

Abstract
Protein arginine methyltransferases (PRMTs) are SAM-dependent enzymes that catalyze the mono- and di-methylation of peptidyl arginine residues. Although all PRMTs produce mono-methyl arginine (MMA), type 1 PRMTs go on to form asymmetrically dimethylated arginine (ADMA), while type 2 enzymes form symmetrically dimethylated arginine (SDMA). PRMT1 is the major type 1 PRMT in vivo, thus it is the primary producer of the competitive NOS inhibitor, ADMA. Hence, potent inhibitors, which are highly selective for this particular isozyme, could serve as excellent therapeutics for heart disease. However, the design of such inhibitors is impeded by a lack of information regarding this enzyme’s kinetic and catalytic mechanisms. Herein we report an analysis of the kinetic mechanism of human PRMT1 using both an unmethylated and a mono-methylated substrate peptide based on the N-terminus of histone H4. The results of initial velocity and product and dead-end inhibition experiments indicate that PRMT1 utilize a rapid equilibrium random mechanism with the formation of dead-end EAP and EBQ complexes. This mechanism is gratifyingly consistent with previous results demonstrating that PRMT1 catalyzes substrate dimethylation in a partially processive manner.
Protein arginine methyltransferase 1 (PRMT1) has recently emerged as a potential new target for the development of a novel therapeutic for heart disease (1–8). This is the case because PRMT1 is overexpressed in the hearts of patients with coronary heart disease (9). Additionally, PRMT1 appears to be responsible for generating the majority of the asymmetrically dimethylated arginine (ADMA; Figure 1) in cells. This is relevant to heart disease because patients with atherosclerosis, hypercholesterolemia, and heart failure have elevated plasma levels of free ADMA (which is released from cells after protein degradation), and these levels correlate with disease severity (4–7). The pathological effects of elevated free ADMA likely relates to its ability to act as an endogenous inhibitor of Nitric Oxide Synthase (NOS) isozymes; and suggest that increased PRMT activity contributes to heart disease by indirectly increasing the levels of free ADMA (Figure 2). Consistent with this hypothesis is the fact that mice genetically engineered to lack an ADMA degrading enzyme, i.e. DDAH1 (Dimethyl arginine dimethylamino hydrolase), have increased levels of serum ADMA, reduced NO signaling, elevated systemic and pulmonary blood pressure, and endothelial dysfunction (33). Thus, one would expect that inhibitors targeting PRMT1 would represent an effective therapy for cardiovascular disease because they would indirectly decrease the ADMA pool. In addition to heart disease, PRMT1-targeted therapeutics may also offer a novel treatment for a number of human cancers because this enzyme acts as a transcriptional coactivator for multiple nuclear receptor (NR) family members (e.g. the androgen and estrogen receptors) (10–12) and dysregulation of nuclear receptor signaling is a hallmark of hormone dependent cancers, e.g. breast cancer (13–16).
Figure 1. Reactions catalyzed by PRMT isozymes.

All PRMT isozymes catalyze the formation of monomethyl-arginine (MMA) and S-adenosyl –L-homocysteine. Type 1 isozymes go onto generate asymmetric dimethyl-arginine (ADMA) after the second round of methylation, whereas type 2 isozymes produce symmetric dimethyl-arginine (SDMA).
Figure 2. Potential role of dysregulated PRMT activity in heart disease.

PRMT1 asymmetrically dimethylates peptidyl-arginine residues to form peptidyl-ADMA. Upon proteolysis, free ADMA can compete with L-arginine to inhibit the activity of niric oxide synthase (NOS). Inhibition of NOS activity causes a decrease in the bioavailability of nitric oxide, and a consequent decrease in vasodilation.
In independent studies, PRMT1 was originally identified as an interacting protein for both the TIS21 and BTG1 proteins (17); as well as the interferon-alpha receptor (18). Since its initial identification and demonstration that it is a bona fide PRMT, this multifunctional enzyme has been implicated in an array of biological processes, including RNA splicing and transport, protein trafficking, and cellular growth (reviewed in (19–22)). Additionally, and as noted above, this enzyme has been shown to act as a coactivator of nuclear receptor mediated gene transcription (10–12) where it can combine with p300/CBP, a histone acetyltransferase, and PRMT4/CARM1(coactivator associated arginine methyltransferase 1) to synergistically coactivate gene transcription from a number of different templates (10, 11). In addition to NRs, PRMT1 can act as a transcriptional coactivator for p53, YY1, and NF-κB (23–25). PRMT1 catalyzed methylation of histone H4 at Arg3 appears to be required for its coactivator activities (10, 11, 19–23).
In addition to these important physiological functions, PRMT1 has served as the prototypical PRMT – principally because it was the first eukaryotic PRMT to be cloned – and as such it has aided the identification of other PRMT family members. These include PRMTs 2 to 8 and 10 that all possess a highly conserved, ~310 amino acid, catalytic core (20–22, 26–28). Individual PRMT isozymes utilize S-adenosyl-L-methionine (SAM) as the methyl donor and catalyze methyl group transfer to the ω-nitrogen of a peptidyl arginine residue, yielding Sadenosyl-L-homocysteine (SAH) and ω-NG-monomethylarginine (MMA) (Figure 1). Specific isozymes can also catalyze the transfer of a second methyl group from SAM to either the same nitrogen atom or to the other ω-nitrogen to generate ω-NG,NG-asymmetrically dimethylated arginine (ADMA) and ω-NG,NG’-symmetrically dimethylated arginine (SDMA), respectively (Figure 1). Because of this difference in methylation specificity, peptidyl-ADMA generating PRMTs are classified as being Type 1 enzymes (PRMTs 1, 3, 4, 6, and 8) whereas those isozymes that catalyze the formation of peptidyl-SDMA are classified as being Type 2 (PRMTs 5 and 7) enzymes. Note that no methyltransferase activity has been observed for PRMTs 2 and 10; thus these enzymes cannot be classified. Also note that two other PRMTs have been identified, i.e. PRMTs 9 and 11 (21, 29); however a recent report (30) suggests that these isozymes may not represent bona fide PRMTs, consistent with the fact that catalytic domains within these enzymes lack significant homology to the catalytic core domain present in other PRMT family members.
To gain insights into the mechanism of PRMT1 catalyzed methyl transfer; and thereby aid the development of PRMT1-specific inhibitors, we recently initiated studies to characterize the substrate specificity and molecular mechanism of PRMT1-catalyzed methyl transfer (31). The results of the substrate specificity studies indicated that a peptide corresponding to the N-terminal 21 amino acids of histone H4, a PRMT1 substrate, was regiospecifically modified at Arg3, an in vivo site of PRMT1-directed methylation, with efficiencies that were comparable to the protein substrate (31). These studies also demonstrated that positively charged residues distal to the site of methylation were required for high affinity interactions between PRMT1 and its substrates (31).
We additionally reported that PRMT1 can catalyze the formation of peptidyl-ADMA in a partially processive fashion (31). The term partially processive was used to explain that PRMT1 catalyzes the production of MMA and ADMA containing peptides in approximately equimolar amounts, even in the presence of an excess of the unmethylated peptide substrate, ruling out a fully processive mechanism in which the production of the ADMA product is obligatory, i.e. the concentration of the intermediate does not rise above the concentration of the enzyme. Also note that MMA containing peptides are poorer substrates for the enzyme (kcat/Kmdecreased 2- to 4- fold; (31)); thereby ruling out a distributive mechanism of ADMA production in which MMA containing peptides possess an intrinsically higher affinity for the enzyme and can rebind even in the presence of an excess of the unmodified peptide (31).
To further investigate the partially processive nature of the PRMT1-catalyzed reaction, a combination of initial velocity, product inhibition, and dead-end analogue inhibition studies, were used to elucidate the kinetic mechanism of human PRMT1 (hPRMT1). Two peptide substrates that are known to be either asymmetrically dimethylated, i.e. the AcH4-21 peptide, or subject to only a single methylation event, i.e. the AcH4-21R3MMA peptide (the sequences of these peptides are depicted in Table 1) were used for these studies. The latter experiments were performed to provide a baseline for interpreting the experiments with peptides that are subject to multiple methylation events. The results of these studies indicate that PRMT1 utilizes a rapid equilibrium random mechanism (RER) of methyl transfer with the formation of dead-end EAP and EBQ complexes. This mechanism is gratifyingly consistent with previous results demonstrating that PRMT1 catalyzes substrate dimethylation in a partially processive manner.
Table 1.
Peptide Substrates
| Peptide | Sequence | Expected Mass (Da) |
Observed Mass (Da) |
|---|---|---|---|
| AcH4-21 | 1-Ac-SGRGKGGKGLGKGGAKRHRKV | 2132 | 2133 |
| AcH4-21R3MMA | 1-Ac-SGR (Me)GKGGKGLGKGGAKRHRKV | 2146 | 2147 |
| AcH4-21R3K | 1Ac-SGKGKGGKGLGKGGAKRHRKV | 2104 | 2105 |
| AcH4-21R3ADMA | 1Ac-SGR(Me)2GKGGKGLGKGGAKRHRKV | 2160 | 2161 |
EXPERIMENTAL PROCEDURES
Chemicals
N-(2-hydroxyethyl)piperazine-N’-(2-ethanesulfonic acid) (HEPES), dithiothreitol (DTT), and ethylenediamine tetraacetic acid (EDTA) were purchased from Sigma-Aldrich. N-α-Fmoc amino acids and pre-loaded Wang based resins were obtained from Novabiochem. Radiolabelled reagents, i.e. 14C-labeled SAM and 14C-labeled bovine serum albumin (BSA), were purchased from Perkin-Elmer Life Sciences.
Protein purification
The purification of hPRMT1 has previously been described (31). Briefly, an expression construct encoding hexa-histidine tagged hPRMT1 was used to express and purify this protein from E. coli. Purification of the recombinant enzyme was achieved through a combination of immobilized metal ion chromatography (using a nickel(II) chelating sepharose column) and strong anion exchange chromatography.
Peptide synthesis
Peptides substrates were synthesized using standard Fmoc chemistry on a Rainin PS3 automated peptide synthesizer. Following their synthesis, peptides were cleaved from the Wang-based resins with Reagent K (19 mL trifluoroacetic acid (TFA), 510 mg of phenol, 500 µL of ddH2O, 500 µL of thioanisole, and 250 µL of 1,2-ethanedithiol). After precipitation with diethyl ether, the peptides were purified by reverse phase HPLC using water/0.05% TFA as the mobile phase. Bound peptides were eluted with a linear acetonitrile/0.05% TFA gradient. The sequences of all the peptides used in the studies described below as well as their expected and observed masses, are depicted in Table 1. All mass spectra were acquired on a Bruker Ultraflex II MALDI-TOF mass spectrometer.
Gel-based activity assay
Activity assays were performed as previously described (31). Briefly, this discontinuous gel-based assay utilizes 14C-labeled SAM as the methyl donor and monitors the incorporation of 14C-labelled methyl groups into arginine containing peptide substrates by phosphorimage analysis after separating the reaction components on 16.5% Tris-Tricine polyacrylamide gels. To quantify the amount of radiolabel incorporated, 14C-labeled BSA is used as an internal reference standard. The Assay Buffer consisted of 50 mM HEPES at pH 8.0, 50 mM NaCl, 1 mM EDTA, and 0.5 mM dithiothreitol. Generally enzyme assays were performed by first pre-incubating the Assay Buffer with SAM and a peptide substrate for 10 min at 37°C and then initiating the reaction by the addition of hPRMT1 (212 nM final). Under these conditions, hPRMT1 activity is linear with respect to both time and enzyme concentration (31). All assays were performed at least in duplicate, the standard deviation was ≤ 20 %, and where appropriate the initial rates obtained from these assays were fit by nonlinear least fit squares to eq 1,
| (1) |
using the GraFit version 5.0.11 software package (32).
Initial velocity studies
Initial velocity patterns were obtained by determining the steady-state kinetic parameters for a substrate at different fixed concentrations of the second substrate. The initial rates for substrate peptides, i.e. the AcH4-21 and AcH4-21 R3MMA peptides, were determined at different fixed concentrations of 14C-SAM (1, 2.5, and 7.5 µM for the AcH4-21 peptide and 2.5, 5, 7.5, 10, and 15 µM for the AcH4-21 R3MMA peptide). For SAM, initial rates were obtained at fixed concentrations of the AcH4-21 peptide (2.5, 5, and 25 µM) and the AcH4-21 R3MMA peptide (2.5, 5, 10, 25, and 125 µM). Initial rates were fit to eq 2,
| (2) |
using the GraFit version 5.0.11 software package (32). Where Kia is the dissociation constant of the varied substrate and Ka and Kb are Michaelis constants for the varied and fixed substrates, respectively.
Inhibition studies
Product inhibition experiments were carried out using the assay methodology outlined above. For these experiments, the AcH4-21R3ADMA peptide and SAH were used as the product inhibitors. Dead-end analogue inhibition experiments were carried out analogously; using the AcH4-21R3K peptide and sinefungin as the dead-end analogues. The initial rates derived from these inhibition experiments were fit to equations representative of linear competitive inhibition (eq 3), linear non-competitive inhibition (eq 4), linear mixed inhibition (eq 5) or linear uncompetitive inhibition (eq 6) by a non-linear least fit squares approach using the GraFit version 5.0.11 software package,
| (3) |
| (4) |
| (5) |
| (6) |
where Kii is the intercept Ki and Kis is the slope Ki. Note that equation 4 is the equation for pure noncompetitive inhibition and the Ki = Kii = Kis. The best fits of the data to eq’s 3–6 were chosen using a combination visual inspection along with a comparison of the standard errors.
RESULTS
Initial velocity studies
To begin to characterize the kinetic mechanism of PRMT1, initial velocity studies were performed. For these studies, the initial rates were determined for SAM at various fixed concentrations of the AcH4-21R3MMA peptide; and the initial velocity patterns assessed by fitting the initial rate data to eq 2 (33). The reciprocal experiments were also performed and the double reciprocal plots of both data sets intersect in the second quadrant. A similar pattern of intersecting lines was also observed when the same experiments were repeated with the AcH4-21 peptide, a peptide that can be asymmetrically dimethylated in a partially processive fashion (31). These results are consistent with a sequential mechanism in which SAM and a peptide substrate bind to the enzyme to form a ternary complex prior to methyl transfer. Note that the intersecting line patterns rule out a ping-pong mechanism for which a parallel pattern of lines is diagnostic. Additionally, these results are inconsistent with a rapid equilibrium ordered mechanism because intersection on the ordinate would have been expected for one of the substrates. The kinetic parameters obtained from these studies are summarized in Table 2.
Table 2.
Steady-state kinetic parameters from initial velocity studies
| Varied Substrate | Fixed Substrate |
kcat (min−1) |
Ki(pep) (µM) |
K(pep) (µM) |
Ki(SAM) (µM) |
K(SAM) |
|---|---|---|---|---|---|---|
| SAM | AcH4-21a | 0.54 ± 0.01 | 20.2±7.3 | 8.67 ± 1.80 | 49.1 ± 11.4 | 21.1 ± 4.0 |
| SAM | AcH4-21 R3MMAb | 1.42 ± 0.02 | 4.01±3.08 | 2.92 ± 0.96 | 20.2 ± 13.4 | 14.7 ± 2.9 |
| AcH4-21 | SAMc | 0.56 ± 0.02 | 42.9 ± 18.5 | 32.5 ± 24.1 | 11.0 ± 9.84 | 8.35 ± 2.11 |
| AcH4-21 R3MMA | SAMd | 0.96 ± 0.01 | 36.1 ± 14.4 | 4.26 ± 3.71 | 24.83± 24.44 | 2.93 ± 0.66 |
2.5, 5, 25 µM
2.5, 5, 10, 25, 125 µM
1, 2.5, 7.5 µM
2.5, 5, 7.5, 10, 15 µM.
Product inhibition studies using SAM and the AcH4-21R3MMA peptide as substrates
To discriminate between the various subtypes of kinetic mechanisms described for two substrate-two product enzymes, product inhibition studies were performed. SAH and the AcH4-21R3ADMA peptide (Table 3), which cannot be further methylated by PRMT1 (31), were used as the product inhibitors for these experiments. The AcH4-21R3MMA peptide was used as the substrate because it is subject to only a single methylation event (31); thereby providing a baseline for interpreting the inhibition patterns obtained with substrates that can be processively methylated. The data from these experiments is summarized in Table 4.
Table 3.
Product and dead-end analogue inhibitors
| Inhibitor | Sequence/Structure |
|---|---|
| AcH4-21R3K | 1Ac-SGKGKGGKGLGKGGAKRHRKV |
| AcH4-21R3ADMA | 1Ac-SGR(Me)2GKGGKGLGKGGAKRHRKV |
| SAH | 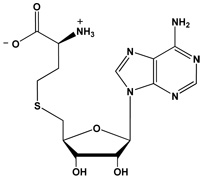 |
| Sinefungin | 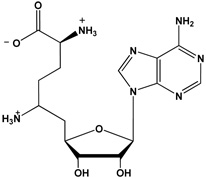 |
Table 4.
Summary of product inhibition results for AcH4-21 R3MMA
| Inhibitor | Varied Substrate | Fixed Substrate | Inhibition Pattern |
Kis (µM) |
Kii (µM) |
Equation |
|---|---|---|---|---|---|---|
| AcH4-21R3ADMA | AcH4-21 R3MMA | SAMa | Competitive | 178 ± 80 | (3) | |
| AcH4-21R3ADMA | SAM | AcH4-21 R3MMAb | Noncompetitive | 238 ± 32 | 238 ± 32 | (4) |
| AcH4-21R3ADMA | SAM | AcH4-21 R3MMAc | No Inhibition | |||
| SAH | AcH4-21 R3MMA | SAMa | Noncompetitive/ Mixed |
10.3 ± 3.3 | 23.3 ± 1.4 | (5) |
| SAH | SAM | AcH4-21 R3MMAb | Competitive | 0.96 ± 0.57 | (3) | |
| SAH | SAM | AcH4-21 R3MMAc | Competitive | 0.11 ± 0.03 | (3) |
[SAM] = 15 µM
[Peptide] = 10 µM
[Peptide] = 500 µM
The results of the product inhibition studies indicate that the AcH4-21R3ADMA peptide acts as a competitive inhibitor when the concentration of the AcH4-21R3MMA peptide is varied, suggesting that these two peptides bind to the same form(s) of the enzyme (Table 4). Interestingly, SAH also acted as a competitive inhibitor when SAM was the varied substrate and the AcH4-21R3MMA peptide was fixed at Km(pep), suggesting that these two compounds also compete for binding to the same form(s) of the enzyme. These results were particularly significant because they effectively narrowed the number of possible kinetic mechanisms to four, i.e.: i) the simple RER mechanism; ii) the RER mechanism with a dead-end EBQ complex; iii) the RER mechanism with dead-end EAP and EBQ complexes; and iv) the Theorell-Chance mechanism, a special case of the ordered bi bi reaction. Note that the latter three possibilities hold true for only two cases; i.e., when either the AcH4-21R3MMA and AcH4-21R3ADMA peptides represent substrate A and product Q and SAM and SAH represent substrate B and product P (Case 1); or when SAM and SAH represent substrate A and product Q; and when the AcH4-21R3MMA and AcH4-21R3ADMA peptides represent substrate B and product P (Case 2).
To begin to discriminate between these various possibilities, the product inhibition pattern afforded by SAH was then obtained at a saturating concentration of the substrate peptide and varied SAM. The observed competitive pattern of inhibition (Table 4) is inconsistent with Case 1 of the RER mechanism with a dead-end EBQ complex because by definition SAH cannot bind to EA (i.e. the E●AcH4-21R3MMA complex) to form an EAP complex (i.e. a E●AcH4-21R3MMA●SAH complex) and no inhibition is expected. Also inconsistent with this particular mechanism is the observation that SAH acts as a noncompetitive inhibitor when the AcH4-21R3MMA peptide is the varied substrate (Table 4) – a competitive pattern of inhibition is expected in a RER mechanism with a dead-end EBQ complex because SAH will compete with the AcH4-21R3MMA peptide for binding only to free enzyme. The fact that SAH is a noncompetitive inhibitor when the AcH4-21R3MMA peptide is the varied substrate also rules out the simple RER mechanism because according to this mechanism SAH would be expected to compete with the AcH4-21R3MMA peptide for binding only to the free enzyme. However note that both of these results are consistent with the RER mechanism with a dead-end EBQ complex if SAM and the AcH4-21R3MMA peptide represent substrates A and B, respectively; and the AcH4-21R3ADMA peptide and SAH represent products P and Q; respectively, i.e. Case 2.
To provide evidence against the latter possibility, the inhibition pattern for the AcH4-21R3ADMA peptide at varied SAM and subsaturating levels of the AcH4-21R3MMA peptide was determined. For a RER mechanism with a dead-end EBQ complex, a competitive pattern of inhibition is expected because by definition the AcH4-21R3ADMA peptide cannot bind to the E●SAM form of the enzyme to form the EAP complex. Thus, the observed noncompetitive pattern of inhibition rules out a RER mechanism with a dead-end EBQ complex. For the reasons discussed above; the observed pattern of inhibition also rules out the simple RER mechanism; thereby leaving only Cases 1 and 2 of both the Theorell-Chance mechanism and the RER mechanism with dead-end EAP and EBQ complexes as possible mechanisms.
The two possible cases of the RER mechanism with dead-end EAP and EBQ complexes can be distinguished by the pattern of inhibition obtained for the AcH4-21R3ADMA peptide at varied SAM and saturating levels of the AcH4-21R3MMA peptide. For Case 1, the AcH4-21R3ADMA peptide should not inhibit the reaction because the high concentrations of the AcH4-21R3MMA peptide will out-compete the AcH4-21R3ADMA peptide for binding to the free form of the enzyme; whereas for Case 2, the net result of AcH4-21R3ADMA binding to both the free enzyme and the E●SAM complex would be a noncompetitive pattern of inhibition. The fact that no inhibition is apparent under these conditions rules out Case 2 of the RER mechanism with dead-end EAP and EBQ complexes. Case 2 of the Theorell-Chance mechanism can also be ruled out because the order of product release, i.e. the release AcH4-21R3ADMA before the release of SAH, would not allow for the processive methylation of a substrate peptide.
Dead-end analogue inhibition studies using SAM and the AcH4-21R3MMA peptide as substrates
Because the above described product inhibition studies cannot distinguish between the Theorell-Chance mechanism and the RER mechanism with dead-end EAP and EBQ complexes (where AcH4-21R3MMA and AcH4-21R3ADMA peptides represent substrate A and product Q and SAM and SAH represent substrate B and product P), dead-end inhibitor studies were performed to differentiate between these two possibilities. Sinefungin and the AcH4-21R3K peptide were used as dead-end analogs in these studies because sinefungin is a SAM analogue that contains a primary amino group in place of the S-methyl group whereas the AcH4-21R3K peptide possesses a lysine residue in place of Arg3, i.e. the site of methylation in the AcH4-21R3MMA peptide, and is not processed by the enzyme (31). The sequence and structure of these compounds are depicted in Table 3.
The expected dead-end inhibition patterns for these two mechanisms are, with one exception, identical. The one difference being the pattern of inhibition obtained for sinefungin when the AcH4-21R3MMA peptide is the varied substrate. Under these conditions, uncompetitive inhibition is expected in a Theorell-Chance mechanism because sinefungin binds to the E●AcH4-21R3MMA complex whereas in a random mechanism noncompetitive inhibition is expected because this compound can bind to the free form of the enzyme as well as the E●AcH4-21R3MMA complex. The results of this particular experiment (Table 5) demonstrate that sinefungin acts as a noncompetitive inhibitor when AcH4-21R3MMA peptide is the varied substrate, consistent with a RER mechanism with dead-end EAP and EBQ complexes (Scheme 1) and ruling out the Theorell-Chance mechanism (Case 1).
Table 5.
Summary of dead-end analog inhibition results for AcH4-21R3MMA
| Inhibitor | Varied Substrate | Fixed Substrate | Inhibition Pattern |
Kis (µM) |
Kii (µM) |
Equation |
|---|---|---|---|---|---|---|
| Sinefungin | AcH4-21R3MMA | SAMa | Noncompetitive/ Mixed |
0.91 ± 0.39 | 0.71 ± 0.06 | (5) |
| Sinefungin | SAM | AcH4-21R3MMAb | Competitive | 0.23 ± 0.09 | (3) | |
| Sinefungin | SAM | AcH4-21R3MMAc | Competitive | 0.16 ± 0.07 | (3) | |
| AcH4-21 R3K | AcH4-21R3MMA | SAMa | Competitive | 44.2 ± 19.4 | (3) | |
| AcH4-21 R3K | SAM | AcH4-21R3MMAb | Noncompetitive | 37.0 ± 14.4 | 37.0 ± 14.4 | (4) |
| AcH4-21 R3K | SAM | AcH4-21R3MMAc | Noncompetitive | 375 ± 36 | 375 ± 36 | (4) |
[SAM] = 15 µM
[Peptide] = 10 µM
[Peptide] = 500 µM
Scheme 1.

Proposed scheme for the PRMT1-catalyzed monomethylation reaction
Note that the remaining dead-end inhibition patterns are consistent with a RER mechanism with dead-end EAP and EBQ complexes. Also note that the noncompetitive pattern of inhibition obtained for the AcH4-21R3K peptide when SAM is the varied substrate further rules out the intellectually unsatisfying Theorell-Chance mechanism (Case 2) where the order of product release, i.e. the release AcH4-21R3ADMA before the release of SAH, precludes the processive methylation of a substrate peptide.
Inhibition studies using SAM and the AcH4-21 peptide as substrates
Having established that PRMT1 utilizes a RER mechanism with dead-end EAP and EBQ complexes for substrates that can be exclusively monomethylated, we then determined the product and dead-end inhibition patterns for a substrate that is known to be asymmetrically dimethylated in a partially processive fashion, i.e. the AcH4-21 peptide (31). These data are summarized in Tables 6 and 7. The results of these studies are qualitatively identical to those obtained for the AcH4-21R3MMA peptide. Thus, regardless of the methylation status of the peptide, the kinetic mechanism utilized by PRMT1 is the same.
Table 6.
Summary of product inhibition results for AcH4-21
| Inhibitor | Varied Substrate | Fixed Substrate | Inhibition Pattern |
Kis (µM) |
Kii (µM) |
Equation |
|---|---|---|---|---|---|---|
| AcH4-21R3ADMA | AcH4-21 | SAMa | Competitive | 39.7 ± 12.4 | (3) | |
| AcH4-21R3ADMA | SAM | AcH4-21b | Noncompetitive/ Mixed |
23.1 ± 6.8 | 102 ± 36 | (5) |
| AcH4-21R3ADMA | SAM | AcH4-21c | No Inhibition | |||
| SAH | AcH4-21 | SAMa | Noncompetitive | 19.7 ± 2.1 | 19.7 ± 2.1 | (4) |
| SAH | SAM | AcH4-21b | Competitive | 0.11 ± 0.03 | (3) | |
| SAH | SAM | AcH4-21c | Competitive | 6.5 ± 1.6 | (3) |
[SAM] = 15 µM
[Peptide] = 10 µM
[Peptide] = 500 µM
Table 7.
Summary of dead-end analog inhibition results for AcH4-21
| Inhibitor | Varied Substrate | Fixed Substrate | Inhibition Pattern |
Kis (µM) |
Kii (µM) |
Equation |
|---|---|---|---|---|---|---|
| Sinefungin | AcH4-21 | SAMa | Noncompetitive/ Mixed |
1.06 ± 0.77 | 0.47 ± 0.19 | (5) |
| Sinefungin | SAM | AcH4-21b | Competitive | 0.16 ± 0.15 | (3) | |
| Sinefungin | SAM | AcH4-21c | Competitive | 0.54 ± 0.17 | (3) | |
| AcH4-21 R3K | AcH4-21 | SAMa | Competitive | 18.3 ± 16.3 | (3) | |
| AcH4-21 R3K | SAM | AcH4-21b | Noncompetitive/ Mixed |
65.9 ± 11.5 | 104 ± 33 | (5) |
| AcH4-21 R3K | SAM | AcH4-21c | Noncompetitive | 264 ± 51 | 264 ± 51 | (4) |
[SAM] = 15 µM
[Peptide] = 10 µM
[Peptide] = 500 µM
DISCUSSION
PRMT1 appears to play important roles in a variety of cellular processes, including nuclear-cytoplasmic protein shuttling, RNA metabolism, and most prominently transcriptional regulation (19–22). However, the molecular details by which PRMT1 activity contributes to the regulation of these diverse processes is incompletely understood. To address this deficiency, we are focused on the development of PRMT1-selective inhibitors (31,34) that can be used to provide a more thorough understanding of in vivo PRMT1 function. We are additionally, focused on this goal because PRMT1 activity appears to be dysregulated in heart disease, and possibly cancer, suggesting it as a novel therapeutic target. To aid the development of such inhibitors, we have previously identified elements present within PRMT1 substrates that are critical for substrate recognition and further demonstrated that PRMT1 catalyzes the formation of asymmetrically dimethylated residues in a partially processive fashion (31). To further aid the design and synthesis of PRMT1 selective inhibitors and gain insights into how PRMT1 can catalyze the sequential methylation of its substrates to generate an asymmetrically dimethylated arginine residue, we determined the kinetic mechanism of hPRMT1.
To begin to characterize the kinetic mechanism of PRMT1, the initial rates were obtained at a fixed concentration of SAM for substrates that are known to be exclusively monomethylated, i.e. the AcH4-21R3MMA peptide, and asymmetrically dimethylated in a partially processive fashion, i.e. the AcH4-21 peptide (31). The sequences of these peptides are based on the N-terminal 21 amino acids of histone H4, a PRMT1 substrate; and are known to be modified with comparable kinetics to the parent protein (31). Additionally, these peptides are methylated at Arg3, an in vivo site of PRMT1-directed methylation, in a regiospecific manner (31). The reciprocal experiments, in which SAM was varied at fixed concentrations of the AcH4-21 R3MMA and AcH4-21 peptides, were also performed.
In all cases, double reciprocal plots of the rate data intersect in the second quadrant, consistent with a sequential mechanism in which SAM and a peptide (or protein) substrate bind to the enzyme to form a ternary complex that would permit the direct transfer of the methyl group of SAM to a ω-nitrogen of a substrate arginine residue. While these results are gratifyingly consistent with non-enzymatic model reactions of SAM-dependent methyl transfer reactions (35), it is interesting to note that a parallel pattern of lines has been observed for a number of Lysine Methyltransferases (KMTs) that catalyze the partially processive methylation of target lysine residues (36). In fact, Dirk et al suggest that the observation of such a pattern is diagnostic for KMTs that methylate their substrates in a partially processive manner (36). The fact that we do not observe a parallel line pattern with PRMT1, suggests that this phenomenon is not universally applicable to all protein methyltransferases.
Having established that PRMT1 utilizes a sequential ternary complex mechanism, product and dead end analogue inhibition experiments were performed. The results of these experiments indicate that PRMT1 utilizes a RER mechanism with dead-end EAP and EBQ complexes (Scheme 1). This was the case regardless of whether the substrate could be exclusively monomethylated or asymmetrically dimethylated in a partially processive fashion. From a mechanistic perspective, the proposed RER mechanism with dead-end EAP and EBQ complexes is gratifyingly consistent with the partially processive nature of the reaction. This is the case because the dissociation of SAH prior to the release of a monomethylated protein would allow for the binding of a second molecule of SAM; and thereby permit the transfer of a second methyl group to the same nitrogen to generate an asymmetrically dimethylated arginine residue (Scheme 2). Conversely, the dissociation of the monomethylated peptide prior to the release of SAH would explain why this product is generated in appreciable amounts. Although our results are consistent with the partially processive nature of the PRMT1 catalyzed reaction observed in vitro, we note that in vivo the apparent processivity of the reaction could be influenced, both negatively and positively, by PRMT1 interacting proteins. Further studies will be required to assess the effect of interacting proteins on the kinetic mechanism of this enzyme.
Scheme 2.

Proposed scheme for the PRMT1-catalyzed dimethylation reaction, accounting for the partially processive nature of the PRMT1-catalyzed reaction.
It should be noted that the reactions catalyzed by PRMT4/CARM1 (Coactivator Associated Arginine Methyltransferase 1) and PRMT6 have been suggested to proceed via an ordered sequential mechanism in which SAM binds the enzyme first and SAH is the last product to dissociate from the enzyme (37, 38). These assignments are based on product inhibition studies with PRMT6 (37) and the observation that the active site of PRMT4/CARM1 undergoes a conformational change upon binding SAH that could facilitate the binding of a protein substrate (38). Although these conclusions differ from our own, it is well established that different isozymes can utilize different kinetic mechanisms. On the other hand, it should be noted that with respect to PRMT6, the difference may be due to the fact that a relatively poor substrate (Km = 500 µM) was used in the initial velocity and product inhibition studies as the lack of affinity for a particular substrate can make a random mechanism appear ordered, e.g. creatine kinase (39, 40).
In summary, our results indicate that PRMT1 utilizes a RER mechanism with dead-end EAP and EBQ complexes. These results are both consistent with and explain the partially processive nature of the PRMT1-catalyzed reaction. The fact that PRMT1 utilizes a ternary complex mechanism also demonstrates the feasibility of linking a substrate peptide to a portion of SAM (or a SAM analog) to develop a potent bisubstrate analog inhibitor that selectively inhibits PRMT1.
ABBREVIATIONS
- DTT
dithiothreitol
- HEPES
N-(2-hydroxyethyl)piperazine-N’-(2-ethanesulfonic acid)
- PRMT
protein arginine methyltransferase
- SAM
S-adenosyl-L-methionine
- CARM1
coactivator associated methyltransferase 1
- SAH
S-adenosyl-L-homocysteine
- MMA
monomethylarginine
- ADMA
asymmetric di-methylarginine
- SDMA
symmetric di-methylarginine
- EDTA
ethylenediamine tetraacetic acid
- MALDI
matrix assisted laser desorption/ionization
- RER
rapid equilibrium random
Footnotes
This work was supported in part by the University Of South Carolina Research Foundation (P.R.T) and from a new investigator grant from the American Heart Association. T.C.O was supported by an NSF GK-12 Fellowship and O.O. by an NIH-PREP fellowship (GM066526).
REFERENCES
- 1.Hong H, Kao C, Jeng MH, Eble JN, Koch MO, Gardner TA, Zhang S, Li L, Pan CX, Hu Z, MacLennan GT, Cheng L. Aberrant expression of CARM1, a transcriptional coactivator of androgen receptor, in the development of prostate carcinoma and androgen-independent status. Cancer. 2004;101:83–89. doi: 10.1002/cncr.20327. [DOI] [PubMed] [Google Scholar]
- 2.Majumder S, Liu Y, Ford OH, Mohler JL, 3rd, Whang YE. Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability. Prostate. 2006;66:1292–1301. doi: 10.1002/pros.20438. [DOI] [PubMed] [Google Scholar]
- 3.Boger RH, Sydow K, Borlak J, Thum T, Lenzen H, Schubert B, Tsikas D, Bode-Boger SM. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: involvement of S-adenosylmethionine-dependent methyltransferases. Circ Res. 2000;87:99–105. doi: 10.1161/01.res.87.2.99. [DOI] [PubMed] [Google Scholar]
- 4.Tran CT, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler. 2003 Suppl 4:33–40. doi: 10.1016/s1567-5688(03)00032-1. [DOI] [PubMed] [Google Scholar]
- 5.Vallance P, Leiper J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler Thromb Vasc Biol. 2004;24:1023–1030. doi: 10.1161/01.ATV.0000128897.54893.26. [DOI] [PubMed] [Google Scholar]
- 6.Leiper J, Murray-Rust J, McDonald N, Vallance P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc Natl Acad Sci U S A. 2002;99:13527–13532. doi: 10.1073/pnas.212269799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572–575. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- 8.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O'Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13:198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Niroomand F, Liu Z, Zankl A, Katus HA, Jahn L, Tiefenbacher CP. Expression of nitric oxide related enzymes in coronary heart disease. Basic Res Cardiol. 2006;101:346–353. doi: 10.1007/s00395-006-0592-5. [DOI] [PubMed] [Google Scholar]
- 10.Koh SS, Chen D, Lee YH, Stallcup MR. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem. 2001;276:1089–1098. doi: 10.1074/jbc.M004228200. [DOI] [PubMed] [Google Scholar]
- 11.Strahl BD, Briggs SD, Brame CJ, Caldwell JA, Koh SS, Ma H, Cook RG, Shabanowitz J, Hunt DF, Stallcup MR, Allis CD. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr Biol. 2001;11:996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Huang Z-Q, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, Briggs SD, Allis CD, Wong J, Tempst P, Zhang Y. Methylation of Histone H4 at Arginine 3 Facilitating Transcriptional Activation by Nuclear Hormone Receptor. Science. 2001;293:853–857. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- 13.Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 14.Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature. 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- 15.Lee SK, Anzick SL, Choi JE, Bubendorf L, Guan XY, Jung YK, Kallioniemi OP, Kononen J, Trent JM, Azorsa D, Jhun BH, Cheong JH, Lee YC, Meltzer PS, Lee JW. A nuclear factor, ASC-2, as a cancer-amplified transcriptional coactivator essential for ligand-dependent transactivation by nuclear receptors in vivo. J Biol Chem. 1999;274:34283–34293. doi: 10.1074/jbc.274.48.34283. [DOI] [PubMed] [Google Scholar]
- 16.Zhu Y, Qi C, Jain S, Le Beau MM, Espinosa R, 3rd, Atkins GB, Lazar MA, Yeldandi AV, Rao MS, Reddy JK. Amplification and overexpression of peroxisome proliferator-activated receptor binding protein (PBP/PPARBP) gene in breast cancer. Proc Natl Acad Sci U S A. 1999;96:10848–10853. doi: 10.1073/pnas.96.19.10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin WJ, Gary JD, Yang MC, Clarke S, Herschman HR. The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J Biol Chem. 1996;271:15034–15044. doi: 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- 18.Abramovich C, Yakobson B, Chebath J, Revel M. A protein-arginine methyltransferase binds to the intracytoplasmic domain of the IFNAR1 chain in the type I interferon receptor. Embo J. 1997;16:260–266. doi: 10.1093/emboj/16.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bachand F. Protein arginine methyltransferases: from unicellular eukaryotes to humans. Eukaryot Cell. 2007;6:889–898. doi: 10.1128/EC.00099-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell. 2005;18:263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S. Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther. 2007;113:50–87. doi: 10.1016/j.pharmthera.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 22.Bedford MT. Arginine methylation at a glance. J Cell Sci. 2007;120:4243–4246. doi: 10.1242/jcs.019885. [DOI] [PubMed] [Google Scholar]
- 23.An W, Kim J, Roeder RG. Ordered Cooperative Functions of PRMT1, p300, and CARM1 in Transcriptional Activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Rezai-Zadeh N, Zhang X, Namour F, Fejer G, Wen YD, Yao YL, Gyory I, Wright K, Seto E. Targeted recruitment of a histone H4-specific methyltransferase by the transcription factor YY1. Genes Dev. 2003;17:1019–1029. doi: 10.1101/gad.1068003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hassa PO, Covic M, Bedford MT, Hottiger MO. Protein arginine methyltransferase 1 coactivates NF-kappaB-dependent gene expression synergistically with CARM1 and PARP1. J Mol Biol. 2008;377:668–678. doi: 10.1016/j.jmb.2008.01.044. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure. 2003;11:509–520. doi: 10.1016/s0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Zhou L, Cheng X. Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. Embo J. 2000;19:3509–3519. doi: 10.1093/emboj/19.14.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiss VH, McBride AE, Soriano MA, Filman DJ, Silver PA, Hogle JM. The structure and oligomerization of the yeast arginine methyltransferase, Hmt1. Nat Struct Biol. 2000;7:1165–1171. doi: 10.1038/82028. [DOI] [PubMed] [Google Scholar]
- 29.Cook JR, Lee JH, Yang ZH, Krause CD, Herth N, Hoffmann R, Pestka S. FBXO11/PRMT9, a new protein arginine methyltransferase, symmetrically dimethylates arginine residues. Biochem Biophys Res Commun. 2006;342:472–481. doi: 10.1016/j.bbrc.2006.01.167. [DOI] [PubMed] [Google Scholar]
- 30.Fielenbach N, Guardavaccaro D, Neubert K, Chan T, Li D, Feng Q, Hutter H, Pagano M, Antebi A. DRE-1: an evolutionarily conserved F box protein that regulates C. elegans developmental age. Dev Cell. 2007;12:443–455. doi: 10.1016/j.devcel.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 31.Osborne TC, Obianyo O, Zhang X, Cheng X, Thompson PR. Protein arginine methyltransferase 1: positively charged residues in substrate peptides distal to the site of methylation are important for substrate binding and catalysis. Biochemistry. 2007;46:13370–13381. doi: 10.1021/bi701558t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leatherbarrow RJ. Grafit Ver 5.0. Staines, UK: Erathicus Software; 2004. [Google Scholar]
- 33.Segel IH. Enzyme kinetics: Behavior and analysis of rapid equilibrium and steady-state enzyme systems. New York: Wiley-Interscience; 1975. [Google Scholar]
- 34.Osborne T, Roska RL, Rajski SR, Thompson PR. In situ generation of a bisubstrate analogue for protein arginine methyltransferase 1. J Am Chem Soc. 2008;130:4574–4575. doi: 10.1021/ja077104v. [DOI] [PubMed] [Google Scholar]
- 35.Coward JK. Chemical mechanisms of methyl transfer reactions: comparison of methylases with nonenzymic 'model reactions'. In: Salvatore F, Borek E, Zappia V, Williams-Ashman HG, editors. The biochemistry of adenosylmethionine. New York, NY: Columbia University Press; pp. 127–144. [Google Scholar]
- 36.Dirk LM, Flynn EM, Dietzel K, Couture JF, Trievel RC, Houtz RL. Kinetic manifestation of processivity during multiple methylations catalyzed by SET domain protein methyltransferases. Biochemistry. 2007;46:3905–3915. doi: 10.1021/bi6023644. [DOI] [PubMed] [Google Scholar]
- 37.Lakowski TM, Frankel A. A kinetic study of human protein arginine N-methyltransferase 6 reveals a distributive mechanism. J Biol Chem. 2008;283:10015–10025. doi: 10.1074/jbc.M710176200. [DOI] [PubMed] [Google Scholar]
- 38.Yue WW, Hassler M, Roe SM, Thompson-Vale V, Pearl LH. Insights into histone code syntax from structural and biochemical studies of CARM1 methyltransferase. EMBO J. 2007;26:4402–4412. doi: 10.1038/sj.emboj.7601856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schimerlik MI, Cleland WW. Inhibition of creatine kinase by chromium nucleotides. J Biol Chem. 1973;248:8418–8423. [PubMed] [Google Scholar]
- 40.Cook PF, Kenyon GL, Cleland WW. Use of pH studies to elucidate the catalytic mechanism of rabbit muscle creatine kinase. Biochemistry. 1981;20:1204–1210. doi: 10.1021/bi00508a023. [DOI] [PubMed] [Google Scholar]