubMed] [Google Scholar]
- Vespa PM, Miller C, McArthur D, Eliseo M, Etchepare M, Hirt D, Glenn TC, Martin N, Hovda D. Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Crit Care Med. 2007;35(12):2830–6. [PMC free article] [PubMed] [Google Scholar]