

Abstract
The mammalian AP-endonuclease (APE1/Ref-1) plays a central role in the repair of oxidized and alkylated bases in mammalian genomes via the base excision repair (BER) pathway. However, APE1, unlike its E. coli prototype Xth, has two unique and apparently distinct transcriptional regulatory activities. APE1 functions as a redox effector factor (Ref-1) for several transcription factors including AP-1, HIF1-α, and p53. APE1 was also identified as a direct trans-acting factor for repressing human parathyroid hormone (PTH) and renin genes by binding to the negative calcium-response element (nCaRE) in their promoters. We have characterized APE1's post-translational modification, namely, acetylation which modulates its transcriptional regulatory function. Furthermore, stable interaction of APE1 with several other trans-acting factors including HIF-1α, STAT3, YB-1, HDAC1, and CBP/p300 and formation of distinct trans-acting complexes support APE1's direct regulatory function for diverse genes. Multiple functions of mammalian APE1, both in DNA repair and gene regulation, warrant extensive analysis of its own regulation and dissection of the mechanisms. In this review, we have discussed APE1's own regulation and its role as a transcriptional coactivator or corepressor by both re-dox-dependent and redox-independent (acetylation-mediated) mechanisms, and explore the potential utility of targeting these functions for enhancing drug sensitivity of cancer cells. Antioxid. Redox Signal. 11, 621–637.
Introduction
The mammalian AP-endonuclease, APE1/Ref-1, is a ubiquitous and remarkably multifunctional protein. It plays a central role in the base excision repair (BER) pathway for damaged bases and DNA single-strand breaks induced by reactive oxygen species (ROS) and alkylating agents, and also in repairing apurinic/apyrimidinic (AP) sites generated spontaneously or after excision of oxidized and alkylated bases by DNA glycosylases (25, 73, 98). APE1 was independently identified as a reductive activator of the AP-1 (c-Jun/Fos heterodimer) transcription factor and named redox effector factor 1 (Ref-1) (149). Subsequently, several other transcription factors (including p53, NF-κB, the hypoxia-inducible factor HIF1-α, PAX 5, PAX 8, and others) were also shown to be activated by APE1, presumably via the same redox process (37, 132). A third and distinct function of APE1 as a trans-acting factor was discovered when APE1 was identified as one of the regulatory proteins that binds to the negative Ca2+ response elements (nCaRE-A and B) in the intracellular calcium [Ca2+]i-dependent downregulation of the parathyroid hormone (PTH) gene (107), and subsequently in the human renin gene (44). We subsequently showed that human APE1 is acetylated at Lys6 and Lys7 by the histone acetyltransferase (HAT) p300, both in vivo and in vitro, and that acetylation enhances APE1's binding to nCaRE-B, leading to repression of the PTH promoter (10). Recently, acetylation of APE1 has been shown to be involved in early growth response (Egr-1)-mediated activation of phosphoinositol phosphatase and tensin homologue (PTEN) expression (39). Our recent study indicated that APE1 stably interacts with Y-box-binding protein 1 (YB-1) and enhances its trans-acting activity, leading to the activation of multidrug resistance (MDR1) gene, which is strongly dependent on APE1 acetylation (unpublished data). Furthermore, stable interaction of APE1 with HlF1-α, STAT3, p300, and HDAC1 further suggests a role for APE's direct trans-acting activity in regulation of diverse genes (10, 54, 161).
In view of its multiple functions, it may not be surprising that APE1-nullizygous mice have early embryonic lethality, and that no APE1-null mouse embryo fibroblast (MEF) line could be established (152). It was, however, still unclear which function(s) of APE1 are absolutely required during early embryogenesis. Because APE1 is the major enzyme responsible for repairing endogenous AP sites, it is not surprising that its DNA repair function is necessary for embryonic development. However, APE1's redox function has recently been shown to be involved in embryonic hematopoiesis and in CD40-mediated B cell activation and thus in regulation of immune responses, indicating that APE1's transcriptional regulatory functions may be also important for early embryonic development (96, 97, 162). Recently, we and others have shown that APE1 inactivation induces apoptosis in MEF conditionally nullizygous for endogenous APE1 (MEFnull) and in many tumor cell lines (47, 70). Using a complementation assay, we also showed that both the active site for repair and acetylation sites (presumably for regulatory function) of APE1 are required to prevent apoptosis of APE1-null MEF (70). Unexpected essentiality of APE1's acetylation-dependent regulatory activity suggests that APE1 is a co-regulator of essential gene(s). Although Cys65 of APE1 was proposed to be the redox active site, lack of embryonic lethality of Cys65Ala knock in mutation in transgenic mice or APE1 conditional null mutation in MEF suggested that the redox function of APE1 is not essential for survival (70, 110, 142). This could alternatively indicate that Cys65 is not involved in APE1's redox function. On the other hand, our novel observation of modulation of APE1's transcriptional function by acetylation and essentiality of acetyl acceptor Lys have provided an opportunity for elucidating the in vivo role of this modification in both negative and positive regulation of diverse genes. Moreover, it is important to identify essential vs. nonessential regulatory functions of APE1, and whether redox activation is linked to acetylation-dependent regulation. In this review, we will summarize APE1's own regulation and its redox-dependent and acetylation-mediated transcriptional function and explore the potential of this protein as a therapeutic target in drug sensitization of cancer cells.
DNA Repair Function of APE1
Multiple oxidative DNA damage such as strand breaks, base loss, and base modifications are caused by reactive oxygen species (ROS) that are generated endogenously or due to environmental stress (2, 15). Nearly all oxidized forms of DNA bases (as well as methylated or inappropriate bases) are repaired via the BER pathway which is initiated with excision of the damaged base by a DNA glycosylase to generate AP site (65, 87, 98). APE1, the second enzyme in the BER pathway, then hydrolyzes the phosphodiester backbone immediately 5′ to an AP site to produce 3′ OH group and 5′ deoxyribose-5-phosphate (25, 31). Following removal of this blocking group via dRP lyase activity of DNA polymerase β repair DNA synthesis, followed by DNA ligase action restores genome integrity (126). Oxidized base-specific DNA glycosylases have intrinsic AP lyase activity and cleaves the DNA strand 3′ to the AP site (65, 87). The resulting 3′ blocking group is removed by APE1 (or in some cases polynucleotide kinases) in the next step of repair (20, 147). APE1's 3′ phophodiesterase activity is also involved in repairing DNA single-strand breaks with 3′ blocking group directly generated by ROS (71). Unrepaired AP sites also lead to DNA strand breaks, apoptosis, and increases cytotoxicity (92). Thus, the DNA repair function of APE1 protects the cell from both endogenous and exogenous DNA damage. All APEs have dual activities as an endonuclease and a 3′phosphodiesterase (25, 31). However, mammalian APE1's endonuclease activity is quite strong relative to its 3′ exonuclease/phosphodiesterase activity (20, 25, 148). Furthermore, unlike Escherichia coli Xth with potent 3′ phosphatase/phosphodiesterase activities, the mammalian APE1 has extremely weak 3′ phosphatase activity that would be required to remove 3′ phosphate directly generated by ROS or due to AP lyase activity of mammalian glycosylases, NEIL1 and NEIL2 (66, 148). We have shown that NEIL-dependent BER utilizes polynucleotide kinase rather than APE1 (148). APE1 also coordinates BER as an assembly factor by interacting with downstream BER protein such as DNA polymerase β, X-ray cross-complementing-1 (XRCC1), proliferating nuclear antigen (PCNA), and flap endonucelase (FEN1) (27, 38, 73). A recent study shows that Bcl2, an anti-apoptotic protein directly interacts with APE1 and inhibits AP site repair by downregulating AP-endonuclease activity of APE1 (160). Exposure of lung cancer cells to the DNA damaging agent promotes Bcl2 accumulation and association with APE1 in the nucleus (160).
Regulation of APE1 Expression
Although APE1 is ubiquitously expressed in cells and tissues, its expression and subcellular localization level appear to be cell-type specific (79, 132). APE1 is regulated at both transcriptional and post-transcriptional levels. Expression of APE1 in mouse NIH3T3 cells was found to be cell cycle dependent with the highest level of APE1 in early or middle S-phase, pointing to a particular function of APE1 in this phase of cell cycle (46). The effects of ROS on APE1 induction have been extensively studied. We and others have shown that hydrogen peroxide (H2O2) and hypochlorous acid (HOCl) acts as inducers of the APE1 gene (56, 120). Subsequently, several in vivo and in vitro studies confirmed APE1 gene activation by oxidative stress (57, 111). This observation is of particular interest, because H2O2 and HOCl are endogenously formed during inflammatory response of macrophages and lymphocytes. Endogenous ROS may elevate the level of DNA damage which then signals an increase in APE1 level, thus enhancing the BER capacity. Indeed, induction of APE1 was found to be accompanied by an adaptive response of cells to the cytotoxic and clastogenic activity of oxidative agents, indicating its physiological relevance of the phenomenon (42, 56, 120). Consistent with this, Helicobacter pylori infection which induces oxidative stress activates APE1 expression in gastric epithelial cells (29, 30). Similarly, induction of oxidative stress was shown to be involved in the enhanced nuclear translocation of thioredoxin (TRX) and APE1 and augmentation of the APE1/NF-κB complex formation in the parenchyma cells of injured lung (52). In many cell types, ROS-mediated activation of APE1 involves two steps. In the first step, APE1 translocates from the cytoplasm to the nucleus. In B-lymphocytes and thyroid cells, such translocation is fairly rapid, within an hour, whereas in HeLa and other cells the process takes many hours (120, 134, 136). The second step involves de novo protein synthesis via transcriptional activation of the APE1 promoter, because various agents that block transcription or protein synthesis, also abolish induction of APE1 (120). Additionally, APE1 induction is associated with an increase in AP-endonuclease activity and cell's resistance to cytotoxic effect of H2O2, methyl methane sulphonate (MMS), bleomycin, and γ-radiation (42, 56, 120). Transiently overexpressed APE1 protects cells against genotoxicity and cell killing provoked by ROS (42). However, whether protection against ROS-induced cell killing by APE1 is due to of its repair or transcriptional regulatory functions or both is still unknown.
ROS activates APE1 promoter-dependent reporter expression in Chinese hamster (CHO) cells. Mutation of a CREB-binding site (CRE) abolished the H2O2-mediated activation of the APE1 promoter (57). Interestingly, a recent study identified an AP-1/CREB binding site in the mouse promoter which is essential for arsenite-induced transcriptional activation of APE1 in mouse 10T1/2 cells (48). This study showed that arsenite induced APE1 mRNA and protein levels in mouse fibroblasts, and that ATF4/c-Jun heterodimer was the responsible transcription factor for this activation. Moreover, suppression of APE1 or of ATF4 enhanced arsenite cytotoxicity in both mouse 10T1/2 and human lymphoblastoid TK6 cells (48). High conservation of this CREB-binding site in the human (hAPE1) and mouse (mAPE1) promoters highlights its importance in oxidative stress-induced transcription activation (63). In this context, the contribution of APE1 to cellular arsenite resistance is consistent with its proposed DNA repair function in BER under oxidative stress and suggests that APE1 activity may become limiting in DNA repair under certain conditions. Another transcription factor, Egr-1, a member of the immediate-early transcription factors that are rapidly and transiently induced by ROS, binds to the APE1 promoter and activates luciferase expression (102, 111). Pines et al. (111) identified a consensus Egr-1 binding site in the proximal APE-1 promoter. Moreover, they showed that APE1 increases Egr-1's cis element-binding activity after H2O2 treatment (111). Thus, activation of both APE1 and Egr-1 after oxidative stress seems to represent a positive autoregulatory loop between APE1 and Egr-1 and that could explain the early transcriptional activation of APE1 expression.
Other external stimuli such as hormones and cytokines modulate APE1 expression. Thyrotropin (TSH) induces APE1 expression in thyroid cells (5, 131, 134). Similarly, human chorionic gonadotropin has been demonstrated to enhance APE1 mRNA synthesis in murine Leydig cells (128). IL-2-dependent APE1 upregulation has also been demonstrated in a murine Pro-B cell line (156). Interestingly, Helicobacter pylori-induced IL-8 activation in gastric epithelial cells was found to be dependent on APE1 (103). Another recent study demonstrated that ATP-mediated purinergic receptor activation upregulates APE1 expression in human tumor thyroid cell line (112).
Although the cloning of the APE1 gene and characterization of its promoter were performed several years ago, the molecular mechanism(s) for APE1's regulation has still not been completely explored (63, 64). The human APE1 gene appears to have multiple positive and negative regulatory elements. Activation of APE1 expression by signal transducer and activator of transcription-3 (STAT3) in the liver and both redox-dependent and independent function of APE1 has been shown for protection against Fas-mediated liver injury (59). Earlier, our laboratory identified three negative calcium-responsive elements (nCaREs: nCaRE-A, nCaRE-B1, and nCaRE-B2) in the distal APE1 promoter (72). Two such elements (nCaRE-A and nCaRE-B) were originally identified in the promoter of PTH gene which is negatively regulated by extracellular calcium and APE1 was identified as one of the trans-acting factors (107). These observations strongly suggest that APE1 may negatively regulate its own expression by binding to these nCaREs, in particular to the nCaRE-B2.
We have recently shown that wild-type (WT) p53 but not mutant p53 negatively regulates APE1 expression in colon carcinoma HCT116 cells (159). Ectopic overexpression of WT p53 but not mutant p53 (V143A, L22G, and T23S) significantly decreased APE1-promoter-dependent luciferase expression in unstressed p53-null HCT116 cells (159). Furthermore, we have shown decrease in the levels of both APE1 mRNA and protein after camptothecin treatment in HCT116 p53(+/+) cells, but not in the isogeneic p53 null cells. Interestingly, the candidate p53-responsive cis element located in the −184 to −143 base pairs (bp) segment of the APE1 promoter lacks a consensus p53-binding site but has one Sp1-binding site (159). However, we showed using chromatin immunoprecipitation (ChIP) assay that p53 was indeed bound to the APE1 promoter, indicating that the APE1 repression by p53 is not mediated via direct cis element binding, but rather involves p53's indirect recruitment to the promoter by other transcription factor (possibly Sp1) (159). Our findings are intriguing because p53 acts as a pro-apoptotic factor in the cellular response to stress, whereas APE1 is a pro-survival protein whose DNA repair function is essential for protecting cells from oxidative DNA damage. It appears that the cell uses p53 to downregulate APE1 expression in response to DNA damage, and promotes apoptosis. Consistent with this idea, several earlier studies showed reduction of APE1 expression during apoptosis in neurons after transient global cerebral ischemia in rats (34, 50, 90, 143). Additional studies are needed to unravel the physiological significance of such downregulation.
Another factor that modulates APE1 expression is hypoxia, which mimics oxygen tension that is encountered by cells in tissues in vivo. Hypoxia induces APE1 mRNA and protein levels in HT29 cells (158). Elevation of APE1 steady-state mRNA levels is an early event following hypoxia, and persists after restoration of cells to normoxia (158). Nuclear run-on analysis demonstrated that induction of transcription is responsible for elevation of APE1 mRNA (158). Changes in APE1 expression in response to hypoxia was correlated with its requirement for enhanced AP-1 binding following hypoxia via redox activation (158). However, another possible role for prolonged expression of APE1 following hypoxia relates to DNA repair function that remains to be elucidated. Although it is not known whether hypoxia-inducible factors (HIFs) bind-specifically to the APE1 promoter or enhancer, APE1 regulates HIF-1α functions in vivo (36, 69). APE1 up-regulation significantly potentiates hypoxia-induced expression of a reporter construct containing the HIF-1α-binding site (36). Moreover, Ema et al. (36) and Carrero et al. (17) showed that APE1 is critical to linking coactivator proteins, CBP/p300 and SRC-1 to HIF-1α. In contrast, Hall et al. showed that hypoxia downregulates APE1 protein level in both calf pulmonary artery endothelial (CPAEC) and human umbilical vein endothelial (HUVEC) cells (60). Such hypoxia-induced decrease of APE1 was associated with significant induction of apoptosis in CPAEC and HUVEC cells (60). Thus, APE1 downregulation may be permissive in promoting apoptosis in endothelial cells in response to hypoxia. Indeed, APE 1 overexpression was shown to protect CPAEC cells from hypoxia-induced apoptosis (60).
Recently, it has been shown that soy isoflavones down-regulate expression of APE1 in PC3 prostate cancer cells (119). Moreover, pretreatment with soy isoflavones inhibits radiation-induced APE1 expression and activation of NF-κB. Although the mechanism by which soy isoflavones down-regulates APE1 expression is not known, downregulation of APE1 and inhibition of NF-κB activation by soy isoflavones was shown to inhibit tumor growth in vivo (119).
Redox Activation Function of APE1
The regulation of protein functions via redox reaction is becoming increasingly evident not only for transcriptional regulation by trans-acting factors but also for other activities such as that of protein phosphatases (23, 113). APE1 enhances DNA-binding of AP-1 via reduction of the conserved Cys272 residue in the DNA-binding domain of c-Jun (149, 150). Oxidation of APE1 significantly diminishes its ability to stimulate AP-1 DNA-binding activity (149). The reduced Ref-1 activity of oxidized APE1 can be restored by treatment with thioredoxin (TRX), suggesting that a redox–sensitive Cys in APE1 is involved in the reductive activation of the AP-1 complex (67). Ionizing radiation induced nuclear translocation of thioredoxin and its stable interaction with APE1, leading to subsequent activation of AP-1 indicated a thiol exchange cycle APE1's Cys reduces oxidized TFs and is subsequently reduced by TRX (67, 138, 146). A large number of stress-inducible transcription factors including NF-κB (101), ATF/CREB (150), p53 (75), Pax5 (135, 136), Pax8 (16, 133), c-Myb (150), HIF1-α (69), and others have been reported to be activated by APE1-mediated redox activation, presumably redox mechanism involving the same Cys residue. Analysis of several deletion mutants of APE1 revealed that N-terminal 1-127 amino acids (aa) residues is sufficient for its redox activation of c-Jun binding in vitro and repair and redox activities of APE1 are encoded by two distinct regions of APE1 (151) (Fig. 1B). Interestingly, N-terminal 61 amino acids residues which are not conserved in E. coli and yeast APN1 are essential for APE1's activation of c-Jun in vitro (142). Site-directed mutagenesis analysis identified Cys65 as the redox active site in APE1 (142). In addition, it was proposed that Cys93 interacts with the Cys65, presumably via intramolecular S–S bridge formation, and is also involved in redox regulation of AP-1 (142). X-ray structure of hAPE1 lacking 40 aa residues, solved by Mol et al. (99) with our collaboration, show Cys65 itself is buried and not easily accessible by other proteins. Moreover, the distance between Cys65 and Cys93 is too far to form a S–S bond unless there is a conformational change occurring upon oxidation (Fig. 2). The human APE1 has seven conserved Cys residues at position 65, 93, 99, 138, 208, 296, and 310 (Fig. 2). The x-ray crystallographic structure suggests that Cys138 located on the surface is easily accessible for oxidation/reduction (Fig. 2). Furthermore, whether the oxidation product of APE1 is intramolecular S–S or sulfenic acid (SOH) has not been established. Reversible oxidation of Cys to intra or intermolecular S–S from two Cys residues is more widely proposed than the single Cys-SH oxidation to Cys-SOH (23, 26, 114). Both SOH and S–S are reduced by reductases, but the key difference is that the single residue SOH does not require a second, appropriately positioned Cys for S–S bridge formation. H2O2 generates Cys-SOH when the pKa of Cys-SH is lowered for ionization in the proximity of basic residues (137). Interestingly, the His–Cys sequence in protein tyrosine phosphatases, which strongly enhances Cys reactivity (91), is also present in APE1 (H309C310).
FIG. 1.
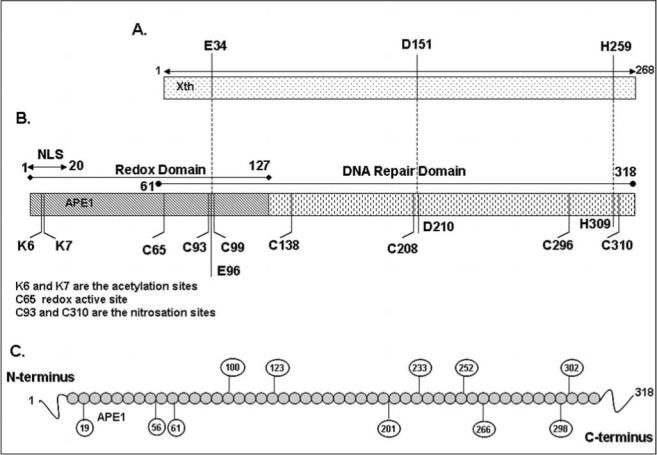
Schematic representation of APE1 polypeptide with key residues. (A and B), N-terminal 1–20 amino acids (aa) residues are putative nuclear localization signal (NLS), 1–61 aa residues are not conserved in E. coli prototype Xth (A) and dispensable for its DNA repair activity, H309 and D210 are active sites, E96 is metal (Mg2+) binding site for AP-endonuclease activity. 1–127 aa residues are sufficient for its redox activity, and Cys65 and Cys93 are putative redox active residues (142). All seven Cys are conserved in mammalian APE1. Lys6 and Ly7 are in vivo acetylation sites (10), Cys93 and Cys310 are S-nitrosation sites (117). (C) Putative phosphorylation sites in APE1 by casein kinase I (CKI), casein kinase II (CKII), and protein kinase C (PKC).
FIG. 2.
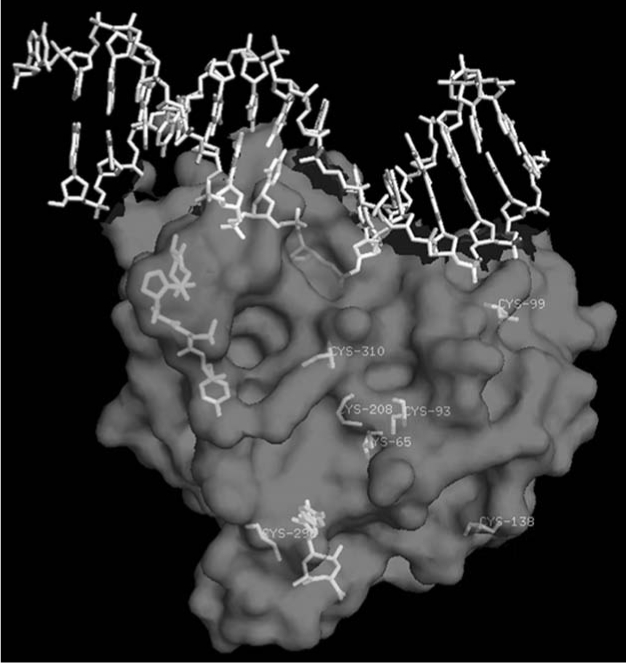
Location of cysteine residues in the three-dimensional structure of APE1 (99). The structure (by computer simulation) of N-terminal 1-40 aa residues (not included in the crystal structures) is shown.
The original suggestion of Cys65 as the active site was supported by the NMR evidence for TRX interaction with Cys65-containing polypeptide (116). Seo et al. (124) showed that the Cys65 mutant acts as a dominant negative inhibitor of the redox function of selenomethionine-containing APE1. Furthermore, apoptosis of neurons after oxidative stress is prevented by ectopic WT APE1 but to a smaller extent by the Cys65Ala mutant (139). Overexpression of either repair defective His309 or Cys65 protected cells to a similar extent upon H2O2 treatment, indicating that both repair function and redox active Cys65 are involved in protecting the cells from oxidative damage (42). Moreover, several studies using transcription factor-dependent reporter expression assays showed that the Cys65 mutant does not behave like the WT APE1 (76, 111, 162). However, this Cys65-mediated proposed redox function of APE1 is recently been challenged by Curran's group (110) who showed that homozygous APE1 Cys64Ala (in human Cys65) knock-in mouse mutants are viable and retain normal levels of AP-1 DNA binding activity. Moreover, the recombinant Cys64Ala APE1 mutant showed normal c-Jun reducing activity like WT APE1 in vitro (110). Taken together, these studies confirm that the APE1 Cys64/65 is not essential for APE1's redox regulatory function. It is thus possible that a second Cys residue could also serve as the redox active residue. Alternatively, either Cys65/64 or Cys93 (both included in the redox active domain of APE1) could serve as a redox active site in the absence of the other. However, given the location of these two Cys in the proposed structure of the APE1 (Fig. 2), we must also consider that neither of the residues is involved in its redox function. However, a recent study showed that small molecule inhibitor 3-[5-(2, 3-dimethoxy-6-methyl-1,4-benzo-quinoyl)]-2 nonyl-2-propionic acid (E3330) specifically inhibits APE1's redox function in ovarian cancer cells (93). Moreover, E3330 also inhibits mouse retinal vascular endothelial cells proliferation and angiogenesis in vitro. Very recently, Ando et al. (3) showed that APE1 stimulates p50 or c-Jun DNA-binding by facilitating their reduction by reducing agents such as glutathione or thioredoxin, and none of the APE1's Cys residues is essential for this activity.
Despite the formation of dimeric APE1 in vitro, there is no strong evidence for favoring S–S formation over S-OH during oxidation of APE1, because the second Cys-SH in the S–S bond has not been identified (53). However, our data show that oxidation of recombinant APE1 with H2O2 inhibits it AP-endonuclease activity in a dose-dependent manner (Fig. 3A). Interestingly, Kelley's group suggested that oxidation of Cys310 residues also affects APE1 DNA repair activity (81). Treatment with 10 mM H2O2 or 3 mM diamide completely abolished DNA repair activity, while high dithiotheritol (DTT) level restored the activity (81). We have shown that none of the Cys→Ser mutation affected APE1's endonuclease activity in low (< 2 mM) Mg2+ (Fig. 3B). However, the Ser99 mutant behaved uniquely because of strong inhibition of its enzymatic activity in higher (10 mM) Mg2+ that is commonly used is activity measurements (94). Furthermore, unlike WT and other Cys→Ser mutants, the Ser99 mutant did not bind to DNA as judged by electrophoretic mobility shift analysis (94). The unexpected involvement of Cys99 (∼16A° from the active site) in APE1's substrate binding and catalysis provides an example of involvement of a residue far from the active site.
FIG. 3.
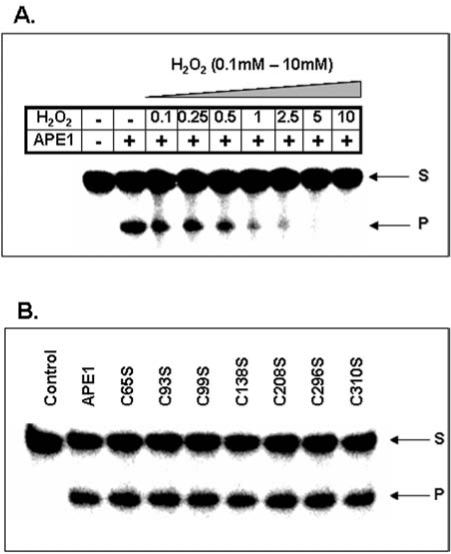
H2O2-induced loss of AP-endonuclease activity in APE1. (A) Human APE1 (2 fmol) was treated with increasing concentration (0.1–10 mM) of H2O2 for 10 min and then incubated with a 43-mer oligonucleotide containing a AP site analog THF (tetrahydro furan) at 37°C for 3 min in a reaction mixture containing 50 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM DTT, 0.1 mM EDTA, 100 ug/ml bovine serum albumin, and 2 mM magnesium chloride (MgCl2). The reaction was stopped with 80% formamide/40 mM NaOH, followed by heating at 95°C for 5 min. The samples then electrophoresed in a denaturing gel of 20% polyacrylamide containing 8 M urea to separate the substrate oligo from the cleaved product. The gel was dried and analyzed in phosphorimager (Molecular Dynamics, Sunnyvale, CA). (B) Dispensability of Cys residues in APE1 for AP-endonuclease activity. The duplex THF-containing 43-mer oligonucleotide was incubated with recombinant WT APE1 or individual Cys mutant polypeptide at 37°C for 3 min during which the reaction rate was linear. The standard AP-endonuclease assay was performed according to the published protocol (18, 94).
Covalent Modifications of APE1
Post-translational modifications of proteins have emerged as the main mechanisms for the control of many biological functions, including signal transduction, gene expression, protein degradation, nucleoplasmic shuttling, and protein–protein interaction (51). It is evident that the activity of a single protein with pleiotropic functions could be “finely tuned” via post-translational modifications, including phosphorylation, acetylation, nitrosation, ubiquitination, and methylation, etc., in order to coordinate specific biological activities. Given the multiple functions, it is not surprising that APE1 is post-translationally modified in vivo. An earlier study by Yacoub et al. (154) showed that APE1 can be phosphorylated in vitro by casein kinase I and II (CKI and CKII). APE1 contains a number of potential phosphorylation sites including consensus sequences for CKI, CKII, and protein kinase C (PKC) (Fig. 1C). CKII-mediated phosphorylation of APE1 abolished DNA repair activity in vitro, while phosphorylation by CKI or PKC has no effect (154). In contrast, a subsequent study by Fritz and Kaina (43) showed that APE1 phosphorylation by CKII enhances redox activation of the AP-1 transcription factor and has no effect on its DNA repair activity. At this time, the reason for these discrepancies is unclear, whereas unequivocal evidence for of APE1's in vivo phosphorylation and identification of phosphorylation sites are clearly warranted. The first in vivo evidence for PKC-mediated phosphorylation of APE1 was shown by Hsieh et al. (68). Although immunoprecipitated APE1 from phorbol 12–myristate 13-acetate (PMA) or hypochlorite treated cell extracts showed enhanced DNA-binding of AP-1 presumably by PKC-mediated phosphorylation of APE1 (68), this study did not identify the specific PKC phosphorylation site nor did address whether APE1's endonuclease activity is also affected by PKC phosphorylation. Thus, an attractive hypothesis is that exposure of cells to genotoixc stress leads to phosphorylation-dependent stimulation of another post-translational modification (which we are currently investigating); this in turn modulates APE1's redox activity for activation of stress-inducible transcription factors such as AP-1, NF-kB, and p53. Since different phosphorylation sites of APE1 have not been functionally characterized in vivo by site-directed mutagenesis, the biological significance of APE1's phosphorylation is still unclear.
We have earlier established APE1's acetylation at Lys6 and Lys7 by the histone acetyltransferase p300 both in vivo and in vitro (10, 12). Our in vitro studies with recombinant WT APE1 or K6R/K7R or K6L/K7L with p300 identified the acetyl-acceptor residues as Lys6 and Lys7 (10). However mass spectroscopic analysis of in vitro acetylated APE1 (AcAPE1) could not detect diacetylated APE1 that could be explained by the possible steric effects of acetyl groups attached to ɛ-amino groups during second acetylation (10). Our data thus indicate that either Lys6 or Lys7 but not both can be acetylated in the same molecule. It is interesting to note that Lys6 and Lys7 are conserved in most mammalian APE1 including human, mouse, and bovine, however Lys7 is not conserved in rat and Chinese hamster, suggesting that acetylation of Lys6 has specific biological function in mammals (Fig. 4). We prepared affinity-purified AcAPE1-specific antibody using a human APE1 peptide with acetylated Lys6 and showed that the AcAPE1 antibodies are highly specific for AcAPE1, and do not cross react with at least 25-fold excess unmodified APE1 as judged by immunoblotting (38, unpublished data). Moreover, this antibody recognizes ectopic FLAG-tagged WT APE1, but not nonacetylable K6R/K7R APE1 in cell extracts confirming its specificity for AcAPE1 (Fig. 5B). Using this antibody we have provided unequivocal evidence for the presence of endogenous AcAPE1 under normal conditions (Fig. 5A).
FIG. 4.

Sequence alignment of human APE1 with other mammalian APEs showing acetylation sites Lys6 and Lys7 are conserved in all mammalian APE, except rat and Chinese hamster where Lys7 was replaced with Arg.
FIG. 5.
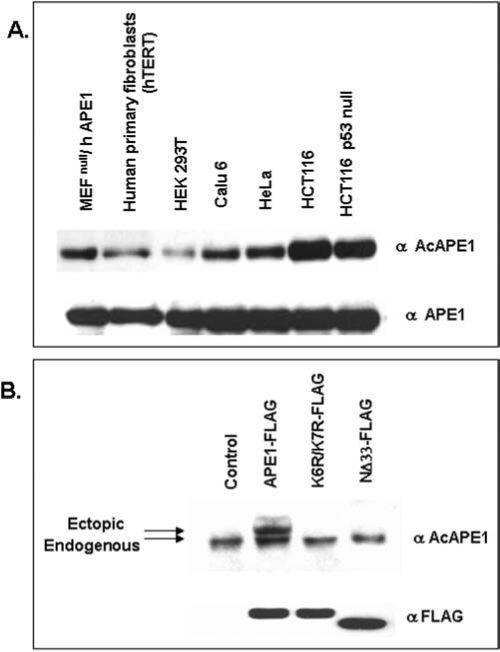
Presence of AcAPE1 in various cell lines. (A) Level of acetylated APE1 (AcAPE1) in various human cell lines. Western blot analysis of cell extracts (25 μg) from various cell lines was carried out with AcAPE1 antibody (upper panel) and followed by reprobing with APE1 antibody (lower panel). (B) Specificity of AcAPE1 antibody. HCT116 cells were transfected with FLAG-tagged WT APE1 or nonacetylable K6R/K7R APE1 mutant or NΔ33-APE1 and cell extracts were immunoblotted with AcAPE1 or FLAG antibodies.
Although the first 20 amino acid residues contain a consensus nuclear localization signal (NLS) sequence (MPKRGKK) that includes the acetylation sites (Fig. 1B), we have shown that acetylation of K6 and K7 of APE1 is not involved in its nuclear localization (74, 129). Although lysine residues are targets for both acetylation and ubiquitination, and acetylation may protect a protein from ubiquitination and degradation (55, 77), both WT and nonacetylable K6R/K7R APE1 mutant showed similar in vivo stability (74). We have shown earlier that acetylation of other DNA repair proteins involved in BER modulates their activity. For example, acetylation of K338 and K341 of 8-oxoguanine-DNA-glycosylase (OGG1) enhances its catalytic activity while acetylation of NEIL2 at the conserved K52 abolished its DNA glycosylase activity (9, 11). In contrast, acetylation of APE1 does not change its AP-endonuclease (DNA repair) activity, but rather modulates its transcriptional regulatory functions (10). We showed earlier that APE1 acetylation stimulates the formation of nCaRE-B complex which also contains hnRNP-L and HDAC1 (10). It appears likely that acetylation-mediated conformational change in the disordered N-terminal segment (encompassing 40 aa residues) which is dispensable for APE1's endonuclease activity, modulates protein–protein interaction. Our recent data indicate that APE1 stably interacts with YB-1 and acetylation enhances its binding with YB-1 both in vivo and in vitro (unpublished data).
Recently, S-nitrosation of APE1, another post-translational modification, has been shown to occur in vivo. Treatment of S-nitroglutathione, an S-nitrosating agent, stimulated nuclear export of APE1 through S-nitrosation of Cys93 and Cys310 in a CRM1-independent manner (117). Immunofluorescence studies using HA-tagged APE1 showed that this translocation process is dependent on Cys93 and Cys310 residues and can be reversed by reductive reagents or antioxidant, however could not be mimicked by oxidative stress (117). Although the precise mechanism by which S-nitrosation controls nuclear export of APE1 is not clear, aa residues 64–80 (close to Cy 93) were found to be critical for the nitrosation-mediated cytosolic translocation of APE1 (117). Moreover, inhibition of this translocation after overexpression of histone deacetylase HDAC2 raised the possibility that acetylation of APE1 (because we have shown that APE1 stably interacts with HDAC2) may cross talk with nitrosation in this process (12, 117). Recently Hara et al. (62) reported S-nitrosation-mediated nuclear translocation of glyceraldehyde 3 phosphate dehydrogenase (GAPDH). Although our earlier studies suggested that APE1 acetylation is not relevant to its nuclear localization, the possible role of acetylation in nuclear export can not be ruled out. Although there is no experimental evidence for APE1's ubiquitylation or sumoylation, its interaction with the ubiquitin conjugating enzyme ubc9 raises the possibility that APE1 may undergo turnover through these modifications (157).
APE1 as a Transcription Factor
APE1 as a trans-acting factor was discovered by Okazaki's group when they identified APE1/Ref-1 as one of the proteins that binds to negative calcium response elements (nCaRE) complex in the human PTH gene promoter (107). In most cell types, rise in intracellular [Ca2+]i triggers activation of many genes' expression and secretion, with the notable exception of parathyroid cells and renin-producing juxtaglomerular cells, where a rise in [Ca2+]i suppresses both renin and PTH gene expression (58, 155). This is mediated by the binding of cognate trans-acting factors to nCaRE in the PTH and renin promoter (107). Two such elements (nCaRE-A and nCaRE-B) in the PTH gene promoter were identified and APE1 was found to be associated with these elements (107). Subsequently, Macfee et al. identified putative nCaREs in some 100 genes, including several genes including calmodulin and β-myosin, that are subject to Ca2+-mediated regulation (95). An nCaRE-B sequence, identical to that in the PTH promoter, at 1.8 kb upstream of the transcription start site in the human renin gene has been identified (44). Involvement of this element in Ca2+-mediated repression of renin promoter-driven reporter expression has been shown in chorio-decidual cells (44). Again, APE1 was identified as a component in the nCaRE-B bound-complex, analogous to the situation in the PTH gene, and calcium-induced nuclear translocation of APE1 was also observed (44). However, APE1 does not directly bind to the nCaRE-B, indicating a requirement for additional factors in the trans-acting complex assembly. The Ku70 (Ku86) proteins were identified in the complex bound to nCaRE-A (21). Subsequently, heterogeneous ribonucleoprotein L (hnRNP-L) and APE1 were identified as major components of the complex bound to nCaRE-B (88). We showed earlier that AcAPE1 does not bind alone to nCaRE-B, although acetylation of APE1 enhances the binding of nuclear extracts to nCaRE-B (10). An increase in extracellular Ca2+ increases the level of acetylated APE1 due to Ca2+-induced activation of p300 HAT activity, presumably by inducing phosphorylation of p300 catalyzed by protein kinase C or Ca2+-dependent calmodulin (CAM) kinase IV (1). Moreover, using ChIP assay, we showed that acetylation of APE1 enhances recruitment of APE1-HDACs complexes to the PTH promoter in a Ca2+-dependent manner and AcAPE1 acts as corepressor. However, the detailed molecular mechanism by which APE1 represses renin gene expression by binding to nCaRE-B is still unclear.
An earlier study showed that oxidative stress-induced activation of APE1 increases Egr-1's DNA-binding activity in osteoblastic HOBIT cells (111). Egr-1, a transcription factor with tumor suppressor function, regulates expression of many genes, including p53 and PTEN, that control cell growth arrest and apoptosis (7, 140). Coimmunoprecipitation studies showed that APE1 stably interacts with Egr-1 and treatment with H2O2 strongly stimulates their association (111). Recently, Tell's group in collaboration with us has shown that Egr-1-mediated activation of PTEN expression is dependent on APE1 acetylation (39). Treatment of HeLa cells with HDAC inhibitors was further shown to increase APE1 acetylation and induced PTEN expression (39). The absence of such induction in APE1 downregulated HeLa cells confirmed APE1's role in regulating inducible PTEN expression. Moreover, activation of Egr-1-dependent PTEN promoter due to overexpression of WT APE1, but not of the nonacetylable K6R/K7R mutant further confirms that acetylation-enhanced direct transcriptional activity of APE1 in Egr-1-dependent PTEN expression (39).
Stable interaction of APE1 with HlF1-α, STAT3, and CBP/p300 in hypoxia-induced expression of vascular endothelial growth factor (VEGF) further suggests a role for APE's direct regulatory role in VEGF expression (54, 161). APE1's interaction with HIF-1α and p300 (but not with ATF/CREB) and its presence was found to be critical for assembly of the hypoxia-inducible transcriptional complex on the hypoxic response element (HRE) in the VEGF gene promoter in the rat pulmonary artery endothelial cells (PAEC) (161). Moreover, immunodepletion of APE1 prevented association of HIF-1 with p300, ATF, and CREB, indicating the critical role of APE1 in complex formation (161). Supporting this, a recent study Gray et al. (54) showed that APE1 stably interacts with STAT3 and HIF-1α, and STAT3, CBP/p300, and APE1 are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas (54). STAT3 and HIF-1α bind simultaneously to the VEGF promoter, where they form a complex with the transcription coactivators CBP/p300 and APE1. In addition to enhancing DNA affinity of transcription factors, APE1 may target recruitment of p300 to the transcription complexes (54) which suggests that APE1 acts as a direct transcriptional coactivator in hypoxia-induced VEGF expression. Why would APE1, a DNA repair protein, with redox function, also be present as a stable component of both constitutive and hypoxia-inducible transcription? In this regard it is worth noting that p300, best known as a scaffold protein that facilitates transcriptional complex assembly, and is also involved in chromatin remodeling via its intrinsic HAT activity (105), also acetylates Lys6 (or Lys7) in APE1. Thus, p300-catalyzed acetylation of APE1, perhaps in concert with other proteins, may be the key to incorporation of APE1 in the constitutive and hypoxia-inducible transcriptional complex. Additional studies will be required to settle this issue.
Recently, using affinity screening, we identified other interacting partners of APE1 including Y-box-binding protein YB-1 that regulates many genes positively or negatively (84, 106). APE1 stably interacts with YB-1 and its acetylation enhances its binding to YB-1, leading to activation of the Y-box-dependent MDR1 promoter (unpublished data). Mutation of Lys6 and Lys7 significantly decreased the binding. The exact mechanism by which acetylation of APE1 activates MDR1 expression is not clear. The likely redox-inactive Cys65Ser and Cys138Ser APE1 mutants behaved like WT APE1 in modulating YB-1-mediated MDR1 promoter activity, suggesting that APE1's redox activity is not involved in MDR1 activation. Using the ChIP assay, we have provided direct evidence that APE1 is present with the Y-box-bound complex in the MDR1 promoter in vivo, and that tricostatin A (TSA, a specific HDAC inhibitor) treatment enhanced (presumably via increased APE1 acetylation) MDR1's promoter occupancy (unpublished data).
Redox-independent transcriptional function of APE1 in modulating HIF-1α, Egr-1, and YB-1 activities is not without precedent. Both redox-dependent and redox-independent transcriptional function of APE1 in modulating p53-DNA-binding was first described by Jayaraman et al. (75). Recombinant APE1 was shown to stimulate DNA-binding of full length p53, which was further strongly stimulated in the presence of DTT, indicating that APE1-mediated activation of P53 might be independent of its function as a redox activator. This was further supported by the observation that while APE1 stimulated binding of full length p53 in the presence of DTT, it was incapable of stimulating binding of p53 deletion mutant (p53Δ30), which lacked the carboxy terminal 30 amino acid residues, suggesting that redox-independent activation of p53 is due to interaction of APE1 with its C-terminal region. This was confirmed in vivo by the observation that APE1 overexpression stimulated transactivation function of full length p53 in a reporter-based assay but not the p53Δ30 mutant (75). Although stable interaction between p53 and APE1 could not be shown, Far Western and coimmunoprecipitation Western assay suggested weak interaction between these proteins (49). It is thus tempting to speculate that APE1 has dual functions with respect to p53 binding: binding to the C-terminus abolishes negative regulation, thereby making the protein accessible to redox regulation. Recently, Hanson et al. (61) showed that APE1 promotes association of p53 dimers into tetramers, and destacking of higher oligomeric forms into tetrameric form in vitro, thereby enhancing p53 binding to target DNA. This novel assembly function of APE1 in promoting tetramer formation of p53 was proposed to be the underlying mechanism for it redox-independent effects on p53.
Although APE1 was identified in diverse trans-acting complexes, APE1 by itself has no affinity for any specific cis element; rather it affects promoter activity by binding to trans-acting factors specific for distinct cis elements. Thus APE's presence in diverse trans-acting complexes requires interaction with diverse partners (Fig. 6). In the case of nCaRE-B, we identified hnRNP-L and HDAC1. Similarly for HRE-binding, APE1 interacts with HIF-1α, STAT3, and p300, and with YB-1 and p300 for Y-box response element (YRE) dependent regulation (Fig. 6). However, how APE1 acts as a coactivator or corepressor in such diverse complexes is not clear.
FIG. 6.
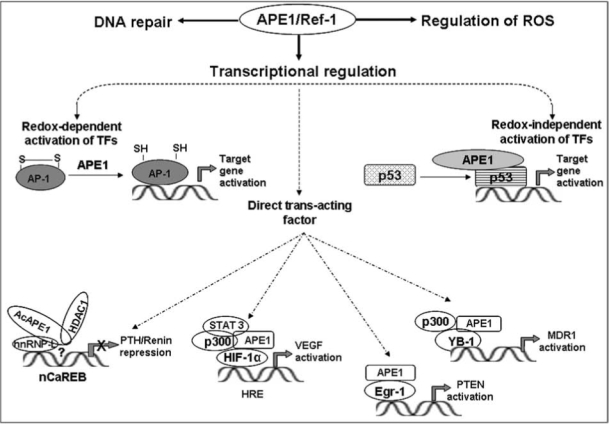
A schematic model for redox-dependent and-independent transcriptional regulatory functions of APE1/Ref-1. APE1 partners with different proteins in diverse trans-acting complexes. APE1 interacts with hnRNP-L, HDAC1 for binding to nCaRE-B (negative calcium response element B) to act as a repressor for Ca2+-mediated downregulation of human PTH and renin genes (10, 44). In HRE (hypoxia response element) bound complex, APE1 interacts with HIF-1α, STAT3, and p300, and activates hypoxia-induced expression of VEGF (54, 161). APE1 interacts with Egr-1 for activation of PTEN (39, 111). APE1 interacts with YB-1 and p300 in Y-box bound complex for activation of MDR1 (unpublished data).
AcAPE1 as a Cancer Therapeutic Target
APE1 overexpression, often observed in tumor cells, is associated with resistance to various anticancer drugs (13, 78, 80, 86, 115, 121, 153); its downregulation sensitizes tumor cells to such agents (14, 122, 144). Several studies have shown that targeted reduction of APE1 protein by specific anti-sense oligo or siRNA sensitized to MMS, H2O2, bleomycin, temozolomide (TMZ), and gemcitabine (14, 89, 109, 141). Whether such enhanced sensitivity is solely due to the loss of APE1's DNA repair activity or also due to the loss of its transcriptional regulatory function or both is still unknown. Because APE1 plays a central role in the repair of endogenous DNA damage and small base adducts induced by alkylating agents via the BER pathway, it is not surprising that APE1 overexpression is associated with tumor cells' resistance to alkylating drugs. However, downregulation of APE1 was shown to sensitize tumor cells to etoposide or cisplatin (86, 153), which cannot be easily explained by the repair function of APE1 as these drugs induce DNA double-strand breaks or DNA intra-strand cross-links that are repaired via the APE1-independent nonhomologous end joining or nucleotide excision repair pathways. As we mentioned earlier, APE1 acetylation enhances its YB-1 binding and activates MDR1 gene expression (unpublished data). Tumor cells frequently develop drug resistance after initial chemotherapy due to elevated level of MDR1 which is often upregulated due to promoter activation after drug treatment (19, 85). Immunostaining analyses of various human cancers including breast cancer and osteosarcoma showed that the elevated nuclear level of YB-1 is closely associated with the acquisition of MDR1-mediated multidrug resistance (6, 45, 104). We have demonstrated that downregulation of APE1 sensitizes MDR1-overexpressing tumor cells to anticancer drugs such as doxorubicin and cisplatin, which suggests a molecular basis by which APE1's transcriptional function and YB-1 could be linked to induce drug resistance of tumor cells. This was further supported by our observation showing a significant correlation between APE1 and MDR1 levels in many lung cancer tissues (unpublished data). Thus, identification of small molecules that blocks p300-mediated APE1 acetylation or inhibition of interaction of AcAPE1 with YB-1 or other transcription factor could be used as potential therapeutic targets for cancer drug sensitization.
APE1 in Neurodegenerative and Cardiovascular Diseases
Ischemia and oxidative stress induced by spinal cord injury (SCI) are known to be associated with death of neuronal cells. Moreover, chronic oxidative stress and DNA damage have been implicated in several neurodegenerative diseases, including Parkinson's (PD), Alzheimer's (AD), Huntington's (HD), and amyotrophic lateral sclerosis (ALS) (8, 22). Isobaric hyperoxia (100% oxygen) stimulated APE1 expression in the hippocampus and basal forebrain of young rats, while aged rats showed no significant changes in APE1 protein levels in all brain areas after hyperoxia (35, 118). In contrast, several studies showed that global cerebral ischemia or traumatic brain injury or cold injury-induced brain trauma (100) induced oxidative stress decreases APE1 expression in the hippocampus and is associated with neuronal apoptosis in rats (34, 50, 143). This specific inhibition of APE1 expression may affect the extent of apoptosis after ischemia. Consistent with this, overexpression of WT APE1 in hippocampal and sensory cells reduced neuronal death after H2O2-mediated oxidative stress (139). C65A repair competent/redox incompetent APE1 conferred only partial cell survival, suggesting redox and repair functions of APE1 are essential for neuronal survival. In spinal cord injury, APE1 expression was noted to be decreased making neurons susceptible to oxidative DNA damage. APE1 is differentially expressed in cells of the nervous system (32, 33). In AD brain, APE1 has been visualized in a subpopulation of senile plaques by immunolocalization, indicating a functional relationship between APE1 and amyloid beta (Aβ) protein, possibly a pivotal role for APE1 in Aβ accumulation (130). APE1 expression was observed to be increased in nuclear extracts of AD patients compared to controls (24). Increased APE1 immunoreactivity was further supported from the hippocampus of AD brains in areas of neuronal injury and plaque-like structures (130). ALS is characterized by degeneration of motor neurons. APE1 expression and activity was observed to be lower in patients with sporadic ALS (83). In contrast, an increase in APE1 was also reported in the brain and spinal cord samples of patients with ALS (125), and reduced DNA repair activity of APE1 protein in familial ALS patients (108) has been shown. It was proposed that action of inducible transcriptional factors such as c-Jun in mammalian neurons is likely to be regulated by constitutively expressed APE1 and protecting neuronal cells after brain injury and oxidative DNA damage.
Although APE1 null mice are embryonic lethal, partial loss of APE1 in the heterozygous mice (APE1+/−) causes cardiovascular diseases such as hypertension (76). Jeon et al. (76) made a remarkable observation that APE1+/− mice have markedly elevated blood pressure, clearly indicating APE1's important role in maintaining blood pressure. This phenotype is accompanied by decreased basal nitric oxide (NO) production in arteries from APE1+/− mice (76). However, expression of endothelial nitric oxide synthase (eNOS) cannot be a major determinant of the hypertensive phenotype of APE1+/− mice, because eNOS levels are actually increased in the vessels of these mice, despite their impaired ability to generate nitric oxide. The redox activator function of APE1 was shown to be involved in moderate upregulation of H-Ras expression and activation of AKT, which potently activates eNOS (76). Therefore, it is critical to emphasize that APE1 may coordinate multiple nuclear events that impact vascular reactivity and blood pressure control through multiple mechanisms in addition to its effects on NO generation. It is too soon to know exactly how APE1 contributes to regulation of blood pressure, in addition to the effects on nitric oxide synthase activity that have been elegantly defined by Irani and colleagues (76). However, it is tempting to speculate that APE1 may play an integrative role in maintaining vascular tone through regulation of master control genes such as nitric oxide synthase, renin, and perhaps others that tune vascular responses.
Vascular endothelial cells can undergo apoptosis in vitro in response to variety of pathophysiological conditions such as hypoxia and pro-inflammatory cytokine (28, 145). Hall et al. (60) showed that decreased APE expression was associated with hypoxia-induced apoptosis in HUVEC and CPACE cells (60), which were significantly inhibited by overexpression of APE1. Deletion of the redox-sensitive domain of APE1 (aa 1–127 residues) abolished the anti-apoptotic function of APE1 in both hypoxia and tumor necrosis factor α (TNF-α) induced endothelial cell death (60). This study suggests that up-regulation of APE1 promotes endothelial cell survival in response to hypoxia and TNF-α through NF-κB-independent and NF-κB-dependent signaling cascades. Interestingly, a recent study shows that APE1-mediated NF-κB nuclear translocation is responsible for lipopolysaccharide-stimulated inducible NO synthase (iNOS) expression in a murine macrophage cell line (127). In contrast, Angkeow et al. (4) showed that cytoplasmic APE1 translocates to the nucleus upon oxidative stress induced by hypoxia/reoxygenation (H/R) in HUVEC cells. APE1 overexpression suppressed H/R-induced oxidative stress, NF-κB activation, and apoptosis (4). It was proposed that cytoplasmic APE1 inhibits oxidative stress (H2O2) generation by inhibiting the rac GTpase-activity and thus prevents H2O2 generation and NF-κB activation (4, 107). This novel cytoplasmic role of APE1 in preventing oxidative stress and apoptosis indicates that both nuclear and cytoplasmic function of APE1 may be important for preventing apoptosis in endothelial cells.
Expression of adhesion molecules accelerates the adhesion and migration of monocytes towards sites of inflammation in response to a variety of stimuli and has been implicated in atherosclerosis (123). A recent study by Kim et al. (82) showed that APE1 overexpression inhibits TNF-α induced VCAM expression in HUVEC cells. Moreover, overexpression of APE1 suppressed monocytes cells (U937) adhesion to HUVEC cells after TNF-α stimulation (82). Moreover, this anti-adhesive property of APE1 is mediated by a NOS-dependent mechanism. It was shown that APE1 may inhibit VCAM by inhibiting superoxide production and p38 MAPK activation (82).
Concluding Remarks and Future Perspective
Our and others studies have confirmed that APE1's DNA repair function is essential for somatic cell survival in mammals. However, the essentiality of APE1's acetylation dependent (redox-independent) transcriptional activity was quite unexpected. Moreover, the observation that the Cys65 residue of APE1 is not essential for redox regulation of AP-1 DNA binding indicated that the exact molecular mechanism underlying the redox activity of APE1 remains unclear. Therefore, definitive identification of redox active Cys residue of APE1 in vivo is essential to settle this issue. Moreover, whether post-translational modification such as phosphorylation, acetylation, or nitrosation is needed for maintaining APE1's redox activation function in vivo is not known. APE1 has been found to stimulate transcriptional activity of numerous factors involved in diverse physiological functions such as cell cycle control, apoptosis, drug resistance, angiogenesis, cellular growth, hematopoesis, and cardiovascular functions (40, 41, 47, 70, 76, 82, 121, 122, 144, 162). It is not clear whether APE1 acts as a direct or indirect sensor in all of these cases for modulating cellular functions. Furthermore, stable interaction of APE1 with several transcription factors and its presence in several trans-acting complexes have raised the possibility of its redox-independent transcriptional functions. We have provided evidence that suggest acetylation of APE1 modulates its coactivator or corespressor functions. However, how APE1 acts as a coactivator or corepressor in such diverse complexes is not clear. One of the major challenges is to elucidate the molecular basis for redox-independent trans-acting activity of APE1 in diverse complexes responsive to repression, activation of many genes. Moreover, there are many still unanswered questions regarding how different post-translational modifications of APE1 control its subcellular localization or trans-acting functions. Given that frequent overexpression of APE1 is associated with tumor cells resistance to various anticancer drugs, elucidating the signaling mechanisms involved in controlling its expression is extremely important from both basic and clinical perspectives. Moreover, attenuation of the APE1's repair or transcriptional functions (both redox and acetylation-dependent) by small molecule inhibitor(s) is a potential approach for reversing drug resistance. Further studies involving APE1 mutants deficient in DNA repair or redox or acetylation dependent function will settle the issues whether repair or transcriptional function or both are involved in tumor cells resistance to drugs.
Acknowledgments
Research supported by United States Public Health Science grants R01 ES08457, R01 CA53791, and P01 ES06676 (SM), American Heart Association grant 0565008Y (KKB). We thank Drs. T. Izumi (Louisiana State University Health Science Center, New Orleans), N. Oezguen, S. Sengupta (University of Texas Medical Branch, Galveston), R. Chattopadhyay (University of Virginia, Charlottesville) for their help with the figures. We also thank other members of Dr. Mitra's laboratory for their helpful discussions.
Abbreviations
aa, amino acids; AcAPE1, acetylated APE1; AD, Alzheimers disease; ALS, amyotrophic lateral sclerosis; AP, apurinic/apyrimidinic; AP-1, activator protein-1; APE1, AP-endonuclease 1; Aβ, amyloid beta; BER, base excision repair; bp, base pair; CAPEC, calf aortic pulmonary endothelial cell; ChIP, chromatin immunoprecipitation; CHO, Chinese hamster; CKI, casein kinase I; CKII, casein kinase II; DTT, dithiotheritol; Egr-1, early growth response protein-1; E3330, 3-[5-(2, 3-dimethoxy-6-methyl-1,4-benzoquinoyl)]-2nonyl-2-propionic acid; eNOS, endothelial nitric oxide synthase; FEN1, flap endonuclease 1; H2O2, hydrogen peroxide; hAPE1, human AP-endonuclease 1; HAT, histone acetyl-transferase; HDAC1, histone deacetylase 1; HIF-1α, hypoxia-inducible factor-1α; ?hnRNP-L, heterogeneous nuclear ribonucleoprotein L; HOCl, hypochlorous acid; HRE, hypoxia response element; HD, Huntington's disease; HUVEC, human umbilical vein endothelial cells; IL, interleukin; IR, ionizing radiation; mAPE1, mouse AP-endonuclease 1; MD, molecular dynamics; MDR, multidrug resistance; MEF, mouse embryonic fibroblast; MMS, methyl methane sulphonate; nCaRE, negative calcium response element; NEIL, nei-like; NF-kB, nuclear factor-kB; NLS, nuclear localization sequence; NO, nitric oxide; OGG1, 8-oxoguanine DNA glycosylase; PAEC, pulmonary artery endothelial cell; PAX, paired beox containing genes; PCNA, proliferating cell nuclear antigen; PD, Parkinson's disease; PKC, protein kinase C; PMA, phorbol 12-myristate 13 acetate; PTH, parathyroid hormone; Ref-1, redox effector factor-1; ROS, reactive oxygen species; SCI, spinal cord injury; SH, sulfhydral; SOH, sulfenic acid; S–S, disulfide; STAT3, signal transducer and activator protein 3; TFs, transcription factors; TNF-α, tumor necrosis factor alpha; TRX, thioredoxin; TSA, trichostatin A; TSH, thyrotropin; VEGF, vascular endothelial growth factor; WT, wild type; XRCC1, x-ray cross-species complementing 1; YB-1, Y-box-binding protein-1; YRE, Y-box response element.
References
- 1.Ait–Si–Ali S. Ramirez S. Barre FX. Dkhissi F. Magnaghi–Jaulin L. Girault JA. Robin P. Knibiehler M. Pritchard LL. Ducommun B. Trouche D. Harel–Bellan A. Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature. 1998;396:184–186. doi: 10.1038/24190. [DOI] [PubMed] [Google Scholar]
- 2.Ames BN. Shigenaga MK. Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ando K. Hirao S. Kabe Y. Ogura Y. Sato I. Yamaguchi Y. Wada T. Handa H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-{kappa}B and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008;36:4327–4336. doi: 10.1093/nar/gkn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angkeow P. Deshpande SS. Qi B. Liu YX. Park YC. Jeon BH. Ozaki M. Irani K. Redox factor-1: an extra-nuclear role in the regulation of endothelial oxidative stress and apoptosis. Cell Death Diff. 2002;9:717–725. doi: 10.1038/sj.cdd.4401025. [DOI] [PubMed] [Google Scholar]
- 5.Asai T. Kambe F. Kikumori T. Seo H. Increase in Ref-1 mRNA and protein by thyrotropin in rat thyroid FRTL-5 cells. Biochem Biophys Res Commun. 1997;236:71–74. doi: 10.1006/bbrc.1997.6906. [DOI] [PubMed] [Google Scholar]
- 6.Bargou RC. Jurchott K. Wagener C. Bergmann S. Metzner S. Bommert K. Mapara MY. Winzer KJ. Dietel M. Dorken B. Royer HD. Nuclear localization and increased levels of transcription factor YB-1 in primary human breast cancers are associated with intrinsic MDR1 gene expression. Nat Med. 1997;3:447–450. doi: 10.1038/nm0497-447. [DOI] [PubMed] [Google Scholar]
- 7.Baron V. Adamson ED. Calogero A. Ragona G. Mercola D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene Ther. 2006;13:115–124. doi: 10.1038/sj.cgt.7700896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 9.Bhakat KK. Hazra TK. Mitra S. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004;32:3033–3039. doi: 10.1093/nar/gkh632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhakat KK. Izumi T. Yang SH. Hazra TK. Mitra S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003;22:6299–6309. doi: 10.1093/emboj/cdg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhakat KK. Mokkapati SK. Boldogh I. Hazra TK. Mitra S. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol Cell Biol. 2006;26:1654–1665. doi: 10.1128/MCB.26.5.1654-1665.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhakat KK. Yang SH. Mitra S. Acetylation of human AP-endonuclease 1, a critical enzyme in DNA repair and transcription regulation. Methods Enzymol. 2003;371:292–300. doi: 10.1016/S0076-6879(03)71022-2. [DOI] [PubMed] [Google Scholar]
- 13.Bobola MS. Blank A. Berger MS. Stevens BA. Silber JR. Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clin Cancer Res. 2001;7:3510–3518. [PubMed] [Google Scholar]
- 14.Bobola MS. Finn LS. Ellenbogen RG. Geyer JR. Berger MS. Braga JM. Meade EH. Gross ME. Silber JR. Apurinic/apyrimidinic endonuclease activity is associated with response to radiation and chemotherapy in medulloblastoma and primitive neuroectodermal tumors. Clin Cancer Res. 2005;11:7405–7414. doi: 10.1158/1078-0432.CCR-05-1068. [DOI] [PubMed] [Google Scholar]
- 15.Breen AP. Murphy JA. Reactions of oxyl radicals with DNA. Free Rad Biol Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- 16.Cao X. Kambe F. Ohmori S. Seo H. Oxidoreductive modification of two cysteine residues in paired domain by Ref-1 regulates DNA-binding activity of Pax-8. Biochem Biophys Res Commun. 2002;297:288–293. doi: 10.1016/s0006-291x(02)02196-4. [DOI] [PubMed] [Google Scholar]
- 17.Carrero P. Okamoto K. Coumailleau P. O'Brien S. Tanaka H. Poellinger L. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol. 2000;20:402–415. doi: 10.1128/mcb.20.1.402-415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chattopadhyay R. Wiederhold L. Szczesny B. Boldogh I. Hazra TK. Izumi T. Mitra S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006;34:2067–2076. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaudhary PM. Roninson IB. Induction of multidrug resistance in human cells by transient exposure to different chemotherapeutic drugs. J Natl Cancer Inst. 1993;85:632–639. doi: 10.1093/jnci/85.8.632. [DOI] [PubMed] [Google Scholar]
- 20.Chen DS. Herman T. Demple B. Two distinct human DNA diesterases that hydrolyze 3’-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res. 1991;19:5907–5914. doi: 10.1093/nar/19.21.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung U. Igarashi T. Nishishita T. Iwanari H. Iwamatsu A. Suwa A. Mimori T. Hata K. Ebisu S. Ogata E. Fujita T. Okazaki T. The interaction between Ku antigen and REF1 protein mediates negative gene regulation by extracellular calcium. J Biol Chem. 1996;271:8593–8598. doi: 10.1074/jbc.271.15.8593. [DOI] [PubMed] [Google Scholar]
- 22.Coyle JT. Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science (New York, NY. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 23.Dalton TP. Shertzer HG. Puga A. Regulation of gene expression by reactive oxygen. Ann Rev Pharmacol Toxicol. 1999;39:67–101. doi: 10.1146/annurev.pharmtox.39.1.67. [DOI] [PubMed] [Google Scholar]
- 24.Davydov V. Hansen LA. Shackelford DA. Is DNA repair compromised in Alzheimer's disease? Neurobiol Aging. 2003;24:953–968. doi: 10.1016/s0197-4580(02)00229-4. [DOI] [PubMed] [Google Scholar]
- 25.Demple B. Harrison L. Repair of oxidative damage to DNA: Enzymology and biology. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 26.den Hertog J. Groen A. van der Wijk T. Redox regulation of protein-tyrosine phosphatases. Arch Biochem Bio-phys. 2005;434:11–15. doi: 10.1016/j.abb.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 27.Dianova II. Bohr VA. Dianov GL. Interaction of human AP endonuclease 1 with flap endonuclease 1 and proliferating cell nuclear antigen involved in long-patch base excision repair. Biochemistry. 2001;40:12639–12644. doi: 10.1021/bi011117i. [DOI] [PubMed] [Google Scholar]
- 28.Dimmeler S. Zeiher AM. Endothelial cell apoptosis in angiogenesis and vessel regression. Circulation Res. 2000;87:434–439. doi: 10.1161/01.res.87.6.434. [DOI] [PubMed] [Google Scholar]
- 29.Ding SZ. Minohara Y. Fan XJ. Wang J. Reyes VE. Patel J. Dirden–Kramer B. Boldogh I. Ernst PB. Crowe SE. Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infect Immun. 2007;75:4030–4039. doi: 10.1128/IAI.00172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding SZ. O'Hara AM. Denning TL. Dirden–Kramer B. Mifflin RC. Reyes VE. Ryan KA. Elliott SN. Izumi T. Boldogh I. Mitra S. Ernst PB. Crowe SE. Helicobacter pylori and H2O2 increase AP endonuclease-1/redox factor-1 expression in human gastric epithelial cells. Gastroenterology. 2004;127:845–858. doi: 10.1053/j.gastro.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 31.Doetsch PW. Cunningham RP. The enzymology of apurinic/apyrimidinic endonucleases. Mutat Res. 1990;236:173–201. doi: 10.1016/0921-8777(90)90004-o. [DOI] [PubMed] [Google Scholar]
- 32.Dragunow M. Ref-1 expression in adult mammalian neurons and astrocytes. Neurosci Lett. 1995;191:189–192. doi: 10.1016/0304-3940(95)11589-o. [DOI] [PubMed] [Google Scholar]
- 33.Duguid JR. Eble JN. Wilson TM. Kelley MR. Differential cellular and subcellular expression of the human multifunctional apurinic/apyrimidinic endonuclease (APE/ref-1) DNA repair enzyme. Cancer Res. 1995;55:6097–6102. [PubMed] [Google Scholar]
- 34.Edwards M. Kent TA. Rea HC. Wei J. Quast M. Izumi T. Mitra S. Perez–Polo JR. APE/Ref-1 responses to ischemia in rat brain. Neuroreport. 1998;9:4015–4018. doi: 10.1097/00001756-199812210-00005. [DOI] [PubMed] [Google Scholar]
- 35.Edwards M. Rassin DK. Izumi T. Mitra S. Perez–Polo JR. APE/Ref-1 responses to oxidative stress in aged rats. J Neurosci Res. 1998;54:635–638. doi: 10.1002/(SICI)1097-4547(19981201)54:5<635::AID-JNR8>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 36.Ema M. Hirota K. Mimura J. Abe H. Yodoi J. Sogawa K. Poellinger L. Fujii–Kuriyama Y. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999;18:1905–1914. doi: 10.1093/emboj/18.7.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans AR. Limp–Foster M. Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 38.Fan J. Wilson DM., 3rd Protein–protein interactions and posttranslational modifications in mammalian base excision repair. Free Radic Biol Med. 2005;38:1121–1138. doi: 10.1016/j.freeradbiomed.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 39.Fantini D. Vascotto C. Deganuto M. Bivi N. Gustincich S. Marcon G. Quadrifoglio F. Damante G. Bhakat KK. Mitra S. Tell G. APE1/Ref-1 regulates PTEN expression mediated by Egr-1. Free Radic Res. 2008;42:20–29. doi: 10.1080/10715760701765616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fishel ML. He Y. Reed AM. Chin–Sinex H. Hutchins GD. Mendonca MS. Kelley MR. Knockdown of the DNA repair and redox signaling protein Ape1/Ref-1 blocks ovarian cancer cell and tumor growth. DNA Repair. 2008;7:177–186. doi: 10.1016/j.dnarep.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fishel ML. Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol Aspects Med. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 42.Fritz G. Grosch S. Tomicic M. Kaina B. APE/Ref-1 and the mammalian response to genotoxic stress. Toxicology. 2003;193:67–78. doi: 10.1016/s0300-483x(03)00290-7. [DOI] [PubMed] [Google Scholar]
- 43.Fritz G. Kaina B. Phosphorylation of the DNA repair protein APE/REF-1 by CKII affects redox regulation of AP-1. Oncogene. 1999;18:1033–1040. doi: 10.1038/sj.onc.1202394. [DOI] [PubMed] [Google Scholar]
- 44.Fuchs S. Philippe J. Corvol P. Pinet F. Implication of Ref-1 in the repression of renin gene transcription by intracellular calcium. J Hypertens. 2003;21:327–335. doi: 10.1097/00004872-200302000-00024. [DOI] [PubMed] [Google Scholar]
- 45.Fujita T. Ito K. Izumi H. Kimura M. Sano M. Nakagomi H. Maeno K. Hama Y. Shingu K. Tsuchiya S. Kohno K. Fujimori M. Increased nuclear localization of transcription factor Y-box binding protein 1 accompanied by up-regulation of P-glycoprotein in breast cancer pretreated with paclitaxel. Clin Cancer Res. 2005;11:8837–8844. doi: 10.1158/1078-0432.CCR-05-0945. [DOI] [PubMed] [Google Scholar]
- 46.Fung H. Bennett RA. Demple B. Key role of a downstream specificity protein 1 site in cell cycle-regulated transcription of the AP endonuclease gene APE1/APEX in NIH3T3 cells. J Biol Chem. 2001;276:42011–42017. doi: 10.1074/jbc.M106423200. [DOI] [PubMed] [Google Scholar]
- 47.Fung H. Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 48.Fung H. Liu P. Demple B. ATF4-dependent oxidative induction of the DNA repair enzyme Ape1 counteracts arsenite cytotoxicity and suppresses arsenite-mediated mutagenesis. Mol Cell Biol. 2007;27:8834–8847. doi: 10.1128/MCB.00974-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaiddon C. Moorthy NC. Prives C. Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J. 1999;18:5609–5621. doi: 10.1093/emboj/18.20.5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gillardon F. Bottiger B. Hossmann KA. Expression of nuclear redox factor ref-1 in the rat hippocampus following global ischemia induced by cardiac arrest. Brain Res. 1997;52:194–200. doi: 10.1016/s0169-328x(97)00237-4. [DOI] [PubMed] [Google Scholar]
- 51.Glozak MA. Sengupta N. Zhang X. Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 52.Gorbunov NV. Das DK. Goswami SK. Gurusamy N. Atkins JL. Spatial coordination of cell-adhesion molecules and redox cycling of iron in the microvascular inflammatory response to pulmonary injury. Antioxid Redox Signal. 2007;9:483–495. doi: 10.1089/ars.2006.1296. [DOI] [PubMed] [Google Scholar]
- 53.Gorman MA. Morera S. Rothwell DG. de La Fortelle E. Mol CD. Tainer JA. Hickson ID. Freemont PS. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997;16:6548–6558. doi: 10.1093/emboj/16.21.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gray MJ. Zhang J. Ellis LM. Semenza GL. Evans DB. Watowich SS. Gallick GE. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24:3110–3120. doi: 10.1038/sj.onc.1208513. [DOI] [PubMed] [Google Scholar]
- 55.Gronroos E. Hellman U. Heldin CH. Ericsson J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell. 2002;10:483–493. doi: 10.1016/s1097-2765(02)00639-1. [DOI] [PubMed] [Google Scholar]
- 56.Grosch S. Fritz G. Kaina B. Apurinic endonuclease (Ref-1) is induced in mammalian cells by oxidative stress and involved in clastogenic adaptation. Cancer Res. 1998;58:4410–4416. [PubMed] [Google Scholar]
- 57.Grosch S. Kaina B. Transcriptional activation of apurinic/apyrimidinic endonuclease (Ape, Ref-1) by oxidative stress requires CREB. Biochem Biophys Res Commun. 1999;261:859–863. doi: 10.1006/bbrc.1999.1125. [DOI] [PubMed] [Google Scholar]
- 58.Hackenthal E. Paul M. Ganten D. Taugner R. Morphology, physiology, and molecular biology of renin secretion. Physiol Rev. 1990;70:1067–1116. doi: 10.1152/physrev.1990.70.4.1067. [DOI] [PubMed] [Google Scholar]
- 59.Haga S. Terui K. Zhang HQ. Enosawa S. Ogawa W. Inoue H. Okuyama T. Takeda K. Akira S. Ogino T. Irani K. Ozaki M. Stat3 protects against Fas-induced liver injury by redox-dependent and -independent mechanisms. J Clin Invest. 2003;112:989–998. doi: 10.1172/JCI17970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hall JL. Wang X. Van A. Zhao Y. Gibbons GH. Over-expression of Ref-1 inhibits hypoxia and tumor necrosis factor-induced endothelial cell apoptosis through nuclear factor-kappab-independent and -dependent pathways. Circ Res. 2001;88:1247–1253. doi: 10.1161/hh1201.091796. [DOI] [PubMed] [Google Scholar]
- 61.Hanson S. Kim E. Deppert W. Redox factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting p53 tetramerization. Oncogene. 2005;24:1641–1647. doi: 10.1038/sj.onc.1208351. [DOI] [PubMed] [Google Scholar]
- 62.Hara MR. Agrawal N. Kim SF. Cascio MB. Fujimuro M. Ozeki Y. Takahashi M. Cheah JH. Tankou SK. Hester LD. Ferris CD. Hayward SD. Snyder SH. Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nature Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 63.Harrison L. Ascione AG. Takiguchi Y. Wilson DM., 3rd Chen DJ. Demple B. Comparison of the promoters of the mouse (APEX) and human (APE) apurinic endonuclease genes. Mutat Res. 1997;385:159–172. doi: 10.1016/s0921-8777(97)00053-0. [DOI] [PubMed] [Google Scholar]
- 64.Harrison L. Ascione G. Menninger JC. Ward DC. Demple B. Human apurinic endonuclease gene (APE): structure and genomic mapping (chromosome 14q11.2-12) Human Mol Genet. 1992;1:677–680. doi: 10.1093/hmg/1.9.677. [DOI] [PubMed] [Google Scholar]
- 65.Hazra TK. Izumi T. Kow YW. Mitra S. The discovery of a new family of mammalian enzymes for repair of oxidatively damaged DNA, and its physiological implications. Carcinogenesis. 2003;24:155–157. doi: 10.1093/carcin/24.2.155. [DOI] [PubMed] [Google Scholar]
- 66.Hazra TK. Kow YW. Hatahet Z. Imhoff B. Boldogh I. Mokkapati SK. Mitra S. Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem. 2002;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- 67.Hirota K. Matsui M. Iwata S. Nishiyama A. Mori K. Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci USA. 1997;94:3633–3638. doi: 10.1073/pnas.94.8.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hsieh MM. Hegde V. Kelley MR. Deutsch WA. Activation of APE/Ref-1 redox activity is mediated by reactive oxygen species and PKC phosphorylation. Nucleic Acids Res. 2001;29:3116–3122. doi: 10.1093/nar/29.14.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang LE. Arany Z. Livingston DM. Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. 1996;271:32253–32259. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 70.Izumi T. Brown DB. Naidu CV. Bhakat KK. Macinnes MA. Saito H. Chen DJ. Mitra S. Two essential but distinct functions of the mammalian abasic endonuclease. Proc Natl Acad Sci USA. 2005;102:5739–5743. doi: 10.1073/pnas.0500986102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Izumi T. Hazra TK. Boldogh I. Tomkinson AE. Park MS. Ikeda S. Mitra S. Requirement for human AP endonuclease 1 for repair of 3′-blocking damage at DNA single-strand breaks induced by reactive oxygen species. Carcinogenesis. 2000;21:1329–1334. [PubMed] [Google Scholar]
- 72.Izumi T. Henner WD. Mitra S. Negative regulation of the major human AP-endonuclease, a multifunctional protein. Biochemistry. 1996;35:14679–14683. doi: 10.1021/bi961995u. [DOI] [PubMed] [Google Scholar]
- 73.Izumi T. Wiederhold LR. Roy G. Roy R. Jaiswal A. Bhakat KK. Mitra S. Hazra TK. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology. 2003;193:43–65. doi: 10.1016/s0300-483x(03)00289-0. [DOI] [PubMed] [Google Scholar]
- 74.Jackson EB. Theriot CA. Chattopadhyay R. Mitra S. Izumi T. Analysis of nuclear transport signals in the human apurinic/apyrimidinic endonuclease (APE1/Ref1) Nucleic Acids Res. 2005;33:3303–3312. doi: 10.1093/nar/gki641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jayaraman L. Murthy KG. Zhu C. Curran T. Xanthoudakis S. Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Devel. 1997;11:558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- 76.Jeon BH. Gupta G. Park YC. Qi B. Haile A. Khanday FA. Liu YX. Kim JM. Ozaki M. White AR. Berkowitz DE. Irani K. Apurinic/apyrimidinic endonuclease 1 regulates endothelial NO production and vascular tone. Circ Res. 2004;95:902–910. doi: 10.1161/01.RES.0000146947.84294.4c. [DOI] [PubMed] [Google Scholar]
- 77.Jeong JW. Bae MK. Ahn MY. Kim SH. Sohn TK. Bae MH. Yoo MA. Song EJ. Lee KJ. Kim KW. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111:709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 78.Kakolyris S. Kaklamanis L. Engels K. Turley H. Hickson ID. Gatter KC. Harris AL. Human apurinic endonuclease 1 expression in a colorectal adenoma-carcinoma sequence. Cancer Res. 1997;57:1794–1797. [PubMed] [Google Scholar]
- 79.Kakolyris S. Kaklamanis L. Giatromanolaki A. Koukourakis M. Hickson ID. Barzilay G. Turley H. Leek RD. Kanavaros P. Georgoulias V. Gatter KC. Harris AL. Expression and subcellular localization of human AP endonuclease 1 (HAP1/Ref-1) protein: a basis for its role in human disease. Histopathology. 1998;33:561–569. doi: 10.1046/j.1365-2559.1998.00541.x. [DOI] [PubMed] [Google Scholar]
- 80.Kelley MR. Cheng L. Foster R. Tritt R. Jiang J. Broshears J. Koch M. Elevated and altered expression of the multifunctional DNA base excision repair and redox enzyme Ape1/ref-1 in prostate cancer. Clin Cancer Res. 2001;7:824–830. [PubMed] [Google Scholar]
- 81.Kelley MR. Parsons SH. Redox regulation of the DNA repair function of the human AP endonuclease Ape1/ref-1. Antioxid Redox Signal. 2001;3:671–683. doi: 10.1089/15230860152543014. [DOI] [PubMed] [Google Scholar]
- 82.Kim CS. Son SJ. Kim EK. Kim SN. Yoo DG. Kim HS. Ryoo SW. Lee SD. Irani K. Jeon BH. Apurinic/apyrimidinic endonuclease1/redox factor-1 inhibits monocyte adhesion in endothelial cells. Cardiovasc Res. 2006;69:520–526. doi: 10.1016/j.cardiores.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 83.Kisby GE. Milne J. Sweatt C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. Neuroreport. 1997;8:1337–1340. doi: 10.1097/00001756-199704140-00004. [DOI] [PubMed] [Google Scholar]
- 84.Kohno K. Izumi H. Uchiumi T. Ashizuka M. Kuwano M. The pleiotropic functions of the Y-box-binding protein, YB-1. Bioessays. 2003;25:691–698. doi: 10.1002/bies.10300. [DOI] [PubMed] [Google Scholar]
- 85.Kohno K. Sato S. Takano H. Matsuo K. Kuwano M. The direct activation of human multidrug resistance gene (MDR1) by anticancer agents. Biochem Biophys Res Commun. 1989;165:1415–1421. doi: 10.1016/0006-291x(89)92761-7. [DOI] [PubMed] [Google Scholar]
- 86.Koukourakis MI. Giatromanolaki A. Kakolyris S. Sivridis E. Georgoulias V. Funtzilas G. Hickson ID. Gatter KC. Harris AL. Nuclear expression of human apurinic/apyrimidinic endonuclease (HAP1/Ref-1) in head-and-neck cancer is associated with resistance to chemoradiotherapy and poor outcome. Int J Radiat Oncol Biol Phys. 2001;50:27–36. doi: 10.1016/s0360-3016(00)01561-3. [DOI] [PubMed] [Google Scholar]
- 87.Krokan HE. Standal R. Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem J. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kuninger DT. Izumi T. Papaconstantinou J. Mitra S. Human AP-endonuclease 1 and hnRNP-L interact with a nCaRE-like repressor element in the AP-endonuclease 1 promoter. Nucleic Acids Res. 2002;30:823–829. doi: 10.1093/nar/30.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lau JP. Weatherdon KL. Skalski V. Hedley DW. Effects of gemcitabine on APE/ref-1 endonuclease activity in pancreatic cancer cells, and the therapeutic potential of antisense oligonucleotides. Br J Cancer. 2004;91:1166–1173. doi: 10.1038/sj.bjc.6602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lewen A. Sugawara T. Gasche Y. Fujimura M. Chan PH. Oxidative cellular damage and the reduction of APE/Ref-1 expression after experimental traumatic brain injury. Neurobiol Disease. 2001;8:380–390. doi: 10.1006/nbdi.2001.0396. [DOI] [PubMed] [Google Scholar]
- 91.Liu TY. Demonstration of the presence of a histidine residue at the active site of streptococcal proteinase. J Biol Chem. 1967;242:4029–4032. [PubMed] [Google Scholar]
- 92.Loeb LA. Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 93.Luo M. Delaplane S. Jiang A. Reed A. He Y. Fishel M. Nyland Ii RL. Borch RF. Qiao X. Georgiadis MM. Kelley MR. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxid Redox Signal. 2008;10:1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mantha AK. Oezguen N. Bhakat KK. Izumi T. Braun W. Mitra S. Unusual role of a cysteine residue in substrate binding and activity of human AP-endonuclease 1. J Mol Biol. 2008;379:28–37. doi: 10.1016/j.jmb.2008.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McHaffie GS. Ralston SH. Origin of a negative calcium response element in an ALU-repeat: implications for regulation of gene expression by extracellular calcium. Bone. 1995;17:11–14. doi: 10.1016/8756-3282(95)00131-v. [DOI] [PubMed] [Google Scholar]
- 96.Merluzzi S. D'Orlando O. Leonardi A. Vitale G. Pucillo C. TRAF2 and p38 are involved in B cells CD40-mediated APE/Ref-1 nuclear translocation: a novel pathway in B cell activation. Mol Immunol. 2008;45:76–86. doi: 10.1016/j.molimm.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 97.Merluzzi S. Moretti M. Altamura S. Zwollo P. Sigvardsson M. Vitale G. Pucillo C. CD40 stimulation induces Pax5/BSAP and EBF activation through a APE/Ref-1-dependent redox mechanism. J Biol Chem. 2004;279:1777–1786. doi: 10.1074/jbc.M305418200. [DOI] [PubMed] [Google Scholar]
- 98.Mitra S. Izumi T. Boldogh I. Bhakat KK. Hill JW. Hazra TK. Choreography of oxidative damage repair in mammalian genomes. Free Radic Biol Med. 2002;33:15–28. doi: 10.1016/s0891-5849(02)00819-5. [DOI] [PubMed] [Google Scholar]
- 99.Mol CD. Izumi T. Mitra S. Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected] Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 100.Morita–Fujimura Y. Fujimura M. Kawase M. Chan PH. Early decrease in apurinic/apyrimidinic endonuclease is followed by DNA fragmentation after cold injury-induced brain trauma in mice. Neuroscience. 1999;93:1465–1473. doi: 10.1016/s0306-4522(99)00231-6. [DOI] [PubMed] [Google Scholar]
- 101.Nishi T. Shimizu N. Hiramoto M. Sato I. Yamaguchi Y. Hasegawa M. Aizawa S. Tanaka H. Kataoka K. Watanabe H. Handa H. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem. 2002;277:44548–44556. doi: 10.1074/jbc.M202970200. [DOI] [PubMed] [Google Scholar]
- 102.Nose K. Shibanuma M. Kikuchi K. Kageyama H. Sakiyama S. Kuroki T. Transcriptional activation of early-response genes by hydrogen peroxide in a mouse osteoblastic cell line. Eur J Biochem/FEBS. 1991;201:99–106. doi: 10.1111/j.1432-1033.1991.tb16261.x. [DOI] [PubMed] [Google Scholar]
- 103.O'Hara AM. Bhattacharyya A. Mifflin RC. Smith MF. Ryan KA. Scott KG. Naganuma M. Casola A. Izumi T. Mitra S. Ernst PB. Crowe SE. Interleukin-8 induction by Helicobacter pylori in gastric epithelial cells is dependent on apurinic/apyrimidinic endonuclease-1/redox factor-1. J Immunol. 2006;177:7990–7999. doi: 10.4049/jimmunol.177.11.7990. [DOI] [PubMed] [Google Scholar]
- 104.Oda Y. Sakamoto A. Shinohara N. Ohga T. Uchiumi T. Kohno K. Tsuneyoshi M. Kuwano M. Iwamoto Y. Nuclear expression of YB-1 protein correlates with P-glycoprotein expression in human osteosarcoma. Clin Cancer Res. 1998;4:2273–2277. [PubMed] [Google Scholar]
- 105.Ogryzko VV. Schiltz RL. Russanova V. Howard BH. Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 106.Ohga T. Koike K. Ono M. Makino Y. Itagaki Y. Tanimoto M. Kuwano M. Kohno K. Role of the human Y box-binding protein YB-1 in cellular sensitivity to the DNA-damaging agents cisplatin, mitomycin C, and ultraviolet light. Cancer Res. 1996;56:4224–4228. [PubMed] [Google Scholar]
- 107.Okazaki T. Chung U. Nishishita T. Ebisu S. Usuda S. Mishiro S. Xanthoudakis S. Igarashi T. Ogata E. A redox factor protein, ref1, is involved in negative gene regulation by extracellular calcium. J Biol Chem. 1994;269:27855–27862. [PubMed] [Google Scholar]
- 108.Olkowski ZL. Mutant AP endonuclease in patients with amyotrophic lateral sclerosis. Neuroreport. 1998;9:239–242. doi: 10.1097/00001756-199801260-00012. [DOI] [PubMed] [Google Scholar]
- 109.Ono Y. Furuta T. Ohmoto T. Akiyama K. Seki S. Stable expression in rat glioma cells of sense and antisense nucleic acids to a human multifunctional DNA repair enzyme, APEX nuclease. Mutation Res. 1994;315:55–63. doi: 10.1016/0921-8777(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 110.Ordway JM. Eberhart D. Curran T. Cysteine 64 of Ref-1 is not essential for redox regulation of AP-1 DNA binding. Mol Cell Biol. 2003;23:4257–4266. doi: 10.1128/MCB.23.12.4257-4266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pines A. Bivi N. Romanello M. Damante G. Kelley MR. Adamson ED. D'Andrea P. Quadrifoglio F. Moro L. Tell G. Cross-regulation between Egr-1 and APE/Ref-1 during early response to oxidative stress in the human osteoblastic HOBIT cell line: evidence for an autoregulatory loop. Free Radic Res. 2005;39:269–281. doi: 10.1080/10715760400028423. [DOI] [PubMed] [Google Scholar]
- 112.Pines A. Perrone L. Bivi N. Romanello M. Damante G. Gulisano M. Kelley MR. Quadrifoglio F. Tell G. Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic Acids Res. 2005;33:4379–4394. doi: 10.1093/nar/gki751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Poole LB. Karplus PA. Claiborne A. Protein sulfenic acids in redox signaling. Ann Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 114.Poole LB. Zeng BB. Knaggs SA. Yakubu M. King SB. Synthesis of chemical probes to map sulfenic acid modifications on proteins. Bioconjug Chem. 2005;16:1624–1628. doi: 10.1021/bc050257s. [DOI] [PubMed] [Google Scholar]
- 115.Puglisi F. Aprile G. Minisini AM. Barbone F. Cataldi P. Tell G. Kelley MR. Damante G. Beltrami CA. Di Loreto C. Prognostic significance of Ape1/ref-1 subcellular localization in non-small cell lung carcinomas. Anticancer Res. 2001;21:4041–4049. [PubMed] [Google Scholar]
- 116.Qin J. Clore GM. Kennedy WP. Kuszewski J. Gronenborn AM. The solution structure of human thioredoxin complexed with its target from Ref-1 reveals peptide chain reversal. Structure. 1996;4:613–620. doi: 10.1016/s0969-2126(96)00065-2. [DOI] [PubMed] [Google Scholar]
- 117.Qu J. Liu GH. Huang B. Chen C. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res. 2007;35:2522–2532. doi: 10.1093/nar/gkl1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Quach N. Chan T. Lu TA. Schreiber SS. Tan Z. Induction of DNA repair proteins, Ref-1 and XRCC1, in adult rat brain following kainic acid-induced seizures. Brain Res. 2005;1042:236–240. doi: 10.1016/j.brainres.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 119.Raffoul JJ. Banerjee S. Singh-Gupta V. Knoll ZE. Fite A. Zhang H. Abrams J. Sarkar FH. Hillman GG. Down-regulation of apurinic/apyrimidinic endonuclease 1/redox factor-1 expression by soy isoflavones enhances prostate cancer radiotherapy in vitro and in vivo. Cancer Res. 2007;67:2141–2149. doi: 10.1158/0008-5472.CAN-06-2147. [DOI] [PubMed] [Google Scholar]
- 120.Ramana CV. Boldogh I. Izumi T. Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci USA. 1998;95:5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Robertson KA. Bullock HA. Xu Y. Tritt R. Zimmerman E. Ulbright TM. Foster RS. Einhorn LH. Kelley MR. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 2001;61:2220–2225. [PubMed] [Google Scholar]
- 122.Robertson KA. Hill DP. Xu Y. Liu L. Van Epps S. Hockenbery DM. Park JR. Wilson TM. Kelley MR. Down-regulation of apurinic/apyrimidinic endonuclease expression is associated with the induction of apoptosis in differentiating myeloid leukemia cells. Cell Growth Differ. 1997;8:443–449. [PubMed] [Google Scholar]
- 123.Ross R. Atherosclerosis—an inflammatory disease. New Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 124.Seo YR. Kelley MR. Smith ML. Selenomethionine regulation of p53 by a ref1-dependent redox mechanism. Proc Natl Acad Sci USA. 2002;99:14548–14553. doi: 10.1073/pnas.212319799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shaikh AY. Martin LJ. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromol Med. 2002;2:47–60. doi: 10.1007/s12017-002-0038-7. [DOI] [PubMed] [Google Scholar]
- 126.Sobol RW. Wilson SH. Mammalian DNA beta-polymerase in base excision repair of alkylation damage. Prog Nucleic Acid Res Mol Biol. 2001;68:57–74. doi: 10.1016/s0079-6603(01)68090-5. [DOI] [PubMed] [Google Scholar]
- 127.Song JD. Lee SK. Kim KM. Kim JW. Kim JM. Yoo YH. Park YC. Redox factor-1 mediates NF-kappaB nuclear translocation for LPS-induced iNOS expression in murine macrophage cell line RAW 264.7. Immunology. 2008;124:58–67. doi: 10.1111/j.1365-2567.2007.02736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Suzuki S. Nagaya T. Suganuma N. Tomoda Y. Seo H. Inductions of immediate early genes (IEGS) and ref-1 by human chorionic gonadotropin in murine Leydig cell line (MA-10) Biochem Mol Biol Intl. 1998;44:217–224. doi: 10.1080/15216549800201242. [DOI] [PubMed] [Google Scholar]
- 129.Takao M. Aburatani H. Kobayashi K. Yasui A. Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res. 1998;26:2917–2922. doi: 10.1093/nar/26.12.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tan Z. Sun N. Schreiber SS. Immunohistochemical localization of redox factor-1 (Ref-1) in Alzheimer's hippocampus. Neuroreport. 1998;9:2749–2752. doi: 10.1097/00001756-199808240-00012. [DOI] [PubMed] [Google Scholar]
- 131.Tell G. Crivellato E. Pines A. Paron I. Pucillo C. Manzini G. Bandiera A. Kelley MR. Di Loreto C. Damante G. Mitochondrial localization of APE/Ref-1 in thyroid cells. Mutation Res. 2001;485:143–152. doi: 10.1016/s0921-8777(00)00068-9. [DOI] [PubMed] [Google Scholar]
- 132.Tell G. Damante G. Caldwell D. Kelley MR. The intracellular localization of APE1/Ref-1: More than a passive phenomenon? Antioxid Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 133.Tell G. Pellizzari L. Cimarosti D. Pucillo C. Damante G. Ref-1 controls pax-8 DNA-binding activity. Biochem Bio-phys Res Commun. 1998;252:178–183. doi: 10.1006/bbrc.1998.9548. [DOI] [PubMed] [Google Scholar]
- 134.Tell G. Pellizzari L. Pucillo C. Puglisi F. Cesselli D. Kelley MR. Di Loreto C. Damante G. TSH controls Ref-1 nuclear translocation in thyroid cells. J Mol Endocrinol. 2000;24:383–390. doi: 10.1677/jme.0.0240383. [DOI] [PubMed] [Google Scholar]
- 135.Tell G. Scaloni A. Pellizzari L. Formisano S. Pucillo C. Damante G. Redox potential controls the structure and DNA binding activity of the paired domain. J Biol Chem. 1998;273:25062–25072. doi: 10.1074/jbc.273.39.25062. [DOI] [PubMed] [Google Scholar]
- 136.Tell G. Zecca A. Pellizzari L. Spessotto P. Colombatti A. Kelley MR. Damante G. Pucillo C. An ‘environment to nucleus' signaling system operates in B lymphocytes: redox status modulates BSAP/Pax-5 activation through Ref-1 nuclear translocation. Nucleic Acids Res. 2000;28:1099–1105. doi: 10.1093/nar/28.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 138.Ueno M. Masutani H. Arai RJ. Yamauchi A. Hirota K. Sakai T. Inamoto T. Yamaoka Y. Yodoi J. Nikaido T. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J Biol Chem. 1999;274:35809–35815. doi: 10.1074/jbc.274.50.35809. [DOI] [PubMed] [Google Scholar]
- 139.Vasko MR. Guo C. Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair. 2005;4:367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 140.Virolle T. Adamson ED. Baron V. Birle D. Mercola D. Mustelin T. de Belle I. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nature Cell Biol. 2001;3:1124–1128. doi: 10.1038/ncb1201-1124. [DOI] [PubMed] [Google Scholar]
- 141.Walker LJ. Craig RB. Harris AL. Hickson ID. A role for the human DNA repair enzyme HAP1 in cellular protection against DNA damaging agents and hypoxic stress. Nucleic Acids Res. 1994;22:4884–4889. doi: 10.1093/nar/22.23.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Walker LJ. Robson CN. Black E. Gillespie D. Hickson ID. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol Cell Biol. 1993;13:5370–5376. doi: 10.1128/mcb.13.9.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walton M. Lawlor P. Sirimanne E. Williams C. Gluckman P. Dragunow M. Loss of Ref-1 protein expression precedes DNA fragmentation in apoptotic neurons. Brain Res. 1997;44:167–170. doi: 10.1016/s0169-328x(96)00291-4. [DOI] [PubMed] [Google Scholar]
- 144.Wang D. Luo M. Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther. 2004;3:679–686. [PubMed] [Google Scholar]
- 145.Wang N. Stemerman MB. Ref-1 and transcriptional control of endothelial apoptosis. Circ Res. 2001;88:1223–1225. doi: 10.1161/hh1201.093162. [DOI] [PubMed] [Google Scholar]
- 146.Wei SJ. Botero A. Hirota K. Bradbury CM. Markovina S. Laszlo A. Spitz DR. Goswami PC. Yodoi J. Gius D. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Res. 2000;60:6688–6695. [PubMed] [Google Scholar]
- 147.Whitehouse CJ. Taylor RM. Thistlethwaite A. Zhang H. Karimi–Busheri F. Lasko DD. Weinfeld M. Caldecott KW. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001;104:107–117. doi: 10.1016/s0092-8674(01)00195-7. [DOI] [PubMed] [Google Scholar]
- 148.Wiederhold L. Leppard JB. Kedar P. Karimi–Busheri F. Rasouli–Nia A. Weinfeld M. Tomkinson AE. Izumi T. Prasad R. Wilson SH. Mitra S. Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Molecular cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 149.Xanthoudakis S. Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Xanthoudakis S. Miao G. Wang F. Pan YC. Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xanthoudakis S. Miao GG. Curran T. The redox and DNA-repair activities of Ref-1 are encoded by nonover-lapping domains. Proc Natl Acad Sci USA. 1994;91:23–27. doi: 10.1073/pnas.91.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xanthoudakis S. Smeyne RJ. Wallace JD. Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci USA. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xu Y. Moore DH. Broshears J. Liu L. Wilson TM. Kelley MR. The apurinic/apyrimidinic endonuclease (APE/ref-1) DNA repair enzyme is elevated in premalignant and malignant cervical cancer. Anticancer Research. 1997;17:3713–3719. [PubMed] [Google Scholar]
- 154.Yacoub A. Kelley MR. Deutsch WA. The DNA repair activity of human redox/repair protein APE/Ref-1 is inactivated by phosphorylation. Cancer Res. 1997;57:5457–5459. [PubMed] [Google Scholar]
- 155.Yamamoto M. Igarashi T. Muramatsu M. Fukagawa M. Motokura T. Ogata E. Hypocalcemia increases and hypercalcemia decreases the steady-state level of parathyroid hormone messenger RNA in the rat. J Clin Invest. 1989;83:1053–1056. doi: 10.1172/JCI113946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yan M. Xu W. Lu L. Sun L. Liu X. Zheng Z. Induction of ref-1 ensures AP-1 activation in intracellular oxidative environment of IL-2-stimulated BA/F3beta cells. Biochem Biophys Res Commun. 2000;278:462–469. doi: 10.1006/bbrc.2000.3826. [DOI] [PubMed] [Google Scholar]
- 157.Yan MD. Xu WJ. Lu LR. Sun LY. Liu XY. Zheng ZC. Ubiquitin conjugating enzyme Ubc9 is involved in protein degradation of redox factor-1 (Ref-1) Sheng wu hua xue yu sheng wu wu li xue bao Acta biochimica et biophysica Sinica. 2000;32:63–68. [PubMed] [Google Scholar]
- 158.Yao KS. Xanthoudakis S. Curran T. O'Dwyer PJ. Activation of AP-1 and of a nuclear redox factor, Ref-1, in the response of HT29 colon cancer cells to hypoxia. Mol Cell. 1994;Biol14:5997–6003. doi: 10.1128/mcb.14.9.5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zaky A. Busso C. Izumi T. Chattopadhyay R. Bassiouny A. Mitra S. Bhakat KK. Regulation of the human AP-endonuclease (APE1/Ref-1) expression by the tumor suppressor p53 in response to DNA damage. Nucleic Acids Res. 2008;36:1555–1566. doi: 10.1093/nar/gkm1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhao J. Gao F. Zhang Y. Wei K. Liu Y. Deng X. Bcl2 inhibits abasic site repair by down-regulating APE1 endonuclease activity. J Biol Chem. 2008;283:9925–9932. doi: 10.1074/jbc.M708345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ziel KA. Campbell CC. Wilson GL. Gillespie MN. Ref-1/Ape is critical for formation of the hypoxia-inducible transcriptional complex on the hypoxic response element of the rat pulmonary artery endothelial cell VEGF gene. FASEB J. 2004;18:986–988. doi: 10.1096/fj.03-1160fje. [DOI] [PubMed] [Google Scholar]
- 162.Zou GM. Luo MH. Reed A. Kelley MR. Yoder MC. Ape1 regulates hematopoietic differentiation of embryonic stem cells through its redox functional domain. Blood. 2007;109:1917–1922. doi: 10.1182/blood-2006-08-044172. [DOI] [PubMed] [Google Scholar]