

Abstract
Nitric oxide (NO) is an important messenger molecule in a variety of physiological systems. NO, a gas, is produced from L-arginine by different isoforms of nitric oxide synthase (NOS) and serves many normal physiologic purposes, such as promoting vasodilation of blood vessels and mediating communication between nervous system cells. In addition to its physiologic actions, free radical activity of NO can cause cellular damage through a phenomenon known as nitrosative stress. Here, we review the role of NO in health and disease, focusing on its role in function and dysfunction of the nervous system. Substantial evidence indicates that NO plays a key role in most common neurodegenerative diseases, and, although the mechanism of NO-mediated neurodegeneration remains uncertain, studies suggest several possibilities. NO has been shown to modify protein function by nitrosylation and nitrotyrosination, contribute to glutamate excitotoxicity, inhibit mitochondrial respiratory complexes, participate in organelle fragmentation, and mobilize zinc from internal stores. In this review, we discuss and analyze the evidence for each of these mechanisms in different neurodegenerative diseases and propose future directions for research of the role of NO in neurodegeneration. Antioxid. Redox Signal. 11, 541–553.
Introduction
In recent years, studies have implicated nitric oxide (NO) as a key mediator of neurodegeneration in numerous diseases of the nervous system, including Parkinson's disease (PD), Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), and ischemic brain injury (stroke) (4, 7, 51, 67, 68, 73, 95, 115, 119, 124, 129, 140, 151). In addition to its many physiologic functions (for example, as a neurotransmitter, neuromodulator, and mediator of blood vessel dilation), NO can convert into highly reactive and toxic molecules that readily react with proteins, DNA, and lipids to alter their function (96). This dual action of NO, as both an important player in normal physiology and a contributor to pathophysiology, makes developing effective treatments to target NO toxicity particularly challenging.
Here, we review the current status of the NO-linked neurodegeneration field, focusing both on the progress that has been made and on identifying areas for future study. First, we present background information on NO, its reactions, its associated reactive species, and its normal physiologic functions. Next, we present the evidence of the participation of NO in neurodegenerative diseases and discuss the proposed mechanisms of NO-mediated neurotoxicity in different diseases. Because significant overlap exists between many protein targets of NO and different neurologic disorders, we organized this section by potential mechanism of pathology rather than by individual disease.
Nitric Oxide Production in Normal Physiology
Nitric oxide (NO) is a gas synthesized from l-arginine in mammals by enzymes known as nitric oxide synthases (NOSs) (88, 109, 116). NO and NOS have been identified in many organ systems including liver, lungs, vascular tissue, skeletal muscle, and smooth muscle (114). In addition, NOS produces NO in all brain cells—both neurons and glia (109, 116). Studies have confirmed the identity of three isoforms of NOS—neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS)—expressed in different cell types and under different cellular conditions (114). More recent studies also suggest the existence of a novel, fourth NOS, mitochondrial NOS (mtNOS) (61).
The NOS isozymes have both similarities and differences in expression pattern and characteristics of enzymatic activity (See Table 1). nNOS is predominantly active in central and peripheral neurons, where production of NO is important for cell communication (114). eNOS produces NO mainly in endothelial tissue of blood vessels, where NO causes vasodilation and endothelial relaxation of muscles and soft tissue (107). iNOS is found primarily in immune cells and glial cells (astrocytes and microglia) and is activated in response to pathogen recognition and cytokine release (104, 107, 114). The primary function of iNOS is to use the oxidative stress of NO to defend against pathogens (114). For example, activated microglia in the nervous system express iNOS, causing neuronal cell death in vivo in mice (41) and in vitro in rat hippocampal cultures (93). Because the NOS isoforms have different physiologic functions, characteristics of their enzymatic activity and subcellular localization also differ. For example, nNOS and eNOS, constitutively active isoforms, produce low concentrations of NO over long periods and are activated by calcium ions (Ca2+) through transient binding to the calcium-binding protein calmodulin, whereas iNOS, the inducible isoform, produces high concentrations of NO for short periods and is Ca2+ independent because calmodulin remains tightly bound to the protein (20, 38, 107). In addition, nNOS and iNOS are both cytosolic, whereas eNOS associates with the membranes of endothelial cells (28, 102, 106).
Table 1.
Nitric Oxide Synthase (NOS) Isoforms
| NOS isoform | Location | Function | Cytosolic/membrane-associated? | Characteristics of enzymatic activity |
|---|---|---|---|---|
| Endothelial NOS (eNOS) | Endothelial tissue of blood vessels | Vasodilation and relaxation of muscles and soft tissue | Membrane-associated | Produces low concentrations of NO over long periods |
| Neuronal NOS (nNOS) | Central and peripheral neurons | Cell-to-cell communication | Cytosolic | Produces low concentrations of NO over long periods |
| Inducible NOS (iNOS) | Immune cells and glial cells | Mediate cell death in response to pathogens | Cytosolic | Produces high concentrations of NO over short periods |
| Mitochondrial NOS (mtNOS) | Mitochondrial inner membrane | Ca2+ regulation | Membrane-associated | Part of Ca2+ feedback loop |
Although the existence of nNOS, iNOS, and eNOS is well documented, whether other NOS isoforms exist and are active in mammals remains a topic of debate. Because the link between mitochondria and NOS activity is well established (60), mitochondria have been an area of intense interest in the search for new NOS isoforms. Consequently, Ghafourifar et al. (61) first reported the presence of constitutively active mtNOS in mitochondria from rat liver. mtNOS associates with the matrix face of the mitochondrial inner membrane and seems to play a role in Ca2+ regulation (61). Ca2+ increases NO formation by mtNOS, which reversibly decreases mitochondrial membrane potential and oxygen consumption (61). Decreased membrane potential increases Ca2+ release from the organelle, which inactivates mtNOS, completing the feedback loop. Although studies have begun to clarify the functions of mtNOS, its identity and relation to other NOSs remain unclear (61).
Reactive Nitrogen Species (RNS) Formation and Nitrosative Stress
NO readily reacts with various molecules within the cellular environment, and the products of these reactions can damage the cell through a variety of mechanisms. The all-encompassing term nitrosative stress describes this ability of NO and its derivatives to damage cellular components such as proteins and DNA. A primary reaction in the production of RNS is the combination of NO and superoxide anions (O2−) to form peroxynitrite (ONOO−) (See Table 2), a highly reactive neurotoxin (15, 43). Mitochondria provide both reactants necessary for the formation of ONOO−, NO from mtNOS and O2− from the electron-transport chain. About 15% of superoxide produced by mitochondria goes toward the formation of peroxynitrite (the other 85% forms hydrogen peroxide) (29, 118). Importantly, ONOO− formation in mitochondria induces cytochrome c release, an indicator of mitochondrial distress and potential inducer of cell death (62), and also irreversibly blocks the respiratory chain by competing with molecular oxygen (107).
Table 2.
NO-Related Chemistry
| Reaction name | Reaction formula |
|---|---|
| NO formation | 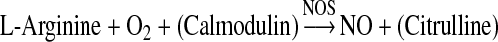 |
| Peroxynitrite formation | NO + O2− → ONOO− |
| Nitrosylation | RS(H) + X-NO → RS-NO + X− + (H+) |
| Nitrotyrosination | Tyr + ONOO· → Tyr-NO2 |
Two other reactions that involve NO modification of proteins, nitrosylation and nitrotyrosination (See Table 2), are important both to the physiologic and pathophysiologic roles of NO. Nitrosylation is the reaction of NO with the amino acid cysteine to form nitrosothiols on interacting proteins (137, 138). Whereas the precise physiological mechanism of nitrosylation is unclear, it has been hypothesized that NO reacts with oxygen to form N2O3, which reacts with glutathione to form the S-nitrosoglutathione (GSNO) group, which is an important intermediate in S-nitrosylation signal transduction (35). Nitrotyrosination is the reaction of the amino acid tyrosine in target proteins with ONOO− to form 3-nitrotyrosine, which may impair cellular function through an as-yet-uncertain mechanism (76). Nitrotyrosination has been used as a marker in neurodegeneration models (9, 18, 135).
Physiologic Functions of NO
Although the primary focus of this review is the pathologic role of NO in neurologic diseases, it is important to understand how NO functions in normal physiology in healthy humans. Studies identifying NO as the endothelium-derived relaxing factor (EDRF) that mediates blood vessel dilation and relaxation were the first to ascribe a physiologic function to NO (55, 74, 75, 117). Studies have also linked NO to neurotransmission and immune cell response (57, 71, 72, 89, 136). These critical functions of NO make combating the negative effects of nitrosative stress difficult because, to be effective, potential treatments must selectively disrupt the pathologic actions of NO.
NO is an unconventional neurotransmitter because it is a gas, is not stored in synaptic vesicles, and is synthesized on demand by neurons (57, 89, 136). NO has many potential roles in the nervous system, some of which are not well understood. For example, NO is likely involved in nerve-mediated relaxation of the gut during digestion (136). Studies have shown that NO-producing agents mimic neuronal relaxation (11, 26), and NOS inhibitors prevent nerve-mediated gut relaxation (11, 26, 46). NO is also likely involved in innervating neural blood vessels, including cerebral arteries (19, 20) and penile arteries in males (27). Finally, modification of cysteine residues by NO in several key proteins, known as S-nitrosylation, enhances neuronal survival. For example, S-nitrosylation of NMDA (N-methyl-d-aspartate) glutamate receptors in neurons increases neuronal survival by locking the receptors in the “closed” position, thus preventing excitotoxicity (34, 82, 96, 97). NO also S-nitrosylates and inhibits caspases, cysteine proteases that play a critical role in apoptosis (83, 94, 100), suggesting that apoptosis may not be a major pathway of neuronal death in NO-linked neurodegenerative diseases.
Another important physiologic function of NO is as a toxic agent in the immune cell response to pathogens. Macrophages activated by bacterial endotoxin show high levels of NOS activity, which is necessary for tumoricidal and bactericidal actions of the macrophages (71, 72, 136). In addition, glial cells of the nervous system, both astrocytes and microglia, rely on NO to perform their immune-like activities (114). In both macrophages and glia, iNOS becomes active on pathogen-mediated activation, and the nitrosative stress created by NO helps defend against pathogens (104, 107, 114).
NO and Neurodegenerative Disease
Although NO has many important and beneficial physiologic functions, it can also play a role in neurodegenerative disease pathology. In this section, we present some of the evidence linking nitrosative stress to various neurodegenerative diseases, including PD, AD, ALS, HD, and stroke. We also discuss the potential mechanisms of NO-related neurotoxicity, such as protein nitrosylation and nitrotyrosination, excitotoxicity, mitochondrial respiratory complex inhibition, organelle fragmentation, and liberation of zinc (Zn2+) from intracellular stores.
Evidence of NO-mediated neurodegeneration
Studies have provided a considerable link between NO and many prevalent neurodegenerative diseases, including PD, AD, ALS, HD, and stroke. First, animal models of 1-methyl 4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity have shown that inhibition of NOS slows progression of disease pathology (67, 95, 119, 129). MPTP is a neurotoxin that inhibits complex I of the mitochondrial respiratory chain and mimics the symptoms of PD by killing substantia nigra neurons (92). Of note, 7-nitroindazole (7-NI), a selective inhibitor of nNOS, blocks MPTP-mediated decrease in striatal dopamine levels in mice (129) and baboons (67) and protects against MPTP-induced neuronal death in mice (119). 7-NI also protects against motor deficits and cognitive decline in the MPTP baboon model (67). In addition to the role of nNOS in MPTP-induced neurotoxicity, iNOS appears to play a role. MPTP treatment of mice causes massive gliosis, a proliferation of glial cells, and upregulation of iNOS, and iNOS-deficient mice are more resistant to MPTP (95). It is important to remember when evaluating the importance of the referenced studies that MPTP neurotoxicity is a phenotypic model of PD based on the clinical observation that MPTP contamination of the designer opioid 1-methyl-4-phenyl-4-propionoxypiperidine (MPPP) caused PD-like symptoms in drug users (91). Thus, the MPTP model of PD does not necessarily have a mechanistic link to sporadic PD in humans. Evidence of nitrosative stress in human PD patients would of course be more convincing, and we highlight a few such studies in the mechanism part of this section.
In addition to the extensive research of NO in PD models, a proteomic study has found a correlation between β-amyloid deposition and nitration in a number of proteins in AD patients (140). Multiple proteins in the AD hippocampus, an area of intense β-amyloid deposition, are nitrated (140). In addition, familial ALS patients with a mutant superoxide dismutase-1 (mSOD1) gene, sporadic ALS patients, and mutant SOD1 transgenic mice show high rates of 3-nitrotyrosination in spinal cord motor neurons (7, 51, 124). Furthermore, mutant huntingtin (mtHTT), the pathologically mutated protein in HD, can complex with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and seven in absentia homolog 1 (SIAH1), a protein complex that reacts with NO (68), in cell cultures (4). Finally, studies have shown that inhibition of nNOS activity is protective against ischemic stroke injury in vivo (73, 115, 151). Taken together, these findings suggest that NO may be involved in many neurodegenerative diseases.
Potential mechanisms of NO-mediated neurodegeneration
As we discussed earlier, two important properties of NO that may contribute to its pathologic functions are its ability to modify proteins through nitrosylation and nitrotyrosination and its ability to react with oxygen to form RNS. Accordingly, studies indicate that nitrosylation and nitrotyrosination of many different proteins play a role in the pathology of multiple neurodegenerative diseases. In addition, NO and RNS appear to play a role in glutamate-mediated excitotoxicity in nerve cells, mitochondrial respiratory complex inhibition, organelle fragmentation, and release of Zn2+ from intracellular stores, all of which have been linked to neurodegenerative disease.
S-Nitrosylation
Nitrosylation of proteins is a well-established mechanism of protein modification and regulation (137). Studies have identified dozens of proteins that become S-nitrosylated (137), several of which are associated with neurodegenerative diseases such as PD, AD, and stroke (See Fig. 1). First, Chung et al. (36) reported that Parkin, an E3 ubiquitin ligase mutated in some familial forms of PD, is S-nitrosylated in an MPTP in vivo mouse model of PD and in postmortem PD patient brain samples, but not in normal, age-controlled brains (36). However, whereas Yao et al. (149) also reported nitrosylation of Parkin, they found an initial increase in E3 ubiquitin ligase activity before a significant decrease in activity. Thus, the ultimate effect of nitrosylation on Parkin and its role in PD pathogenesis remains a subject for further investigation.
FIG. 1.

NO S-nitrosylates multiple proteins in neurodegenerative diseases. NO S-nitrosylates Parkin and peroxiredoxin 2 (PRX2) in PD (shown in blue); protein-disulfide isomerase (PDI) in PD and AD (shown in green); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in PD and HD (shown in red); matrix metalloproteinase-9 (MMP-9) in stroke (shown in yellow); and heat-shock protein 90 (HSP90) in AD (shown in pink). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Next, peroxiredoxin 2 (PRX2), the member of a family of abundant antioxidants most commonly found in neurons that reduce intracellular peroxides, becomes S-nitrosylated by reaction with NO (50). S-nitrosylation of PRX2 prevents its reaction with peroxides and inhibits its enzymatic activity and protective function against oxidative stress (50). Studies of human postmortem brains revealed an increase in S-nitrosylated PRX2 in human PD patients (50). A third nitrosylated protein linked to both PD and AD is protein-disulfide isomerase (PDI). PDI is an endoplasmic reticulum (ER)-associated chaperone protein that prevents neurotoxicity caused by ER stress and protein misfolding (143) and can also function as an NO receptor or donor, depending on the cellular context (134). Both PD and AD patient postmortem brains exhibit increased levels of nitrosylated PDI as compared with normal-aged brains (143). PDI nitrosylation prevents PDI-mediated ER stress reduction and allows protein misfolding (143). Heat-shock protein 90 (HSP90), a chaperone protein and coactivator of eNOS, is another protein associated with AD that undergoes nitrosylation (103). Postmortem brain samples from patients with AD exhibit increased levels of HSP90 (48, 80), and it has been suggested that inactivation of HSP90 may allow accumulation of tau and amyloid-β aggregates in the AD brain (113). Accordingly, S-nitrosylation of HSP90 abolishes ATPase activity that is necessary for its chaperone function (103).
GAPDH, another protein that may play a role in multiple neurodegenerative diseases, also undergoes S-nitrosylation in neurons (68). In addition to its well-known role in glycolysis, GAPDH contributes to nuclear signaling in apoptosis (77, 125). S-nitrosylation of GAPDH terminates its enzymatic activity and allows binding of GAPDH to SIAH1, an E3 ubiquitin ligase. SIAH1 has a nuclear localization signal and carries GAPDH to the nucleus. GAPDH stabilizes SIAH1 in the nucleus and allows degradation of nuclear proteins through ubiquitination (68). Interestingly, deprenyl, a drug that slows the progression of early-stage PD (113), prevents S-nitrosylation of GAPDH, binding of GAPDH to SIAH1, and nuclear translocation of GAPDH in an MPTP mouse model of PD (69). This action of deprenyl may account for its neuroprotective properties. Another potential link between GAPDH and neurodegenerative disease is the observed complex between mtHTT and GAPDH/SIAH1 in cell cultures (4). Furthermore, GAPDH accumulates in the nucleus of HD mice, suggesting a relation between the mtHTT–GAPDH–SIAH1 complex and the ability of mtHTT to enter the nucleus and kill neurons (131). Thus, the GAPDH/SIAH1 pathway of ubiquitination and cell death may be a common pathway in the pathology of multiple neurodegenerative diseases.
Finally, matrix metalloproteinase-9 (MMP-9), a protein involved in degradation of extracellular matrix proteins, is the target of S-nitrosylation during ischemic brain injury (stroke) (65). Studies have found increased expression of MMP-9 during ischemic stroke in the human brain (108, 123). MMP-9 colocalizes with nNOS in cerebral ischemia, facilitating activation of the enzyme activity (65). S-nitrosylation and further irreversible oxidation of the protein thiol activates MMP-9 and activates its pathologic function in ischemic stroke (65).
3-Nitrotyrosination
When ONOO−, formed from the reaction of NO and O2−, attacks proteins, it often results in 3-nitrotyrosination of tyrosine residues (8, 122). As discussed earlier, ALS patients and transgenic mice exhibit increased concentrations of 3-nitrotyrosine in spinal cord neurons (7, 51, 124). In addition, postmortem human AD brain samples show increased levels of 3-nitrotyrosinated proteins (135), and nitrotyrosination is a common event in the MPTP mouse model of PD (3).
Glutamate excitotoxicity
Glutamate excitotoxicity is caused by overstimulation of synaptic glutamate receptors that results in excessive Ca2+ influx and subsequent neuronal injury (98). Excitotoxicity is a common event in many neurodegenerative disorders, including ischemic stroke, HD, ALS, and perhaps AD (98).
As mentioned earlier, NO can protect cells from excitotoxicity by blocking NMDA-receptor opening (34, 82, 96, 97). This protective function of NO requires its conversion to the nitrosonium ion (NO+) by the loss of one electron (96). (See Fig. 2) However, under different conditions, NO can also exacerbate neuronal injury resulting from excitotoxicity (44, 45). Increased intracellular Ca2+ levels, resulting from activation of glutamate receptors such as NMDA receptors, stimulate nNOS to produce more NO (44). (See Fig. 2) High concentrations of NO can interfere with S-nitrosylation of NMDA-receptor thiols (114). Thus, the continual influx of Ca2+ through the open NMDA receptors compounds the stress on mitochondria, which attempt to sequester and buffer Ca2+. As mitochondrial membrane potential decreases because of the overflow of Ca2+ (2, 22, 23, 101), mitochondria reverse their ATP synthase in an attempt to restore it (2). Eventually, the excess Ca2+ uptake causes mitochondrial membrane potential loss, mitochondrial swelling, opening of the mitochondrial permeability transition pore, outer membrane rupture, and spill of Ca2+ and apoptogenic factors into the cytoplasm, which ultimately results in neuronal death (114).
FIG. 2.
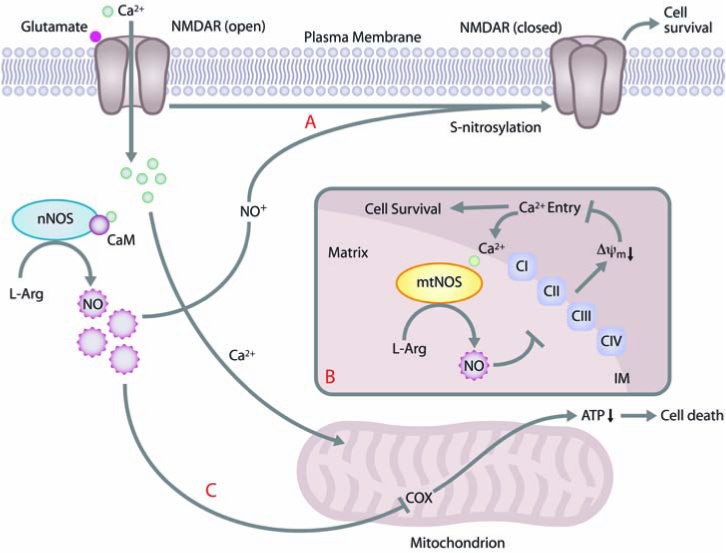
NO can both protect and sensitize cells to excitotoxic cell death through a mitochondrial pathway. (A) NO plays a key role in excitotoxic pathways mediated by NMDA Ca2+ channels. Under normal physiologic conditions, NO (after converting to NO+) S-nitrosylates NMDA receptors, blocks Ca2+ influx, and promotes cell survival. (B) When mtNOS is responsible for the majority of NOS activity (as is the case in immature neurons), Ca2+ enters mitochondria and stimulates NO production by mtNOS. NO inhibits the respiratory chain, which reduces the mitochondrial membrane potential, collapses the ion gradient, and decreases entry of Ca2+ into the mitochondria. Decreased Ca2+ influx causes a decrease in mtNOS activity and NO production, promoting cell survival. (C) When cytosolic nNOS is the primary producer of NO (as is the case in mature neurons), Ca2+ entry through overactive NMDA channels stimulates nNOS, and NO can then enter the mitochondria and directly inhibit complex IV (cytochrome c oxidase; COX) of the respiratory chain, which leads to a block of ATP production and eventual cell death due to energetic failure. In contrast to pathway B, no feedback loop is present to protect the cell from damage. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
A study by Marks and co-workers (101) recently showed that developmental differences can determine whether NO is protective or harmful to neurons after excitotoxic insults. (See Fig. 2.) In immature hippocampal neurons (from postnatal day 5 rats), Ca2+ influx after NMDA-receptor activation results in increased NOS activity—hence increased NO production and loss of membrane potential in mitochondria. Loss of membrane potential decreases Ca2+ uptake by mitochondria, which decreases mitochondrial NO production, salvages mitochondrial function, and protects neurons from NMDA toxicity (101). However, in mature hippocampal neurons (from postnatal day 19 rats), NO production after NMDA activation does not depend on mitochondrial membrane potential because the active NOS is localized to the cytosol rather than the mitochondria. Thus, the feedback effect of NO on mtNOS is not relevant and cannot protect the neurons from Ca2+ (101). These findings raise the question of how cells regulate the different NOS enzymes at different stages of development and potentially support the existence of a mtNOS that is distinct from nNOS.
In sum, both the protective and detrimental effects of NO in glutamate signaling are well established. This suggests that, although blocking NOS activity (128) or selectively downregulating NMDA receptors (113) might be potential treatment options for neurodegenerative disease, the application of such treatments will be very complex and will require a careful balancing act.
Mitochondrial respiratory complex inhibition
Because neurons have high energy demands and do not readily use glycolysis, mitochondria must provide most of the required energy through oxidative phosphorylation (87). Thus, disruption of the mitochondrial respiratory chain that causes decreased ATP production can be very damaging to neurons. NO and ONOO− have been shown to inhibit mitochondrial respiratory complexes, particularly complex IV (also known as cytochrome c oxidase or COX), where NO competes with O2 to bind at the enzyme active site, in many cell types including neurons (14, 24, 25, 37, 130). (See Fig. 3.) Interestingly, the cortex of human postmortem AD brains exhibits a loss of COX activity (42, 84, 110) and increased levels of iNOS (66, 70).
FIG. 3.

ONOO− directly inhibits the mitochondrial respiratory chain. NO combines with O2− produced by the mitochondrial respiratory chain to form ONOO−, which inhibits the respiratory chain at complex IV, and when reduced glutathione (GSH) levels are low, complex I. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Studies also have shown inhibition of mitochondrial respiratory complex I in isolated brain mitochondria and intact neurons by ONOO− under certain conditions (12, 30, 99, 121). In these studies, ONOO− did not inhibit complex I unless mitochondrial integrity was disrupted (121) or levels of reduced glutathione (GSH) were low (5). This suggests a potential role for GSH, a major antioxidant in mammalian cells, in neuroprotection in PD (107). Interestingly, presymptomatic PD patients are deficient in both GSH in the substantia nigra (79) and complex I activity (126, 127).
Organelle fragmentation
Fragmentation of two important cellular organelles, the mitochondrion and the Golgi apparatus, may play an important role in neurodegeneration. To help maintain bioenergetic functionality and to facilitate equal energy transmission throughout neurons, mitochondria are dynamic organelles that actively migrate, divide, and fuse (86). A family of large, dynamin-related GTPases directs mitochondrial division (fission) and fusion. Three important members of the mitochondrial fission and fusion machinery are dynamin-related protein 1 (DRP1), a fission protein, and mitofusins 1 and 2 (MFN1, MFN2), outer membrane fusion proteins. (See Fig. 4)
FIG. 4.

Mitochondrial fission and fusion. (A) DRP1, in coordination with other factors including the protein FIS1 and possibly BAX, directs fission of mitochondria. NO can stimulate mitochondrial fission by an unknown mechanism. (B) MFNs mediate fusion of the mitochondrial outer membrane. NO might inhibit this process. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
To remain functional, cells must balance fission and fusion events. It is becoming increasingly clear that a shift in the fission/fusion balance toward continuous fission (fragmentation) can cause neurodegeneration, although the precise mechanism is unclear (16, 86). We recently showed that NO triggers mitochondrial fission and cell death in cortical neurons (6). In addition, NO causes spotlike cluster formation of the proapoptotic protein BAX on mitochondria at potential fission sites (81, 152). The p38 mitogen-activating protein (MAP) kinase stimulates BAX translocation to neurons (63). Occasionally, mitochondrial fission caused by NO was reversible, and neurons survived (6, 152). Interestingly, fission can be asymmetric, in which one division product is intact while its fission partner exhibits profound ultrastructural damage. Thus, mitochondrial fission caused by NO may resemble a normal stress response and repair mechanism. In support of this idea, we observed that autophagosomes engulf damaged mitochondria after fission caused by NO. Whereas fission caused by NO alone did not cause cell death, we found that fission associated with BAX cluster formation on mitochondria triggered irreversible fission and cell death (110). Thus, NO-mediated mitochondrial fission and autophagy may be a neuroprotective mechanism, yet if the induction of additional signal-transduction pathways (such as BAX clustering to fission sites) overwhelms the fission and autophagy system, neuronal cell death can result. Interestingly, expression of MFN1 and dominant-negative mutant DRP1 inhibits fission and BAX foci formation on mitochondria (6, 152). This builds on another study that found that MFN2 protects cerebellar granule neurons against oxidative stress (78). Collectively, these studies suggest that mitochondrial fragmentation initiates NO-mediated neurodegeneration.
The precise mechanism of NO-mediated mitochondrial fission is presently unclear. Of note, dynamin, which is involved in endocytosis, is activated by NO (144). Thus, one can speculate that NO directly regulates DRP1 and/or MFN1/MFN2 activity. Alternatively, it is becoming increasingly clear that these large GTPases are regulated by phosphorylation and dephosphorylation (31, 39, 105, 142). Thus, NO may regulate kinases and phosphatases that control DRP1 and/or MFN activity. Studies have shown S-nitrosylation of G protein–coupled receptor kinases (148) and protein tyrosine phosphatases (32). Whether kinases and phosphatases, which regulate mitochondrial fission and fusion components, such as protein kinase A (PKA) and cyclin-dependent kinase 1 (Cdk1), are targets of S-nitrosylation is a subject for future investigation.
The Golgi apparatus is another cellular organelle that undergoes fragmentation under both physiologic and pathophysiologic conditions. Golgi disassembly and fragmentation is an important event in mitosis (as is mitochondrial fission) (141, 145) and has been observed in vivo in AD and ALS (64). In addition, NO induces Golgi fragmentation in cortical neuron cultures, and nitro-l-arginine, an NOS inhibitor, significantly inhibited NMDA-induced fragmentation (112). Interestingly, addition of MFN1 or dominant-negative mutant DRP1 decreased Golgi fragmentation after NMDA or NO exposure, suggesting that mitochondrial fragmentation occurs upstream of Golgi fragmentation (112). Collectively, these studies suggest that the roles of Golgi and mitochondrial fragmentation in NO-mediated neurodegeneration are important areas for further investigation.
Liberation of Zn2+ from intracellular ligands
Similar to NO, Zn2+ is a neuromodulator under normal physiologic conditions. Zn2+ is stored in presynaptic vesicles of glutaminergic neurons and is released on action potential firing. Zn2+ can modulate glutamate receptors by binding to the NR2A subunit (120) and thus plays a role in long-term potentiation and memory. Most of the intracellular Zn2+ is not free and is instead bound to high-affinity ligands, such as Zn2+ finger–binding proteins or metallothionein.
Also similar to NO, Zn2+ has been implicated in neurodegenerative disorders, in particular acute brain injuries such as ischemic stroke, prolonged seizures, and trauma (33, 53, 56, 90, 147). During these brain injuries, a dramatic increase occurs in free Zn2+. It has been suggested that free Zn2+ then triggers neuronal death. Supporting this belief, Zn2+ was found to be particularly toxic to neurons (150). However, the mechanism responsible for the massive accumulation of Zn2+ is unknown. One possible mechanism is that Zn2+ released from presynaptic vesicles crosses from the synaptic cleft, through the plasma membrane, and enters the postsynaptic neuron via voltage-gated ion channels (133, 139, 146). However, we and others have provided another possibility for the accumulation of free Zn2+ ions during brain injury. Nitrosative stress or oxidative stress evokes the liberation of Zn2+ from its intracellular ligands, such as metallothionein, which in turn blocks the mitochondrial electron-transport chain, decreases mitochondrial membrane potential, increases free radical production, evokes cytochrome c release, and eventually leads to neuronal cell death (1, 17, 40, 52, 54). (See Fig. 5.) Thus, these studies have established an important connection between NO and Zn2+ pathways in neuronal injury. In addition, Zn2+ is a potent inhibitor of proteins involved in energy production and defense against oxidative stress, including pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and the Rieske Fe-S protein complex (58, 59), and inhibition of these proteins may set off a vicious cycle of free radical production and bioenergetic failure. Thus, chelation of free Zn2+ might in part block NO-mediated neuronal injury.
FIG. 5.

Nitric oxide–mediated zinc (Zn2+) causes mitochondrial dysfunction and cell death. NO, after conversion to ONOO−, causes release of Zn2+ from metallothionein (MT). Free Zn2+ inhibits the mitochondrial respiratory chain, causing increased cytochrome c (Cyt c) release, increased reactive oxygen species (ROS) production, decreased mitochondrial membrane potential (ΔΨm), decreased respiration, and ultimately, cell death. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article at www.liebertonline.com/ars).
Nonneuronal Cell–Type Involvement
Whereas neurodegenerative disease research typically focuses on neurons, and rightfully so, because neuronal injury is the ultimate cause of the disease symptoms, it is becoming increasingly clear that other cells of the nervous system, including astrocytes and microglia, also play important roles in disease pathology. For example, much of the recent research in ALS has focused on the role of astrocytes in disease onset and progression (85). Studies have reported that activated astrocytes expressing the mutant superoxide dismutase 1 (mSOD1) gene, which causes a form of familial ALS, release a soluble factor that contributes to motor neuron degeneration and death (47, 111). NO is one candidate factor that activated astrocytes may release. One important normal function of glial cells in the nervous system is to protect neurons from NO (13, 49). For example, concentrations of NO that are nontoxic to intact neurons (i.e., with glial cells present) inhibit ATP synthesis in neurons cultured alone (i.e., without glia) (21). Thus, the possibility of a link between NO and astrocyte-mediated neuronal injury in ALS is a question that deserves further exploration. More generally, how NO affects glial cells in other neurodegenerative diseases is a question that requires more research.
Outlook
As the field of NO research continues to expand, a growing appreciation exists of the many and varied functions of NO in human health and disease. Because NO activity is crucial for normal physiologic function, particularly of the nervous system where NO acts as a neurotransmitter and can protect against excitotoxicity and caspase activation under certain conditions, attempting to target selectively the deleterious actions of NO for treatment of neurodegenerative diseases is a difficult task. The NO system has many “moving parts,” many of which have likely yet to be identified, and many of the reactions of NO and its reactive derivatives are nonspecific. Thus, attempting to understand how NO will behave, as a mediator of healthy function or as an inducer of neurodegeneration by nitrosylating and nitrotyrosinating proteins, increasing excitotoxic vulnerability, inhibiting mitochondrial respiration, fragmenting organelles, or mobilizing intracellular Zn2+ will require continued intensive and painstaking research. For example, two recent studies found that the antioxidant enzymes known as thioredoxins (TRX1 in the cytosol and TRX2 in mitochondria) denitrosylate caspase-3 (10, 132) and other low-molecular-weight proteins (132). Denitrosylation of caspase-3 activates the protease, and this activation is a key event in the apoptotic cascade. Although the link between caspase activation and neurodegenerative disease is controversial, this type of mechanistic insight is crucial for our general understanding of NO-mediated pathology. We hope that further progress in unraveling the mechanisms of NO reactions and identifying more of the players involved will allow us to develop more specific and effective ways to treat neurodegenerative diseases through the shared pathway of nitrosative stress.
Acknowledgments
We appreciate the support of NIH grants R01 EY016164, R01 NS047456, and R01 NS055193, and the Hereditary Disease Foundation. We thank Adam Wilson for the illustrations.
Abbreviations
7-NI; 7-nitroindazole; AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; BAX, BCL-2–associated X protein; Ca2+, calcium ion; COX, cytochrome c oxidase; DRP1, dynamin-related protein 1; EDRF, endothelium-derived relaxing factor; eNOS, endothelial nitric oxide synthase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GSH, glutathione (reduced); GSNO, S-nitrosoglutathione; HD, Huntington disease; iNOS, inducible nitric oxide synthase; MAP, mitogen-activating protein; MFN1, mitofusin 1; MFN2, mitofusin 2; MMP-9, matrix metalloproteinase-9; MPPP, 1-methyl-4-phenyl-4-propionoxypiperidine; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; mSOD1, mutant superoxide dismutase 1; mtHTT, mutant Huntingtin; mtNOS, mitochondrial nitric oxide synthase; N2O3, dinitrogen trioxide; NMDA, N-methyl-d-aspartate; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NOS, nitric oxide synthase; NR2A, NMDA-receptor subunit 2A; O2−, superoxide radical; ONOO−, peroxynitrite; PD, Parkinson disease; PRX2, peroxiredoxin 2; RNS, reactive nitrogen species; ROS, reactive oxygen species; SIAH1, seven in absentia homolog 1 (Drosophila); Zn2+, zinc ion.
References
- 1.Aizenman E. Stout AK. Hartnett KA. Dineley KE. McLaughlin B. Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- 2.Almeida A. Bolanos JP. A transient inhibition of mitochondrial ATP synthesis by nitric oxide synthase activation triggered apoptosis in primary cortical neurons. J Neurochem. 2001;77:676–690. doi: 10.1046/j.1471-4159.2001.00276.x. [DOI] [PubMed] [Google Scholar]
- 3.Ara J. Przedborski S. Naini AB. Jackson-Lewis V. Trifiletti RR. Horwitz J. Ischiropoulos H. Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Proc Natl Acad Sci U S A. 1998;95:7659–63. doi: 10.1073/pnas.95.13.7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bae BI. Hara MR. Cascio MB. Wellington CL. Hayden MR. Ross CA. Ha HC. Li XJ. Snyder SH. Sawa A. Mutant huntingtin: nuclear translocation and cytotoxicity mediated by GAPDH. Proc Natl Acad Sci U S A. 2006;103:3405–3409. doi: 10.1073/pnas.0511316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barker JE. Bolanos JP. Land JM. Clark JB. Heales SJ. Glutathione protects astrocytes from peroxynitrite-mediated mitochondrial damage: implications for neuronal/astrocytic trafficking and neurodegeneration. Dev Neurosci. 1996;18:391–396. doi: 10.1159/000111432. [DOI] [PubMed] [Google Scholar]
- 6.Barsoum MJ. Yuan H. Gerencser AA. Liot G. Kushnareva Y. Graber S. Kovacs I. Lee WD. Waggoner J. Cui J. White AD. Bossy B. Martinou JC. Youle RJ. Lipton SA. Ellisman MH. Perkins GA. Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GT-Pases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beal MF. Ferrante RJ. Browne SE. Matthews RT. Kowall NW. Brown RH., Jr Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997;42:644–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- 8.Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 9.Beckman JS. Carson M. Smith CD. Koppenol WH. ALS, SOD and peroxynitrite. Nature. 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 10.Benhar M. Forrester MT. Hess DT. Stamler JS. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boeckxstaens GE. Pelckmans PA. Bult H. De Man JG. Herman AG. Van Maercke YM. Non-adrenergic non-cholinergic relaxation mediated by nitric oxide in the canine ileocolonic junction. Eur J Pharmacol. 1990;190:239–246. doi: 10.1016/0014-2999(90)94132-h. [DOI] [PubMed] [Google Scholar]
- 12.Bolanos JP. Heales SJ. Land JM. Clark JB. Effect of peroxynitrite on the mitochondrial respiratory chain: differential susceptibility of neurones and astrocytes in primary culture. J Neurochem. 1995;64:1965–1972. doi: 10.1046/j.1471-4159.1995.64051965.x. [DOI] [PubMed] [Google Scholar]
- 13.Bolanos JP. Heales SJ. Peuchen S. Barker JE. Land JM. Clark JB. Nitric oxide-mediated mitochondrial damage: a potential neuroprotective role for glutathione. Free Radic Biol Med. 1996;21:995–1001. doi: 10.1016/s0891-5849(96)00240-7. [DOI] [PubMed] [Google Scholar]
- 14.Bolanos JP. Peuchen S. Heales SJ. Land JM. Clark JB. Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem. 1994;63:910–916. doi: 10.1046/j.1471-4159.1994.63030910.x. [DOI] [PubMed] [Google Scholar]
- 15.Bonfoco E. Krainc D. Ankarcrona M. Nicotera P. Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bossy-Wetzel E. Barsoum MJ. Godzik A. Schwarzenbacher R. Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 17.Bossy-Wetzel E. Talantova MV. Lee WD. Schölzke MN. Harrop A. Mathews E. Gotz T. Han J. Ellisman MH. Perkins GA. Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- 18.Bredt DS. Endogenous nitric oxide synthesis: biological functions and pathophysiology. Free Radic Res. 1999;31:577–596. doi: 10.1080/10715769900301161. [DOI] [PubMed] [Google Scholar]
- 19.Bredt DS. Glatt CE. Hwang PM. Fotuhi M. Dawson TM. Snyder SH. Nitric oxide synthase protein and mRNA are discretely localized in neuronal populations of the mammalian CNS together with NADPH diaphorase. Neuron. 1991;7:615–624. doi: 10.1016/0896-6273(91)90374-9. [DOI] [PubMed] [Google Scholar]
- 20.Bredt DS. Hwang PM. Glatt CE. Lowenstein C. Reed RR. Snyder SH. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 21.Brorson JR. Schumacker PT. Zhang H. Nitric oxide acutely inhibits neuronal energy production: the Committees on Neurobiology and Cell Physiology. J Neurosci. 1999;19:147–158. doi: 10.1523/JNEUROSCI.19-01-00147.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brorson JR. Sulit RA. Zhang H. Nitric oxide disrupts Ca2+ homeostasis in hippocampal neurons. J Neurochem. 1997;68:95–105. doi: 10.1046/j.1471-4159.1997.68010095.x. [DOI] [PubMed] [Google Scholar]
- 23.Brorson JR. Zhang H. Disrupted [Ca2+]i homeostasis contributes to the toxicity of nitric oxide in cultured hippocampal neurons. J Neurochem. 1997;69:1882–1889. doi: 10.1046/j.1471-4159.1997.69051882.x. [DOI] [PubMed] [Google Scholar]
- 24.Brown GC. Bolanos JP. Heales SJ. Clark JB. Nitric oxide produced by activated astrocytes rapidly and reversibly inhibits cellular respiration. Neurosci Lett. 1995;193:201–204. doi: 10.1016/0304-3940(95)11703-y. [DOI] [PubMed] [Google Scholar]
- 25.Brown GC. Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 26.Bult H. Boeckxstaens GE. Pelckmans PA. Jordaens FH. Van Maercke YM. Herman AG. Nitric oxide as an inhibitory non-adrenergic non-cholinergic neurotransmitter. Nature. 1990;345:346–347. doi: 10.1038/345346a0. [DOI] [PubMed] [Google Scholar]
- 27.Burnett AL. Lowenstein CJ. Bredt DS. Chang TS. Snyder SH. Nitric oxide: a physiologic mediator of penile erection. Science. 1992;257:401–403. doi: 10.1126/science.1378650. [DOI] [PubMed] [Google Scholar]
- 28.Busconi L. Michel T. Endothelial nitric oxide synthase: N-terminal myristoylation determines subcellular localization. J Biol Chem. 1993;268:8410–8413. [PubMed] [Google Scholar]
- 29.Cadenas E. Poderoso JJ. Antunes F. Boveris A. Analysis of the pathways of nitric oxide utilization in mitochondria. Free Radic Res. 2000;33:747–756. doi: 10.1080/10715760000301271. [DOI] [PubMed] [Google Scholar]
- 30.Cassina A. Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- 31.Chang CR. Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282:21583–21587. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 32.Chen YY. Huang YF. Khoo KH. Meng TC. Mass spectrometry-based analyses for identifying and characterizing S-nitrosylation of protein tyrosine phosphatases. Methods. 2007;42:243–249. doi: 10.1016/j.ymeth.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Choi DW. Yokoyama M. Koh J. Zinc neurotoxicity in cortical cell culture. Neuroscience. 1988;24:67–79. doi: 10.1016/0306-4522(88)90312-0. [DOI] [PubMed] [Google Scholar]
- 34.Choi YB. Tenneti L. Le DA. Ortiz J. Bai G. Chen HS. Lipton SA. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat Neurosci. 2000;3:15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- 35.Chung KK. Say NO to neurodegeneration: role of S-nitrosylation in neurodegenerative disorders. Neurosignals. 2006;15:307–313. doi: 10.1159/000109071. [DOI] [PubMed] [Google Scholar]
- 36.Chung KK. Thomas B. Li X. Pletnikova O. Troncoso JC. Marsh L. Dawson VL. Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 37.Cleeter MW. Cooper JM. Darley-Usmar VM. Moncada S. Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide: implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 38.Crane BR. Arvai AS. Ghosh DK. Wu C. Getzoff ED. Stuehr DJ. Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 39.Cribbs JT. Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cuajungco MP. Lees GJ. Nitric oxide generators produce accumulation of chelatable zinc in hippocampal neuronal perikarya. Brain Res. 1998;799:118–129. doi: 10.1016/s0006-8993(98)00463-6. [DOI] [PubMed] [Google Scholar]
- 41.Cunningham C. Wilcockson DC. Campion S. Lunnon K. Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis RE. Miller S. Herrnstadt C. Ghosh SS. Fahy E. Shinobu LA. Galasko D. Thal LJ. Beal MF. Howell N. Parker WD., Jr Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:4526–4531. doi: 10.1073/pnas.94.9.4526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Dawson VL. Dawson TM. Nitric oxide in neurodegeneration. Prog Brain Res. 1998;118:215–229. doi: 10.1016/s0079-6123(08)63210-0. [DOI] [PubMed] [Google Scholar]
- 44.Dawson VL. Dawson TM. Bartley DA. Uhl GR. Snyder SH. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13:2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dawson VL. Dawson TM. London ED. Bredt DS. Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Desai KM. Sessa WC. Vane JR. Involvement of nitric oxide in the reflex relaxation of the stomach to accommodate food or fluid. Nature. 1991;351:477–9. doi: 10.1038/351477a0. [DOI] [PubMed] [Google Scholar]
- 47.Di Giorgio FP. Carrasco MA. Siao MC. Maniatis T. Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dou F. Netzer WJ. Tanemura K. Li F. Hartl FU. Takashima A. Gouras GK. Greengard P. Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 50.Fang J. Nakamura T. Cho DH. Gu Z. Lipton SA. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson's disease. Proc Natl Acad Sci U S A. 2007;104:18742–18747. doi: 10.1073/pnas.0705904104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferrante RJ. Shinobu LA. Schulz JB. Matthews RT. Thomas CE. Kowall NW. Gurney ME. Beal MF. Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann Neurol. 1997;42:326–334. doi: 10.1002/ana.410420309. [DOI] [PubMed] [Google Scholar]
- 52.Frazzini V. Rockabrand E. Mocchegiani E. Sensi SL. Oxidative stress and brain aging: is zinc the link? Biogerontology. 2006;7:307–314. doi: 10.1007/s10522-006-9045-7. [DOI] [PubMed] [Google Scholar]
- 53.Frederickson CJ. Koh JY. Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- 54.Frederickson CJ. Maret W. Cuajungco MP. Zinc and excitotoxic brain injury: a new model. Neuroscientist. 2004;10:18–25. doi: 10.1177/1073858403255840. [DOI] [PubMed] [Google Scholar]
- 55.Furchgott RF. Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 56.Galasso SL. Dyck RH. The role of zinc in cerebral ischemia. Mol Med. 2007;13:380–387. doi: 10.2119/2007-00044.Galasso. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garthwaite J. Charles SL. Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- 58.Gazaryan IG. Krasinskaya IP. Kristal BS. Brown AM. Zinc irreversibly damages major enzymes of energy production and antioxidant defense prior to mitochondrial permeability transition. J Biol Chem. 2007;282:24373–34380. doi: 10.1074/jbc.M611376200. [DOI] [PubMed] [Google Scholar]
- 59.Gazaryan IG. Krasnikov BF. Ashby GA. Thorneley RN. Kristal BS. Brown AM. Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J Biol Chem. 2002;277:10064–10072. doi: 10.1074/jbc.M108264200. [DOI] [PubMed] [Google Scholar]
- 60.Ghafourifar P. Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci. 2005;26:190–195. doi: 10.1016/j.tips.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 61.Ghafourifar P. Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- 62.Ghafourifar P. Schenk U. Klein SD. Richter C. Mitochondrial nitric-oxide synthase stimulation causes cytochrome c release from isolated mitochondria: evidence for intramitochondrial peroxynitrite formation. J Biol Chem. 1999;274:31185–31188. doi: 10.1074/jbc.274.44.31185. [DOI] [PubMed] [Google Scholar]
- 63.Ghatan S. Larner S. Kinoshita Y. Hetman M. Patel L. Xia Z. Youle RJ. Morrison RS. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150:335–347. doi: 10.1083/jcb.150.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gonatas NK. Stieber A. Gonatas JO. Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J Neurol Sci. 2006;246:21–30. doi: 10.1016/j.jns.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 65.Gu Z. Kaul M. Yan B. Kridel SJ. Cui J. Strongin A. Smith JW. Liddington RC. Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 66.Haas J. Storch-Hagenlocher B. Biessmann A. Wildemann B. Inducible nitric oxide synthase and argininosuccinate synthetase: co-induction in brain tissue of patients with Alzheimer's dementia and following stimulation with beta-amyloid 1-42 in vitro. Neurosci Lett. 2002;322:121–125. doi: 10.1016/s0304-3940(02)00095-2. [DOI] [PubMed] [Google Scholar]
- 67.Hantraye P. Brouillet E. Ferrante R. Palfi S. Dolan R. Matthews RT. Beal MF. Inhibition of neuronal nitric oxide synthase prevents MPTP-induced parkinsonism in baboons. Nat Med. 1996;2:1017–1021. doi: 10.1038/nm0996-1017. [DOI] [PubMed] [Google Scholar]
- 68.Hara MR. Agrawal N. Kim SF. Cascio MB. Fujimuro M. Ozeki Y. Takahashi M. Cheah JH. Tankou SK. Hester LD. Ferris CD. Hayward SD. Snyder SH. Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 69.Hara MR. Thomas B. Cascio MB. Bae BI. Hester LD. Dawson VL. Dawson TM. Sawa A. Snyder SH. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci U S A. 2006;103:3887–3889. doi: 10.1073/pnas.0511321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heneka MT. Feinstein DL. Expression and function of inducible nitric oxide synthase in neurons. J Neuroimmunol. 2001;114:8–18. doi: 10.1016/s0165-5728(01)00246-6. [DOI] [PubMed] [Google Scholar]
- 71.Hibbs JB., Jr Taintor RR. Vavrin Z. Rachlin EM. Nitric oxide: a cytotoxic activated macrophage effector molecule. Biochem Biophys Res Commun. 1988;157:87–94. doi: 10.1016/s0006-291x(88)80015-9. [DOI] [PubMed] [Google Scholar]
- 72.Hibbs JB., Jr Vavrin Z. Taintor RR. L-arginine is required for expression of the activated macrophage effector mechanism causing selective metabolic inhibition in target cells. J Immunol. 1987;138:550–565. [PubMed] [Google Scholar]
- 73.Huang Z. Huang PL. Panahian N. Dalkara T. Fishman MC. Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 74.Ignarro LJ. Buga GM. Wood KS. Byrns RE. Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ignarro LJ. Byrns RE. Buga GM. Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res. 1987;61:866–879. doi: 10.1161/01.res.61.6.866. [DOI] [PubMed] [Google Scholar]
- 76.Ischiropoulos H. Zhu L. Chen J. Tsai M. Martin JC. Smith CD. Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 77.Ishitani R. Chuang DM. Glyceraldehyde-3-phosphate dehydrogenase antisense oligodeoxynucleotides protect against cytosine arabinonucleoside-induced apoptosis in cultured cerebellar neurons. Proc Natl Acad Sci U S A. 1996;93:9937–9941. doi: 10.1073/pnas.93.18.9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jahani-Asl A. Cheung EC. Neuspiel M. MacLaurin JG. Fortin A. Park DS. McBride HM. Slack RS. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J Biol Chem. 2007;282:23788–23798. doi: 10.1074/jbc.M703812200. [DOI] [PubMed] [Google Scholar]
- 79.Jenner P. Dexter DT. Sian J. Schapira AH. Marsden CD. Oxidative stress as a cause of nigral cell death in Parkinson's disease and incidental Lewy body disease: the Royal Kings and Queens Parkinson's Disease Research Group. Ann Neurol. 1992;32(suppl):S82–S87. doi: 10.1002/ana.410320714. [DOI] [PubMed] [Google Scholar]
- 80.Kakimura J. Kitamura Y. Takata K. Umeki M. Suzuki S. Shibagaki K. Taniguchi T. Nomura Y. Gebicke-Haerter PJ. Smith MA. Perry G. Shimohama S. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 2002;16:601–603. doi: 10.1096/fj.01-0530fje. [DOI] [PubMed] [Google Scholar]
- 81.Karbowski M. Lee YJ. Gaume B. Jeong SY. Frank S. Nechushtan A. Santel A. Fuller M. Smith CL. Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim WK. Choi YB. Rayudu PV. Das P. Asaad W. Arnelle DR. Stamler JS. Lipton SA. Attenuation of NMDA receptor activity and neurotoxicity by nitroxyl anion, NO. Neuron. 1999;24:461–469. doi: 10.1016/s0896-6273(00)80859-4. [DOI] [PubMed] [Google Scholar]
- 83.Kim YM. Talanian RV. Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 84.Kish SJ. Bergeron C. Rajput A. Dozic S. Mastrogiacomo F. Chang LJ. Wilson JM. DiStefano LM. Nobrega JN. Brain cytochrome oxidase in Alzheimer's disease. J Neurochem. 1992;59:776–779. doi: 10.1111/j.1471-4159.1992.tb09439.x. [DOI] [PubMed] [Google Scholar]
- 85.Knott AB. Bossy-Wetzel E. ALS: astrocytes take center stage, but must they share the spotlight? Cell Death Differ. 2007;14:1985–1988. doi: 10.1038/sj.cdd.4402241. [DOI] [PubMed] [Google Scholar]
- 86.Knott AB. Bossy-Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann NY Acad Sci. doi: 10.1196/annals.1427.030. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Knott AB. Perkins G. Schwarzenbacher R. Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Knowles RG. Moncada S. Nitric oxide synthases in mammals. Biochem J. 1994;298:249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Knowles RG. Palacios M. Palmer RM. Moncada S. Formation of nitric oxide from L-arginine in the central nervous system: a transduction mechanism for stimulation of the soluble guanylate cyclase. Proc Natl Acad Sci U S A. 1989;86:5159–5162. doi: 10.1073/pnas.86.13.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Koh JY. Choi DW. Zinc toxicity on cultured cortical neurons: involvement of N-methyl-D-aspartate receptors. Neuroscience. 1994;60:1049–1057. doi: 10.1016/0306-4522(94)90282-8. [DOI] [PubMed] [Google Scholar]
- 91.Langston JW. Ballard P. Tetrud JW. Irwin I. Chronic Parkinsonism in humans due to a product of meperidineanalog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 92.Langston JW. Irwin I. MPTP: current concepts and controversies. Clin Neuropharmacol. 1986;9:485–507. [PubMed] [Google Scholar]
- 93.Lee HK. Choi SS. Han EJ. Han KJ. Suh HW. Role of glutamate receptors and an on-going protein synthesis in the regulation of phosphorylation of Ca2+/calmodulin-dependent protein kinase II in the CA3 hippocampal region in mice administered with kainic acid intracerebroventricularly. Neurosci Lett. 2003;348:93–96. doi: 10.1016/s0304-3940(03)00752-3. [DOI] [PubMed] [Google Scholar]
- 94.Li J. Billiar TR. Talanian RV. Kim YM. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem Biophys Res Commun. 1997;240:419–424. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- 95.Liberatore GT. Jackson-Lewis V. Vukosavic S. Mandir AS. Vila M. McAuliffe WG. Dawson VL. Dawson TM. Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 96.Lipton SA. Choi YB. Pan ZH. Lei SZ. Chen HS. Sucher NJ. Loscalzo J. Singel DJ. Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 97.Lipton SA. Choi YB. Takahashi H. Zhang D. Li W. Godzik A. Bankston LA. Cysteine regulation of protein function: as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002;25:474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- 98.Lipton SA. Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 99.Lizasoain I. Moro MA. Knowles RG. Darley-Usmar V. Moncada S. Nitric oxide and peroxynitrite exert distinct effects on mitochondrial respiration which are differentially blocked by glutathione or glucose. Biochem J. 1996;314:877–880. doi: 10.1042/bj3140877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mannick JB. Schonhoff C. Papeta N. Ghafourifar P. Szibor M. Fang K. Gaston B. S-Nitrosylation of mitochondrial caspases. J Cell Biol. 2001;154:1111–1116. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marks JD. Boriboun C. Wang J. Mitochondrial nitric oxide mediates decreased vulnerability of hippocampal neurons from immature animals to NMDA. J Neurosci. 2005;25:6561–6575. doi: 10.1523/JNEUROSCI.1450-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marletta MA. Nitric oxide synthase: aspects concerning structure and catalysis. Cell. 1994;78:927–930. doi: 10.1016/0092-8674(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 103.Martinez-Ruiz A. Villanueva L. Gonzalez de Orduna C. Lopez-Ferrer D. Higueras MA. Tarin C. Rodriguez-Crespo I. Vazquez J. Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci U S A. 2005;102:8525–8530. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Merrill JE. Murphy SP. Mitrovic B. Mackenzie-Graham A. Dopp JC. Ding M. Griscavage J. Ignarro LJ. Lowenstein CJ. Inducible nitric oxide synthase and nitric oxide production by oligodendrocytes. J Neurosci Res. 1997;48:372–384. [PubMed] [Google Scholar]
- 105.Meuer K. Suppanz IE. Lingor P. Planchamp V. Goricke B. Fichtner L. Braus GH. Dietz GP. Jakobs S. Bahr M. Weishaupt JH. Cyclin-dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ. 2007;14:651–661. doi: 10.1038/sj.cdd.4402087. [DOI] [PubMed] [Google Scholar]
- 106.Michel T. Li GK. Busconi L. Phosphorylation and subcellular translocation of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1993;90:6252–6256. doi: 10.1073/pnas.90.13.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moncada S. Bolanos JP. Nitric oxide, cell bioenergetics and neurodegeneration. J Neurochem. 2006;97:1676–1689. doi: 10.1111/j.1471-4159.2006.03988.x. [DOI] [PubMed] [Google Scholar]
- 108.Montaner J. Alvarez-Sabin J. Molina C. Angles A. Abilleira S. Arenillas J. Gonzalez MA. Monasterio J. Matrix metalloproteinase expression after human cardioembolic stroke: temporal profile and relation to neurological impairment. Stroke. 2001;32:1759–1766. doi: 10.1161/01.str.32.8.1759. [DOI] [PubMed] [Google Scholar]
- 109.Murphy S. Simmons ML. Agullo L. Garcia A. Feinstein DL. Galea E. Reis DJ. Minc-Golomb D. Schwartz JP. Synthesis of nitric oxide in CNS glial cells. Trends Neurosci. 1993;16:323–328. doi: 10.1016/0166-2236(93)90109-y. [DOI] [PubMed] [Google Scholar]
- 110.Mutisya EM. Bowling AC. Beal MF. Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. J Neurochem. 1994;63:2179–2184. doi: 10.1046/j.1471-4159.1994.63062179.x. [DOI] [PubMed] [Google Scholar]
- 111.Nagai M. Re DB. Nagata T. Chalazonitis A. Jessell TM. Wichterle H. Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nakagomi S. Barsoum MJ. Bossy-Wetzel E. Sutterlin C. Malhotra V. Lipton SA. A Golgi fragmentation pathway in neurodegeneration. Neurobiol Dis. 2008;29:221–231. doi: 10.1016/j.nbd.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nakamura T. Lipton SA. S-Nitrosylation and uncompetitive/fast off-rate (UFO) drug therapy in neurodegenerative disorders of protein misfolding. Cell Death Differ. 2007;14:1305–1314. doi: 10.1038/sj.cdd.4402138. [DOI] [PubMed] [Google Scholar]
- 114.Nelson EJ. Connolly J. McArthur P. Nitric oxide and S-nitrosylation: excitotoxic and cell signaling mechanism. Biol Cell. 2003;95:3–8. doi: 10.1016/s0248-4900(03)00004-2. [DOI] [PubMed] [Google Scholar]
- 115.Nowicki JP. Duval D. Poignet H. Scatton B. Nitric oxide mediates neuronal death after focal cerebral ischemia in the mouse. Eur J Pharmacol. 1991;204:339–340. doi: 10.1016/0014-2999(91)90862-k. [DOI] [PubMed] [Google Scholar]
- 116.Palmer RM. Ashton DS. Moncada S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. 1988;333:664–666. doi: 10.1038/333664a0. [DOI] [PubMed] [Google Scholar]
- 117.Palmer RM. Ferrige AG. Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 118.Poderoso JJ. Lisdero C. Schopfer F. Riobo N. Carreras MC. Cadenas E. Boveris A. The regulation of mitochondrial oxygen uptake by redox reactions involving nitric oxide and ubiquinol. J Biol Chem. 1999;274:37709–37716. doi: 10.1074/jbc.274.53.37709. [DOI] [PubMed] [Google Scholar]
- 119.Przedborski S. Jackson-Lewis V. Yokoyama R. Shibata T. Dawson VL. Dawson TM. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci U S A. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rachline J. Perin-Dureau F. Le Goff A. Neyton J. Paoletti P. The micromolar zinc-binding domain on the NMDA receptor subunit NR2B. J Neurosci. 2005;25:308–317. doi: 10.1523/JNEUROSCI.3967-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Radi R. Rodriguez M. Castro L. Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys. 1994;308:89–95. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- 122.Reiter CD. Teng RJ. Beckman JS. Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J Biol Chem. 2000;275:32460–32466. doi: 10.1074/jbc.M910433199. [DOI] [PubMed] [Google Scholar]
- 123.Rosell A. Ortega-Aznar A. Alvarez-Sabin J. Fernandez-Cadenas I. Ribo M. Molina CA. Lo EH. Montaner J. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- 124.Rosen DR. Siddique T. Patterson D. Figlewicz DA. Sapp P. Hentati A. Donaldson D. Goto J. O'Regan JP. Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 125.Sawa A. Khan AA. Hester LD. Snyder SH. Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc Natl Acad Sci U S A. 1997;94:11669–11674. doi: 10.1073/pnas.94.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schapira AH. Cooper JM. Dexter D. Clark JB. Jenner P. Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 127.Schapira AH. Mann VM. Cooper JM. Dexter D. Daniel SE. Jenner P. Clark JB. Marsden CD. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson's disease. J Neurochem. 1990;55:2142–2145. doi: 10.1111/j.1471-4159.1990.tb05809.x. [DOI] [PubMed] [Google Scholar]
- 128.Schulz JB. Matthews RT. Jenkins BG. Ferrante RJ. Siwek D. Henshaw DR. Cipolloni PB. Mecocci P. Kowall NW. Rosen BR. Beal MF. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J Neurosci. 1995;15:8419–8429. doi: 10.1523/JNEUROSCI.15-12-08419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schulz JB. Matthews RT. Muqit MM. Browne SE. Beal MF. Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. J Neurochem. 1995;64:936–939. doi: 10.1046/j.1471-4159.1995.64020936.x. [DOI] [PubMed] [Google Scholar]
- 130.Schweizer M. Richter C. Nitric oxide potently and reversibly deenergizes mitochondria at low oxygen tension. Biochem Biophys Res Commun. 1994;204:169–175. doi: 10.1006/bbrc.1994.2441. [DOI] [PubMed] [Google Scholar]
- 131.Senatorov VV. Charles V. Reddy PH. Tagle DA. Chuang DM. Overexpression and nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase in a transgenic mouse model of Huntington's disease. Mol Cell Neurosci. 2003;22:285–297. doi: 10.1016/s1044-7431(02)00013-1. [DOI] [PubMed] [Google Scholar]
- 132.Sengupta R. Ryter SW. Zuckerbraun BS. Tzeng E. Billiar TR. Stoyanovsky DA. Thioredoxin catalyzes the denitrosation of low-molecular mass and protein S-nitrosothiols. Biochemistry. 2007;46:8472–8483. doi: 10.1021/bi700449x. [DOI] [PubMed] [Google Scholar]
- 133.Sensi SL. Yin HZ. Carriedo SG. Rao SS. Weiss JH. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci U S A. 1999;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sliskovic I. Raturi A. Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. J Biol Chem. 2005;280:8733–8741. doi: 10.1074/jbc.M408080200. [DOI] [PubMed] [Google Scholar]
- 135.Smith MA. Richey Harris PL. Sayre LM. Beckman JS. Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Snyder SH. Nitric oxide: first in a new class of neurotransmitters. Science. 1992;257:494–496. doi: 10.1126/science.1353273. [DOI] [PubMed] [Google Scholar]
- 137.Stamler JS. Lamas S. Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 138.Stamler JS. Toone EJ. Lipton SA. Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 139.Suh SW. Chen JW. Motamedi M. Bell B. Listiak K. Pons NF. Danscher G. Frederickson CJ. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 2000;852:268–273. doi: 10.1016/s0006-8993(99)02095-8. [DOI] [PubMed] [Google Scholar]
- 140.Sultana R. Poon HF. Cai J. Pierce WM. Merchant M. Klein JB. Markesbery WR. Butterfield DA. Identification of nitrated proteins in Alzheimer's disease brain using a redox proteomics approach. Neurobiol Dis. 2006;22:76–87. doi: 10.1016/j.nbd.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 141.Sutterlin C. Hsu P. Mallabiabarrena A. Malhotra V. Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell. 2002;109:359–369. doi: 10.1016/s0092-8674(02)00720-1. [DOI] [PubMed] [Google Scholar]
- 142.Taguchi N. Ishihara N. Jofuku A. Oka T. Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 143.Uehara T. Nakamura T. Yao D. Shi ZQ. Gu Z. Ma Y. Masliah E. Nomura Y. Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 144.Wang G. Moniri NH. Ozawa K. Stamler JS. Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc Natl Acad Sci U S A. 2006;103:1295–1300. doi: 10.1073/pnas.0508354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Warren G. Membrane partitioning during cell division. Annu Rev Biochem. 1993;62:323–348. doi: 10.1146/annurev.bi.62.070193.001543. [DOI] [PubMed] [Google Scholar]
- 146.Weiss JH. Sensi SL. Ca2+-Zn2+ permeable AMPA or kainate receptors: possible key factors in selective neurodegeneration. Trends Neurosci. 2000;23:365–371. doi: 10.1016/s0166-2236(00)01610-6. [DOI] [PubMed] [Google Scholar]
- 147.Weiss JH. Sensi SL. Koh JY. Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci. 2000;21:395–401. doi: 10.1016/s0165-6147(00)01541-8. [DOI] [PubMed] [Google Scholar]
- 148.Whalen EJ. Foster MW. Matsumoto A. Ozawa K. Violin JD. Que LG. Nelson CD. Benhar M. Keys JR. Rockman HA. Koch WJ. Daaka Y. Lefkowitz RJ. Stamler JS. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 149.Yao D. Gu Z. Nakamura T. Shi ZQ. Ma Y. Gaston B. Palmer LA. Rockenstein EM. Zhang Z. Masliah E. Uehara T. Lipton SA. Nitrosative stress linked to sporadic Parkinson's disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yokoyama M. Koh J. Choi DW. Brief exposure to zinc is toxic to cortical neurons. Neurosci Lett. 1986;71:351–355. doi: 10.1016/0304-3940(86)90646-4. [DOI] [PubMed] [Google Scholar]
- 151.Yoshida T. Limmroth V. Irikura K. Moskowitz MA. The NOS inhibitor, 7-nitroindazole, decreases focal infarct volume but not the response to topical acetylcholine in pial vessels. J Cereb Blood Flow Metab. 1994;14:924–929. doi: 10.1038/jcbfm.1994.123. [DOI] [PubMed] [Google Scholar]
- 152.Yuan H. Gerencser AA. Liot G. Lipton SA. Ellisman M. Perkins GA. Bossy-Wetzel E. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 2007;14:462–471. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]