

Abstract
Gellan Gum (GG) has been recently proposed for tissue engineering applications. GG hydrogels are produced by physical crosslinking methods induced by temperature variation or by the presence of divalent cations. However, physical crosslinking methods may yield hydrogels that become weaker in physiological conditions due to the exchange of divalent cations by monovalent ones. Hence, this work presents a new class of GG hydrogels crosslinkable by both physical and chemical mechanisms. Methacrylate groups were incorporated in the GG chain, leading to the production of a methacrylated gellan gum (MeGG) hydrogel with highly tunable physical and mechanical properties. The chemical modification was confirmed by proton nuclear magnetic resonance (1H-NMR) and Fourier transform infrared spectroscopy (FTIR-ATR). The mechanical properties of the developed hydrogel networks, with Young’s modulus values between 0.15 and 148 kPa, showed to be tuned by the different crosslinking mechanisms used. The in vitro swelling kinetics and hydrolytic degradation rate was dependent on the crosslinking mechanisms used to form the hydrogels. Three-dimensional (3D) encapsulation of NIH-3T3 fibroblast cells in MeGG networks demonstrated in vitro biocompatibility confirmed by high cell survival. Given the highly tunable mechanical and degradation properties of MeGG, it may be applicable for a wide range of tissue engineering approaches.
1. Introduction
Hydrogels have received much attention in the field of tissue engineering, mainly due to their ability to be used as vehicles for drug delivery and as cell carriers [1-5]. Hydrogels are similar to natural extracellular matrix (ECM) molecules in that they have the ability to retain great quantities of water. One of the main advantages of hydrogels is their capacity to be formed in situ through a minimally invasive procedure. Both physical and chemical methods can be applied for the development of hydrogel networks [2, 6]. In situ forming physical hydrogels are usually spontaneously formed by weak secondary forces such as hydrogen bonding, van der Waals interactions and ionic bonding [6-8]. After injection, they instantaneously change from sol to gel state under physiological temperature, pH and ionic conditions. On the other hand, in situ forming chemical hydrogels can have their mechanical strength, integrity and swelling properties fine tuned by the degree of chemical crosslinking. These hydrogels have mainly been formed by the photo-irradiation of the vinyl groups of the injected pre-polymer [9, 10]. Physical and chemical crosslinking methods have been studied for several natural-origin hydrogels, such as hyaluronic acid (HA) [11-13], gelatin [14, 15] and alginate [16, 17]. Recently, the FDA approved Gellan Gum (GG) has been receiving particular attention for tissue engineering applications, namely cartilage regeneration [18-22], mainly due to its good mechanical properties and promising results as an ophthalmic drug vehicle [23, 24]. GG is an anionic microbial polysaccharide composed of a tetrasaccharide repeating unit of two β-D-glucose, one β-D-glucuronic acid and one α-L-rhamnose [25]. Upon temperature decrease, a random coil-helix transition occurs with further aggregation of the helices leading to the formation of junction zones [26]. The sol-gel transition of GG is ionotropic, as in alginate. Therefore, the presence of cations is necessary for the formation of a stable hydrogel. However, the gelation is greatly affected by the chemical nature of the cations present, being stronger with divalent ones. The rheological and physical properties of GG have been widely investigated [27-30]. Recently, Masakuni et al. proposed a gelation mechanism of deacylated GG in aqueous solution, with and without Ca2+ cations [31]. However, these physically crosslinked hydrogels tend to lose their stability in vivo after implantation due to the exchange of divalent cations with monovalent ones that are present in higher concentrations in physiological environment. In order to surpass this disadvantage, some chemical modifications to the GG backbone have been proposed. For instance, a chemical scissoring process has been used to adjust the molecular weight of GG [20]. Due to the properties of these formed GG hydrogels, they have been mainly proposed for cartilage tissue applications [20].
Although hydrogels closely mimic ECM and have great potential for tissue engineering applications, the major drawback is their weak mechanical properties. Thus, apart from in vivo stability, other important attributes of hydrogels for tissue engineering are their mechanical properties and degradation rate. Different tissues have different tissue regeneration rates and different mechanical properties. For instance, stiffness of solid tissues in the body can range from 1 kPa for the liver until 100 kPa for the collagenous bone [32]. Therefore, the materials used within a tissue engineering approach must have their degradation and mechanical properties finely tuned to match those of the native tissues.
In this present work, we present a simple method to functionalize GG with double bonds. We further used the photocrosslinkable methacrylated GG to prepare hydrogels using combination of physical and chemical crosslinking methods. It was hypothesized that physical and mechanical properties of hydrogels can be tuned by the crosslinking mechanisms applied. The described photocrosslinkable hydrogels can be used in various applications of tissue engineering due to their wide range of mechanical properties.
2. Experimental Section
2.1. Synthesis of Methacrylated Gellan Gum
Methacrylated Gellan Gum (MeGG) was synthesized by reacting Gellan Gum (GG, Gelrite®, Sigma, Mw=1.000.000) with methacrylic anhydride (MA, Sigma). Briefly, 1 g of GG was dissolved in 100 mL of deionized water at 90 °C for 20-30 minutes, as described elsewhere [22]. To this solution, either 2 or 8 mL of MA was added at 50 °C, in order to synthesize MeGG with low (Low-MeGG) or high (High-MeGG) degrees of methacrylation, respectively. The reaction was continued for 6 hours. Periodically, pH (8.0) was adjusted with 5.0 M NaOH solution. The modified MeGG solution was purified by dialysis (Fisher Scientific, membrane with molecular weight cutoff of 11-14 kD, USA) for at least 3 days against distilled water to remove the excess of MA. Purified MeGG was obtained by lyophilization and stored at a dry place protected from light.
2.2. Characterization of Methacrylated Gellan Gum
The chemical modification to GG was assessed by proton nuclear magnetic resonance (1H-NMR) spectroscopy. 1H-NMR spectra were recorded with a Varian Inova 500 NMR equipped with a variable temperature system. Lyophilized materials were dissolved in D2O at a concentration of 10 mg/mL and at temperature of 50 °C. Chemical shifts were referred to the methyl group of rhamnose as an internal standard, which is at δ 1.45 ppm [33]. The degree of substitution (DS, fraction of modified hydroxyl groups per repeating unit) was determined by the relative integration of the double bond proton peak (IDB) of the methacrylate groups to the methyl protons of the internal standard (ICH3rham), according to Eq (1), as described elsewhere [33]. nHDB and nHCH3rham corresponds to the number of protons in the double bond and the methyl group of rhamnose, respectively. nOHmonomer corresponds to the number of reactive −OH sites in the GG structure.
| (1) |
The chemistry of the modified materials was further analyzed by Fourier transform infrared spectroscopy with attenuated total reflection (FTIR-ATR). The infrared spectra were recorded on a spectrophotometer Bruker Alpha FT-IR with a resolution of 2 cm-1. The results are presented as the average of 32 scans.
2.3. Preparation of Hydrogels
The lyophilized Low-MeGG and High-MeGG were dissolved at 0.5, 1 and 2% (w/v) in deionized water under constant stirring at 50 °C for 10 minutes. Plain GG was dissolved at the same concentrations, as described elsewhere [22]. Briefly, GG was dissolved in deionized water under constant stirring. The solution was progressively heated to 90°C, under which complete and homogeneous dispersion of the material was obtained after 20-30 minutes. Afterwards, calcium chloride (CaCl2, Sigma, USA) and/or 0.5% (w/v) 2-hydroxy-1-[4-(2-hydroxyethoxy)phenyl]-2-methyl-1-propanone (Irgacure 2959, Ciba Specialty Chemicals) were added to the dissolved materials to fabricate hydrogels with different crosslinking mechanisms. The final concentration of CaCl2 solution was 0.08 mg/mL, unless mentioned otherwise, yielding a ratio of calcium equivalent/GG carboxylate moieties of 0.052 for 1% (w/v) solution of GG. After dissolution, the temperature of Low-MeGG, High-MeGG and plain GG was progressively decreased to 45 °C. GG and physically crosslinked MeGG hydrogels were produced by casting the solutions into molds and allowing the temperature to cool to room temperature. Chemically crosslinked hydrogels were obtained by exposing to light (320-500 nm, 7.14 mW/cm2, EXFO OmniCure S2000) for 60 sec. For all described experiments, polymer disks (1 mm thick, 4 mm in diameter) were punched from hydrogel slabs, unless stated otherwise.
2.4. Characterization of hydrogels
2.4.1. Swelling kinetics
To study the swelling kinetics of the developed hydrogels, three solutions with different ionic content were used: distilled water (no ions); phosphate buffered saline (PBS, Gibco, with monovalent ions); and media (Dulbecco’s Modified Eagle’s medium, DMEM, Gibco, with mono and divalent ions). Hydrogel samples at 1% (w/v) were immersed in 2 mL of each solution at 37 °C, under mild shaking. At different time points, the hydrogels (n=3) were removed from the solutions and gel surfaces were quickly blotted on a filter paper. Their wet weight was measured (wt) and compared to the initial wet weight (w0). The swelling ratio (Sk) was defined according to Eq (2).
| (2) |
2.4.2. Mechanical properties
To assess the effect of the degree of methacrylation, crosslinking mechanisms and polymer concentration on the mechanical properties of the developed hydrogels, compression tests were performed on an Instron 5542 mechanical tester. Freshly prepared GG and MeGG hydrogels (1 mm thick, 8 mm in diameter, n=3) were compressed in the direction normal to the circular face of the cylindrical samples at a rate of 0.2 mm per minute until failure of the hydrogel. The Young’s modulus was defined as the slope of the linear region of the stress-strain curve in the 5-15% of the strain range. Ultimate stress and ultimate strain values were taken as the point where failure of the hydrogel occurred.
2.4.3. In vitro degradation
The effect of the degree of methacrylation, crosslinking mechanisms and polymer concentration over the degradation profile of MeGG hydrogels was studied. Weighed hydrogels (n=3) were hydrolytically degraded in a 0.1 mM NaOH solution at 37 °C for 24 hours on a shaker. The samples were removed, freeze dried and weighed again to determine mass loss.
2.4.4. In vitro cell encapsulation
The effect of methacrylation and photocrosslinking on cell viability was assessed by encapsulating NIH-3T3 fibroblasts. The cells were cultured in DMEM (Sigma) supplemented with 10% of heat-inactivated fetal bovine serum (FBS, Gibco) and 1% penicillin-streptomycin (Gibco) at 37 °C, in a humified atmosphere with 5 % of CO2. A cell suspension of NIH-3T3 (107 cells/mL) was prepared by trypsinization (trypsin/EDTA solution, Gibco) and incorporated into the GG, Low-MeGG and High-MeGG polymer solutions prepared as described previously and stabilized at 40 °C. After resuspension, the cell/polymer mixture was casted into molds. GG hydrogels with encapsulated cells were placed at room temperature for 1-2 minutes to form a solid gel. MeGG cell laden hydrogels were exposed to light (7.14 mW/cm2) for 60 sec. Cell viability of cells encapsulated in the hydrogels under study was characterized 1h after preparation and after 1, 3 and 7 days of culture, by incubating cells with a Live/Dead (Invitrogen) assay (calcein AM/ethidium homodimer-1 in DPBS) for 20 min. Tissue culture polystyrene (TCPS) was used as a control.
2.5. Statistical Analysis
All the data were subjected to statistical analysis and were reported as a mean ± standard deviation. Statistical differences (***p<0.0001, **p<0.001, *p<0.01) were determined using Student’s paired t-test between two groups and two-way ANOVA followed by Bonferroni post-hoc test for multiple comparisons in swelling and mechanical data.
3. Results and Discussion
3.1. MeGG synthesis and characterization
GG hydrogels have been recently used in tissue engineering applications [18]. However, these hydrogels are produced only by means of physical crosslinking mechanisms. This crosslinking mechanisms yield to hydrogels that become weaker in physiological conditions due to the exchange of divalent cations by monovalent ones. To surpass this disadvantage we synthesized a new class of GG hydrogels crosslinkable by both physical and chemical mechanisms. MeGG was prepared by the methacrylation of GG polymer (Fig. 1A). The spectra of GG obtained at 50 °C (Fig. 1B) showed the presence of four characteristic peaks that correspond to -CH of rhamnose (δ 5.29 ppm), -CH of glucuronic acid (δ 5.11 ppm), -CH of glucose (δ 4.88 ppm) and -CH3 of rhamnose (δ 1.45 ppm). 1H-NMR spectroscopy confirmed the methacrylation of GG by the appearance of distinctive peaks in the double bond region (δ 5.50-7.00 ppm) and a sharp peak that corresponds to the -CH3 of the methacrylate groups (δ 2.09 ppm) on the modified GG spectra. Fig. 1C and Fig. 1D show the 1H-NMR spectra of methacrylated GG with low and high degrees of methacrylation, respectively. Degree of methacrylation for Low-MeGG and High-MeGG was calculated by the ratio of average intensity of the methyl proton peaks of the methacrylate groups over the average intensity of the methyl groups of the rhamnose. For the Low-MeGG (Fig. 1C), for each repeating unit on the polymer chain there is 0.15 methacrylate groups covalently linked to the polymer. On the other hand, 1.24 methacrylate groups were found in each repeating unit for the High-MeGG of methacrylation material (Fig. 1D). With the integral of the double bond peaks of the methacrylate group protons and the methyl protons of rhamnose, it is possible to approximate a percentage of degree of substitution (DS), as mentioned in Eq (1). Therefore, the average DS increased from 1.2% (Low-MeGG) to 11.25% (High-MeGG), when the volume of methacrylic anhydride added to the reaction was quadruplicated.
Fig. 1.

(A) Schematic illustration of the synthesis of MeGG. 1H-NMR spectra (T=50 °C) of (B) GG, (C) Low-MeGG and (D) High-MeGG recorder in D2O. Methyl group of the MA (ii) was located at δ 2.09 ppm and vinyl groups of the MA (iii) were identified around δ 5.5-7 ppm.
The methacrylation of GG was also confirmed by FTIR-ATR by the appearance of the carbon double bond peak at 1640 cm-1, known to be present in methacrylate groups but not in GG chains (Fig. 2). Moreover, the characteristic C=O peak of the ester bond appeared around 1770-1680 cm-1 and increased in intensity with increasing degree of methacrylation.
Fig. 2.

FTIR-ATR spectra of GG, Low-MeGG and High-MeGG. The shoulder appearing around 1640 cm-1 corresponds to the C=C bond of the MA and is present on both Low-MeGG and High-MeGG. The absorption band present around 1770-1680 cm-1 corresponds to the C=O bond of the ester introduced in the chain.
3.2. Fabrication of MeGG hydrogels
GG is capable of physical gelation. As the temperature of a solution of the GG in water is decreased, GG chains undergo a conformational thermo-reversible change from random coils to double helices [26]. Then, the aggregation of the double helical elements occurs to form a three-dimensional (3D) network by hydrogen bonding with water. The gelation of GG is strongly affected by the presence of specific cations, namely Na+, K+, Ca2+ and Mg2+ although divalent cations promote a more efficient gelation than monovalent cations [29]. It was observed that after the methacrylation, Low-MeGG was still able to form a hydrogel in response to temperature decrease and in the presence of cations. Besides these physical crosslinking procedures, MeGG was also chemically crosslinked by UV exposure. Fig. 3 represents a schematics of the crosslinking mechanisms used in this study to produce each hydrogel: P for physical crosslinking by addition of Ca2+; C for chemical crosslinking by UV exposure; and PC when samples were both physically and chemically crosslinked by addition of Ca2+ ions and by UV exposure. The physical crosslinking by temperature decrease was present for all samples.
Fig. 3.

Possible crosslinking mechanisms for GG, Low-MeGG and High-MeGG hydrogels. MeGG schematically represents both degrees of methacrylations. For all the materials, the physical crosslinking by temperature decrease was used. Upon temperature decrease, GG and MeGG chains randomly transit from a coil to a helix form. Physical crosslinking occurs when cations are present and a more stable hydrogel is formed through ionic bonding. Chemical crosslinking occurs if hydrogels are exposed to UV, allowing the double bonds of the methacrylate groups to react with each other. Physical and chemical crosslinking will occur when these mechanisms are combined.
3.3. Swelling kinetics of MeGG to ionic solutions
The swelling kinetics of MeGG hydrogels in vitro was assessed by immersing the hydrogels in solutions with different amounts and types of cations (Fig. 4). Crosslinked hydrogels were prepared with different types of crosslinking by combining the presence of cations and the exposure to UV. The difference in their wet weight over time reflects changes in their chemical and physical structure. Samples of plain GG were disintegrated after 30 min. Distilled water has no ions. Therefore, the less crosslinked hydrogel networks quickly uptake water, losing their integrity over time. On the other hand, modified hydrogels with the least crosslinked network (Low-MeGG-C) presented the highest swelling of almost 750% after 30 min of immersion in water (Fig. 4A) (***p<0.0001). The High-MeGG hydrogels have more hydrophobic methacrylate groups in the chain. This, combined with a more crosslinked network due to photocrosslinking, led to a significantly lower water uptake for these hydrogels (***p<0.0001).
Fig. 4.

Swelling kinetics of GG and MeGG hydrogels in solutions with different ionic content: (A) water (no ions), (B) PBS (monovalent ions) and (C) DMEM with 10% FBS (mono and divalent ions). GG was not stable in water and dissolves in 30 min. Hence, swelling could not be carried out. (D) Confirmation of the ionic nature of the hydrogel shrinking behavior when immersed in PBS. High-MeGG hydrogels were used has an example. Samples were immersed in water for 30 min, then placed in a PBS solution for 20 hours and again immersed in water for 30 min. The weight and size of the samples varied with the presence or absence of ions in the solution.
To study the effect of ionic solutions on MeGG and GG hydrogel swelling kinetics, hydrogels were immersed in both PBS (Fig. 4B) and DMEM (Fig. 4C) for 28 and 24 hours, respectively. In contrast to their behavior in water, all hydrogels rapidly shrank when immersed in solutions with cations. The presence of ions in these solutions increases the double helix formation and the establishment of junction zones, leading to the formation of more crosslinked networks [34]. When immersed in PBS, High-MeGG samples present a significant shrinking (***p<0.0001) as compared with the Low-MeGG samples, possibly because of the higher amount of hydrophobic methacrylic groups in the High-MeGG repulses water molecules from the bulk to outside of the hydrogel. Similarly, Qiang Li et al reported [35] a lower swelling ratio with increasing acrylate content in polyphosphoester hydrogels. A sharper decrease was registered for High-MeGG-PC, presenting a significant deswelling of nearly 60% after 30 min of immersion in PBS (**p<0.001 when compared to High-MeGG-C, ***p<0.0001 when compared to all other samples). Interestingly, when immersed in DMEM, where both mono and divalent cations are present, hydrogels showed lower shrinking (deswelling) than in PBS. This may be a result of the higher concentration of monovalent cations in media. Therefore, the divalent cations used to keep hydrogels physically crosslinked will be exchanged by monovalent ones, leading to the formation of less crosslinked networks, as described elsewhere [36].
We hypothesize that this is a result of an ionic crosslinking of the hydrogels with the cations present in the solutions. A similar result was reported with alginate hydrogels [36] where shrinking of the hydrogels occurred when immersed in a medium containing higher Ca2+ concentration. To further confirm the ionic nature of the hydrogel shrinking, samples were immersed in water for 30 min, followed by immersion in PBS for 20 hours to allow hydrogel crosslinking. Shrunken samples were again placed in water for 30 min allowing them to regain their original shape (Fig. 4D). It was observed that hydrogels swelled in water, deswelled when immersed in PBS and swelled again when immersed in water. This experiment confirmed the ionic nature of swelling-deswelling behavior of MeGG hydrogels. It also shows the possibility to control the physical properties of the developed MeGG hydrogels, by changing the solution in which they are immersed.
3.4. Tunable mechanical properties
The tissue stiffness can vary greatly depending on the function of the tissue in the body [37]. Therefore, when designing a biomaterial for enhancing tissue regeneration, it is of utmost importance to match the material stiffness with that of natural tissues. The use of both physical and chemical mechanisms to crosslink hydrogels gives the possibility to tune this property.
It is known that ionically crosslinked hydrogels exhibit weak mechanical properties [38]. This limitation can be overcome by the introduction of acrylate groups followed by photocrosslinking. Here we combined physical and chemical crosslinking mechanisms to fabricate MeGG with wide range of mechanical properties. The influence of three parameters over the mechanical properties was evaluated: ion concentration in the hydrogel fabrication (Fig. 5A), type of crosslinking mechanism (Fig. 5B) and polymer concentration (Fig. 5C), while keeping the other conditions constant. To study the mechanical properties of fabricated hydrogels, compression testing was performed on GG and MeGG hydrogels. Despite stable hydrogel formation, High-MeGG-P samples were too weak to test mechanically.
Fig. 5.

Mechanical properties of GG and MeGG hydrogels: Young’s Modulus, Ultimate stress and Ultimate strain. The influence of three parameters over the mechanical properties of the hydrogels was evaluated: (A) Concentration of ions in the hydrogel fabrication; (B) Type of crosslinking mechanism (P, C or PC); and (C) Polymer concentration. The mechanical properties of the developed hydrogels showed to be highly dependent on the ionic content, type of crosslinking, methacrylation degree and polymer concentration. Statistical analysis through two-way ANOVA and Bonferroni post-hoc test showed significant differences (***p<0.0001, **p<0.001, *p<0.01) between the analyzed groups. NP: not processable.
3.4.1. Effect of Ca2+ in the hydrogel fabrication
As assessed by the swelling kinetics assay, the hydrogel response was strongly influenced by the presence of cations in the solution. Hence, we tested the effect of varying the concentration of Ca2+ ions present during hydrogel fabrication on the resulting gel’s mechanical properties. Fig. 5A shows the influence of the concentration of ions over the entanglement of the hydrogel network. For Low-MeGG hydrogels, the increase in concentration of ions from 0.08 mg/mL to 0.12 mg/mL showed no influence on the Young’s modulus but a significant increase on the ultimate stress (**p<0.001) and ultimate strain (*p<0.01) of these hydrogels. On the other hand, a higher amount of Ca2+ ions used in the fabrication of the High-MeGG hydrogels significantly decreased the Young’s modulus. Similarly, a marked decrease in the mechanical properties was observed in calcium-alginate hydrogels immersed in physiological baths of NaCl and CaCl2 [39]. Moreover, the same ionic concentration on the fabrication process resulted in hydrogels with different stiffness depending on the degree of methacrylation. A significantly stiffer (***p<0.0001) hydrogel was obtained for High-MeGG in the presence of ions as compared to Low-MeGG. Interestingly, High-MeGG hydrogels were significantly less resistant (***p<0.0001) to fracture and to elongation compared to the Low-MeGG hydrogels. This might result from an excessive crosslinking of hydrogels due to their higher degree of methacrylation, leading to the formation of a brittle hydrogel [40]. These results demonstrate that the use of ions as a crosslinking agent affected Low-MeGG and High-MeGG hydrogels in opposite ways.
3.4.2. Effect of crosslinking mechanism
As previously described [41, 42], the crosslinking mechanisms used to fabricate the hydrogels significantly influenced their mechanical properties (Fig. 5B). When only the physical mechanism was employed, the value of Young’s modulus and ultimate stress of hydrogels significantly decreased (***p<0.0001) with increase in the degree of methacrylation of the materials. For higher degree of methacrylation, the sites available for ionic interactions between polymer chains are less, since the chain is occupied by methacrylate groups, as schematized in Fig. 3. Therefore, the ionic bonding may be weaker for the polymer chains with higher content of methacrylate groups.
The chemical crosslinking alone led to the development of hydrogels with significantly higher Young’s modulus (**p<0.001) and ultimate stress (*p<0.01) values, when compared to those fabricated only with physical mechanism. Since GG is not chemically crosslinkable, only physical crosslinked GG is represented in Fig. 5B. The coupling effect of physical and chemical crosslinking mechanisms significantly decreased the stiffness of High-MeGG hydrogels, and did not influence the stiffness of the Low-MeGG hydrogels. A similar result was observed when the concentration of ions in the hydrogel production is increased (Fig. 5A). Though, similar tendency was observed when the mechanisms are used alone, a significantly higher Young’s modulus and lower ultimate stress and strain values (***p<0.0001) were observed with the increase in the methacrylation degree.
3.4.3. Effect of polymer concentration
It is known that the polymer concentration exerts a great effect over the mechanical properties of hydrogels [41]. Therefore, three different concentrations (0.5, 1, 2% w/v) were tested for the PC samples of both low and high degree of methacrylation (Fig. 5C). Stable hydrogels of 0.5% (w/v) of Low-MeGG could not be produced. The Young’s modulus of the hydrogels significantly increased as the amount of polymer in the hydrogel network increased. Also, the capacity of High-MeGG samples to resist fracture significantly increased whereas significantly decreasing for the Low-MeGG hydrogels (**p<0.001). This increase of polymer concentration also led to a significant decrease on the elongation capacity of the materials (**p<0.001). Polymer concentration is known to influence the helix content in GG solutions [43]. As the concentration of GG polymer increases in solution, the helix content increases, leading to more intensive aggregation of the chains and to a stronger physical and chemical crosslinking.
Overall, combination of physical and chemical crosslinking mechanisms resulted in MeGG hydrogels with a wide range of mechanical properties. Therefore, hydrogels with biologically relevant stiffness values in the range of 0.15 to 148 kPa could be fabricated by means of physiologically feasible crosslinking mechanisms. Ultimate stress varied from 11.7 kPa to 889.5 kPa whereas ultimate strain values were in the range of 34.2 to 96.1%. Such tunable mechanical properties of hydrogels could be useful for tissue engineering applications ranging from soft tissues, as the brain to stiff tissues, as the collagenous bone [32].
3.5. Tunable degradation properties
The rate of tissue regeneration is dependent on the microenvironment and the tissue function [44]. Therefore, the development of a biomaterial that can degrade at a similar rate at which the new tissue is being formed is of utmost importance. Understanding the degradation behavior of polymers aimed to be used in tissue engineering applications is important to predict and ultimately tune their behavior in the in vivo environment. To our knowledge, so far no previous study has looked into the degradation mechanism of GG in vitro. However, hydrolytic degradation of polysaccharides as GG and MeGG can be usually achieved in vivo through the action of enzymes such as lysozyme or amylase. Moreover, the in vivo degradation of GG hydrogels was already assessed, showing that GG degrades in vivo [18]. Also, it was already reported that methacrylated hydrogels can be degraded in vivo [11]. In contrast with other hydrogels [45], GG and MeGG are quite stable in PBS due to the ionic crosslinking, as described in a previous section, where we did not find any degradation in PBS. Hence, to compare the relative differences in mass loss over time between the different hydrogel networks the hydrolytic degradation was performed in accelerated condition with a sodium hydroxide 0.1 mM solution, as described previously [46]. Fig. 6 shows the in vitro accelerated hydrolytic degradation of GG and MeGG hydrogels. As expected, the crosslinking mechanisms used to produce the hydrogel led to different degradation profiles. Specifically, the physical crosslinking combined with chemical crosslinking mechanisms prolonged the rate of degradation as compared to only chemically crosslinked hydrogels. Moreover, High-MeGG hydrogels showed a slower degradation profile in comparison to Low-MeGG hydrogels, as reported by Oju Jeon et al for photocrosslinked alginate hydrogels [47].
Fig. 6.
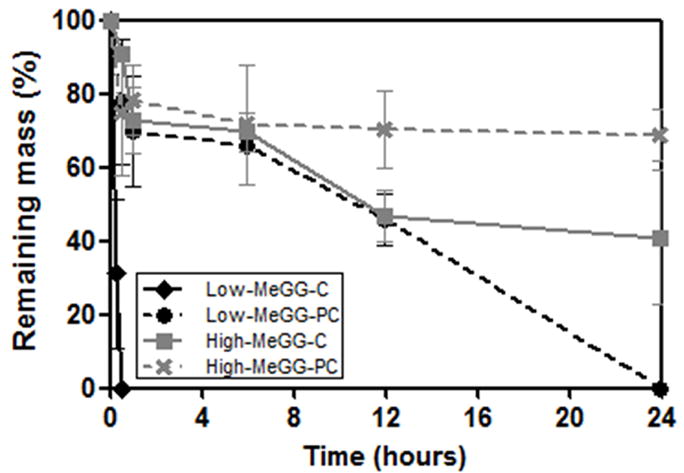
In vitro hydrolytic degradation kinetics of the hydrogels in NaOH (0.1 mM) at 37 °C.
3.6. In vitro cell viability of NIH-3T3 cells encapsulated in MeGG
Previous studies [18, 21, 48] have demonstrated that GG hydrogels can adequately support the growth and ECM deposition of human articular chondrocytes in vivo. Here we studied the viability of photoencapsulated NIH-3T3 cells in the MeGG hydrogels by Live/Dead assay (Fig. 7). We evaluated the cell viability immediately after the fabrication to assess the effect of the crosslinking mechanisms on the cell viability. The UV exposure and the toxicity of the methacrylate groups of the modified GG showed no significant effect on the viability of encapsulated cells. These results further confirmed the biocompatibility of MeGG for cell encapsulation.
Fig. 7.

Representative fluorescence micrographs of live (green) and dead (red) encapsulated NIH-3T3 cells in GG, Low-MeGG and High-MeGG immediately after encapsulation (day 0) and cultured in vitro for 3 days. The scale bars indicate 100 μm.
4. Conclusions
MeGG hydrogels were synthesized by reacting GG with different amounts of methacrylic anhydride, yielding to the development of hydrogels with two different degrees of methacrylation. The combination of physical crosslinking methods (temperature and the addition of cations) with chemical crosslinking approaches (through photocrosslinking), enabled the development of MeGG hydrogels with highly tunable physical and mechanical properties without affecting their biocompatibility. In summary, physiologically compatible crosslinking methods were used for the development of highly tunable hydrogels applicable for a wide range of tissue engineering applications.
Supplementary Material
Acknowledgments
This research was funded by the US Army Engineer Research and Development Center, the Institute for Soldier Nanotechnology, the NIH (HL092836, DE019024, EB007249), and the National Science Foundation CAREER award (AK). This work was partially supported by FCT, through funds from the POCTI and/or FEDER programs and from the European Union under the project NoE EXPERTISSUES (NMP3-CT-2004-500283). DFC acknowledges the Foundation for Science and Technology (FCT), Portugal and the MIT-Portugal Program for personal grant SFRH/BD/37156/2007. HS was supported by a Samsung Scholarship. SS acknowledges the postdoctoral fellowship awarded by Fonds de Recherche sur la Nature et les Technologies (FQRNT), Quebec, Canada. We would like to thank Dr. Che Hutson for scientific discussions.
Footnotes
Author Contribution DFC, SS, AK designed the study; DFC synthesized the MeGG and produced the hydrogels; DFC and HS performed the 1H-NMR and FTIR-ATR experiments; DFC performed the swelling, mechanical and cell experiments; DFC and SS performed the degradation study. DFC and JTO produced the plain GG. DFC, SS and AK wrote the paper. MG, NN, RR revised the paper. All authors discussed the results and commented on the manuscript.
Appendix Supplementary data associated with this article can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Qiu Y, Park K. Environment-sensitive hydrogels for drug delivery. Adv Drug Deliv Rev. 2001;53(3):321–339. doi: 10.1016/s0169-409x(01)00203-4. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman AS. Hydrogels for biomedical applications. Adv Drug Deliv Rev. 2002;54(1):3–12. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 3.Caldorera-Moore M, Peppas NA. Micro- and nanotechnologies for intelligent and responsive biomaterial-based medical systems. Adv Drug Deliv Rev. 2009;61(15):1391–1401. doi: 10.1016/j.addr.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv Mater. 2006;18(11):1345–1360. [Google Scholar]
- 5.Rabanel J, Bertrand N, Sant S, Louati S, Hildgen P. Polysaccharide hydrogels for the preparation of immunoisolated cell delivery systems. In: Marchessault RH, Ravenelle F, Zhu X, editors. Polysaccharides for drug delivery and pharmaceutical applications. Washington: Amer Chemical Soc; 2006. p. 305. [Google Scholar]
- 6.Chung HJ, Park TG. Self-assembled and nanostructured hydrogels for drug delivery and tissue engineering. Nano Today. 2009;4(5):429–437. [Google Scholar]
- 7.Jeong B, Kim SW, Bae YH. Thermosensitive sol-gel reversible hydrogels. Adv Drug Deliv Rev. 2002;54(1):37–51. doi: 10.1016/s0169-409x(01)00242-3. [DOI] [PubMed] [Google Scholar]
- 8.Hennink WE, van Nostrum CF. Novel crosslinking methods to design hydrogels. Adv Drug Deliv Rev. 2002;54(1):13–36. doi: 10.1016/s0169-409x(01)00240-x. [DOI] [PubMed] [Google Scholar]
- 9.Van Tomme SR, Storm G, Hennink WE. In situ gelling hydrogels for pharmaceutical and biomedical applications. Int J Pharm. 2008;355(1-2):1–18. doi: 10.1016/j.ijpharm.2008.01.057. [DOI] [PubMed] [Google Scholar]
- 10.Ifkovits JL, Burdick JA. Review: Photopolymerizable and degradable biomaterials for tissue engineering applications. Tissue Eng. 2007;13(10):2369–2385. doi: 10.1089/ten.2007.0093. [DOI] [PubMed] [Google Scholar]
- 11.Hahn SK, Park JK, Tomimatsu T, Shimoboji T. Synthesis and degradation test of hyaluronic acid hydrogels. Int J Biol Macromol. 2007;40(4):374–380. doi: 10.1016/j.ijbiomac.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 12.Khademhosseini A, Eng G, Yeh J, Fukuda J, Blumling J, Langer R, et al. Micromolding of photocrosslinkable hyaluronic acid for cell encapsulation and entrapment. J Biomed Mater Res A. 2006;79A(3):522–532. doi: 10.1002/jbm.a.30821. [DOI] [PubMed] [Google Scholar]
- 13.Oudshoorn MHM, Rissmann R, Bouwstra JA, Hennink WE. Synthesis of methacrylated hyaluronic acid with tailored degree of substitution. Polymer. 2007;48(7):1915–1920. [Google Scholar]
- 14.Van den Bulcke AI, Bogdanov B, De Rooze N, Schacht EH, Cornelissen M, Berghmans H. Structural and rheological properties of methacrylamide modified gelatin hydrogels. Biomacromolecules. 2000;1(1):31–38. doi: 10.1021/bm990017d. [DOI] [PubMed] [Google Scholar]
- 15.Benton JA, DeForest CA, Vivekanandan V, Anseth KS. Photocrosslinking of gelatin macromers to synthesize porous hydrogels that promote valvular interstitial cell function. Tissue Eng Part A. 2009;15(11):3221–3230. doi: 10.1089/ten.tea.2008.0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chou AI, Akintoye SO, Nicoll SB. Photo-crosslinked alginate hydrogels support enhanced matrix accumulation by nucleus pulposus cells in vivo. Osteoarthritis Cartilage. 2009;17(10):1377–1384. doi: 10.1016/j.joca.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou AI, Nicoll SB. Characterization of photocrosslinked alginate hydrogels for nucleus pulposus cell encapsulation. J Biomed Mater Res A. 2009;91A(1):187–194. doi: 10.1002/jbm.a.32191. [DOI] [PubMed] [Google Scholar]
- 18.Oliveira JT, Santos TC, Martins L, Picciochi R, Marques AP, Castro AG, et al. Gellan gum injectable hydrogels for cartilage tissue engineering applications: In vitro studies and preliminary in vivo evaluation. Tissue Eng Part A. 2009;16(1):343–353. doi: 10.1089/ten.TEA.2009.0117. [DOI] [PubMed] [Google Scholar]
- 19.Oliveira JT, Santos TC, Martins L, Silva MA, Marques AP, Castro AG, et al. Performance of new gellan gum hydrogels combined with human articular chondrocytes for cartilage regeneration when subcutaneously implanted in nude mice. J Tissue Eng Regen Med. 2009;3(7):493–500. doi: 10.1002/term.184. [DOI] [PubMed] [Google Scholar]
- 20.Gong YH, Wang CM, Lai RC, Su K, Zhang F, Wang DA. An improved injectable polysaccharide hydrogel: Modified gellan gum for long-term cartilage regeneration in vitro. J Mater Chem. 2009;19(14):1968–1977. [Google Scholar]
- 21.Oliveira JT, Gardel L, Martins L, Rada T, Gomes ME, Reis RL. Injectable gellan gum hydrogels with autologous cells for the treatment of rabbit articular cartilage defects. Journal of Orthopedic Research. 2010 doi: 10.1002/jor.21114. in press. [DOI] [PubMed] [Google Scholar]
- 22.Oliveira JT, Martins L, Picciochi R, Malafaya PB, Sousa RA, Neves NM, et al. Gellan gum: A new biomaterial for cartilage tissue engineering applications. J Biomed Mater Res A. 2010 doi: 10.1002/jbm.a.32574. [DOI] [PubMed] [Google Scholar]
- 23.Carlfors J, Edsman K, Petersson R, Jornving K. Rheological evaluation of gelrite (r) in situ gels for ophthalmic use. Eur J Pharm Sci. 1998;6(2):113–119. doi: 10.1016/s0928-0987(97)00074-2. [DOI] [PubMed] [Google Scholar]
- 24.Rozier A, Mazuel C, Grove J, Plazonnet B. Gelrite®: A novel, ion-activated, in-situ gelling polymer for ophthalmic vehicles. Effect on bioavailability of timolol. Int J Pharm. 1989;57(2):163–168. [Google Scholar]
- 25.Jansson PE, Lindberg B, Sandford PA. Structural studies of gellan gum, an extracellular polysaccharide elaborated by pseudomonas-elodea. Carbohydr Res. 1983;124(1):135–139. [Google Scholar]
- 26.Miyoshi E, Takaya T, Nishinari K. Rheological and thermal studies of gel-sol transition in gellan gum aqueous solutions. Carbohydr Polym. 1996;30:109–119. [Google Scholar]
- 27.Matricardi P, Cencetti C, Ria R, Alhaique F, Coviello T. Preparation and characterization of novel gellan gum hydrogels suitable for modified drug release. Molecules. 2009;14(9):3376–3391. doi: 10.3390/molecules14093376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caggioni M, Spicer PT, Blair DL, Lindberg SE, Weitz DA. Rheology and microrheology of a microstructured fluid: The gellan gum case. J Rheol. 2007;51(5):851–865. [Google Scholar]
- 29.Singh BN, Kim KH. Effects of divalent cations on drug encapsulation efficiency of deacylated gellan gum. J Microencapsul. 2005;22(7):761–771. doi: 10.1080/02652040500273704. [DOI] [PubMed] [Google Scholar]
- 30.Ogawa E, Takahashi R, Yajima H, Nishinari K. Thermally induced coil-to-helix transition of sodium gellan gum with different molar masses in aqueous salt solutions. Biopolymers. 2005;79(4):207–217. doi: 10.1002/bip.20349. [DOI] [PubMed] [Google Scholar]
- 31.Tako M, Teruya T, Tamaki Y, Konishi T. Molecular origin for rheological characteristics of native gellan gum. Colloid Polym Sci. 2009;287(12):1445–1454. [Google Scholar]
- 32.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 33.Hamcerencu M, Desbrieres J, Khoukh A, Popa M, Riess G. Synthesis and characterization of new unsaturated esters of gellan gum. Carbohydr Polym. 2008;71(1):92–100. [Google Scholar]
- 34.Miyoshi E, Takaya T, Nishinari K. Rheological and thermal studies of gel-sol transition in gellan gum aqueous solutions. Carbohydr Polym. 1996 Jun-Jul;30(2-3):109–119. [Google Scholar]
- 35.Li Q, Wang J, Shahani S, Sun D, Sharma B, Elisseeff J, et al. Biodegradable and photocrosslinkable polyphosphoester hydrogel. Biomaterials. 2006;27(7):1027–1034. doi: 10.1016/j.biomaterials.2005.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuo CK, Ma PX. Maintaining dimensions and mechanical properties of ionically crosslinked alginate hydrogel scaffolds in vitro. J Biomed Mater Res A. 2008;84A(4):899–907. doi: 10.1002/jbm.a.31375. [DOI] [PubMed] [Google Scholar]
- 37.Nemir S, West JL. Synthetic materials in the study of cell response to substrate rigidity. Ann Biomed Eng. 2009;38(1):2–20. doi: 10.1007/s10439-009-9811-1. [DOI] [PubMed] [Google Scholar]
- 38.Jagur-Grodzinski J. Polymeric gels and hydrogels for biomedical and pharmaceutical applications. Polym Adv Technol. 21(1):27–47. [Google Scholar]
- 39.LeRoux MA, Guilak F, Setton LA. Compressive and shear properties of alginate gel: Effects of sodium ions and alginate concentration. J Biomed Mater Res. 1999;47(1):46–53. doi: 10.1002/(sici)1097-4636(199910)47:1<46::aid-jbm6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 40.Tang Q, Sun X, Li Q, Wu J, Lin J. Fabrication of a high-strength hydrogel with an interpenetrating network structure. Colloids Surf A Physicochem Eng Asp. 2009;346(1-3):91–98. [Google Scholar]
- 41.Brigham MD, Bick A, Lo E, Bendali A, Burdick JA, Khademhosseini A. Mechanically robust and bioadhesive collagen and photocrosslinkable hyaluronic acid semi-interpenetrating networks. Tissue Eng Part A. 2009;15(7):1645–1653. doi: 10.1089/ten.tea.2008.0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anseth KS, Bowman CN, BrannonPeppas L. Mechanical properties of hydrogels and their experimental determination. Biomaterials. 1996;17(17):1647–1657. doi: 10.1016/0142-9612(96)87644-7. [DOI] [PubMed] [Google Scholar]
- 43.Dai L, Liu XX, Liu YL, Tong Z. Concentration dependence of critical exponents for gelation in gellan gum aqueous solutions upon cooling. Euro Polym J. 2008 Dec;44(12):4012–4019. [Google Scholar]
- 44.Liu CZ, Czernuszka JT. Development of biodegradable scaffolds for tissue engineering: A perspective on emerging technology. Mater Sci Technol. 2007;23(4):379–391. [Google Scholar]
- 45.Meyvis TKL, De Smedt SC, Demeester J, Hennink WE. Influence of the degradation mechanism of hydrogels on their elastic and swelling properties during degradation. Macromolecules. 2000 Jun;33(13):4717–4725. [Google Scholar]
- 46.Nijst CLE, Bruggeman JP, Karp JM, Ferreira L, Zumbuehl A, Bettinger CJ, et al. Synthesis and characterization of photocurable elastomers from poly(glycerol-co-sebacate) Biomacromolecules. 2007;8(10):3067–3073. doi: 10.1021/bm070423u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeon O, Bouhadir KH, Mansour JM, Alsberg E. Photocrosslinked alginate hydrogels with tunable biodegradation rates and mechanical properties. Biomaterials. 2009;30(14):2724–2734. doi: 10.1016/j.biomaterials.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 48.Oliveira JT, Santos TC, Martins L, Picciochi R, Marques AP, Castro AG, et al. Gellan gum injectable hydrogels for cartilage tissue engineering applications: In vitro studies and preliminary in vivo evaluation. Tissue Engineering: Part A. 2009:15. doi: 10.1089/ten.TEA.2009.0117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.