

Abstract
PtII azido complexes [Pt(bpy)(N3)2] (1), [Pt(phen)(N3)2] (2) and trans-[Pt(N3)2(py)2] (3) incorporating the bidentate diimine ligands 2,2′-bipyridine (bpy), 1,10-phenanthroline (phen) or the monodentate pyridine (py) respectively, have been synthesised from their chlorido precursors and characterised by x-ray crystallography; complex 3 shows significant deviation from square-planar geometry (N3–Pt–N3 angle 146.7°) as a result of steric congestion at the Pt centre. The novel PtIV complexes trans, cis-[Pt(bpy)(OAc)2(N3)2] (4), trans, cis-[Pt(phen)(OAc)2(N3)2] (5), trans, trans, trans-[Pt(OAc)2(N3)2(py)2] (6), were obtained from 1–3 via oxidation with H2O2 in acetic acid followed by reaction of the intermediate with acetic anhydride. Complexes 4–6 exhibit interesting structural and photochemical properties that were studied by x-ray, NMR and UV-vis spectroscopy and TDDFT. These PtIV complexes exhibit greater absorption at longer wavelengths (ε = 9756 M−1cm−1 at 315 nm for 4; ε = 796 M−1cm−1 at 352 nm for 5; ε = 16900 M−1cm−1 at 307 nm for 6, in aqueous solution) than previously reported PtIV azide complexes, due to the presence of aromatic amines, and 4–6 undergo photoactivation with both UVA (365 nm) and visible green light (514 nm). The UV-vis spectra of complexes 4–6 were calculated using TD-DFT; the nature of the transitions contributing to the UV-vis bands provide insight into the mechanism of production of the observed photoproducts. The UV-vis spectra of 1–3 were also simulated by computational methods and comparison between PtII and PtIV electronic and structural properties allowed further elucidation of the photochemistry of 4–6.
Introduction
PtII pyridyl compounds are well-known to exhibit a rich photochemistry, which can be tuned through peripheral modification of the ligands bound to the PtII centre.1,2 The low energy spectroscopic absorption band in the visible region and long-lived excited states of these complexes lead to a variety of applications, including their use in biosensors,3 light-emitting devices4 and dye-sensitised solar cells.5 The biological properties of PtII amines have been extensively investigated, with particular emphasis on potential antitumor activity following the success in the clinic of the PtII drug cisplatin, cis-[Pt(NH3)2Cl2], and related complexes.6
PtIV complexes are much more inert to reaction than PtII species, and tuning their reactivity to achieve selective reduction to PtII in vivo provides a strategy for reducing the side-reactions associated with PtII antitumor drugs.7,8 Although reports of PtIV bipyridine complexes, for example, have been largely structural,9 Lippert and co-workers have investigated the photoreduction of trans-[Pt(bpy)(MeNH2)2(OH)2]Cl2; after irradiation for several days (dose not stated), free MeNH3+ was detected, indicating that amine dissociation had occured.7,10
Photoactivation of metal complexes can be used for triggering specific interactions between the metal complex and target macromolecules of biological relevance (such as DNA, RNA or proteins).11 Upon irradiation, photoactivable metal complexes can undergo ligand dissociation from the excited state, forming labile solvated species that are more reactive towards the target macromolecule.12 Light activation offers the advantage of temporal- and spatial-control of the active species in tissues, potentially reducing undesired secondary effects due to the toxicity of such species. It is notable that several complexes of d8 metal ions with α-diimine ligands have been reported to photosensitise the production of singlet oxygen (1O2)13-15 and so there is scope for the use of photoactivatable metal complexes in photodynamic therapy16 and for virucidal applications.17,18
The efficacy of photoactivatable metal complexes as potential anticancer agents depends strongly on their excited-state properties. The relative energies of singlet and triplet excited states influence the choice of the excitation wavelength, and determine the nature of the photoproducts, their mechanism of formation and the yield of ligand photodissociation.19 Density functional theory (DFT) and time-dependent DFT (TDDFT) are fundamental tools for rational design of metal-based drugs with tunable properties, since they can provide a description of electronic structures and excited states of metal complexes.20
Our recent approach has been to develop PtIV complexes which can be reduced to cytotoxic PtII by photoactivation, selectively at a tumour site.21,22,23,24 For effective photochemotherapy of non-surface tumours it is desirable to activate complexes with relatively long wavelengths of light since the penetration of light into tissue is wavelength-dependent.25 Furthermore, the use of visible light is less likely to cause damage of biological tissue.25c,26 Current photochemotherapy typically employs red light (~ 620 nm) which is suitable for activation of established photodynamic agents.27 We are therefore investigating methods to achieve photoactivation of PtIV-azido complexes at a variety of wavelengths. Here we describe the synthesis and characterisation of new PtIV azido complexes 4–6 and their synthetic PtII precursors 1–3 (Figure 1); although complexes 1 and 2 have been previously reported18,28 we present here further characterisation, including x-ray crystallographic structures.
Figure 1.

Structures of [Pt(bpy)(N3)2] (1), [Pt(phen)(N3)2] (2), trans-[Pt(N3)2(py)2] (3), trans, cis-[Pt(OAc)2(N3)2(bpy)] (4), trans, cis-[Pt(OAc)2(N3)2(phen)] (5) and trans, trans, trans-[Pt(OAc)2(N3)2(py)2] (6).
Results
Dichlorido PtII compounds [Pt(bpy)Cl2], [Pt(phen)Cl2] and trans-[PtCl2(py)2] were prepared by literature methods.29,30 The chloride ligands were then substituted by azido ligands by stirring the complexes with an excess of sodium azide in DMF. The reaction of complexes 1–3 with H2O2 in acetic acid resulted in oxidation.31,32 The main products in all three cases were the monohydroxido, monoacetato species [Pt(L)(N3)2(OAc)(OH)] (L = bpy, phen, (py)2), which were then readily converted to the diacetato complexes 4–6 by stirring in acetic anhydride.
Crystal Structures
Square-planar PtII bipyridine and phenanthroline complexes (1 and 2) both crystallised in a monoclinic crystal system. In both systems deviation from square-planar geometry is observed; the angle(s) which the platinum azide group Pt-Nα makes with the plane defined by N(ring)-Pt-N(ring) is 1.54 ° for 1 (which contains a two-fold axis of symmetry) and 3.02 ° and 2.24 ° for 2. For 1, angles at the coordinated azide group (Pt–Nα–Nβ) are 119.3(2)° with Pt–Nα and Pt–N(ring) bond lengths of 2.034(3) Å and 2.008(3) Å, respectively. For 2, angles at the coordinated azide group are 121.8(4)° and 125.1(4)° with Pt–Nα bond lengths of 2.022(6) Å and 2.037(5) Å and Pt–N(ring) lengths of 2.011(6) Å and 2.011(5) Å, respectively. Complex 3 also crystallised in a monoclinic crystal system containing a two-fold axis of symmetry. The geometry around the platinum centre is significantly distorted from square-planar geometry and the pyridine rings demonstrate positional disorder. The pyridine ring is disordered approximately equally over two orientations. The two components were restrained to have similar bond distances and angles. For 3, angles at the coordinated azide group (Pt–Nα–Nβ) are 125.4(5)° with Nα–Pt–Nα angles and Pt–Nα bond lengths of 146.7(4)° and 2.036(7) Å respectively. For the structures and crystallographic parameters of 1, 2 and 3 see Figure S1 and Table 1 respectively.
Table 1.
Crystallographic data for [Pt(bpy)(N3)2] (1), [Pt(phen)(N3)2] (2), trans-[Pt(N3)2(py)2] (3), trans, cis-[Pt(bpy)(OAc)2(N3)2] (4), trans, cis-[Pt(phen)(OAc)2(N3)2] (5) and trans, trans, trans-[Pt(OAc)2(N3)2(py)2] (6).
| Parameter | Complex |
|||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
|
| ||||||
| Empirical formula | C10H8N8Pt | C12H8N8Pt | C10H10N8Pt | C14H14N8O4Pt | C16H14N8O4Pt | C14H16N8O4Pt |
| Formula weight | 435.32 | 459.34 | 437.33 | 553.42 | 577.43 | 555.42 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic | Triclinic |
| Space group | C2/c | C2/c | I121 | Pbcn | P2/n | P-1 |
| a (Å) | 10.9346(5) | 19.3584(7) | 11.6121(5) | 8.0488(4) | 7.0303(1) | 6.9480(3) |
| b (Å) | 15.3328(8) | 10.1802(4) | 3.8371(2) | 14.1103(7) | 11.7849(2) | 8.2197(4) |
| c (Å) | 7.0453(4) | 13.8692(4) | 14.1714(9) | 15.3280(7) | 11.3271(2) | 8.6473(4) |
| α(°) | 90 | 90 | 90 | 90 | 90 | 92.136(3) |
| β (°) | 94.717(3) | 113.816(2) | 102.756(2) | 90 | 101.236(1) | 111.574(3) |
| γ(°) | 90 | 90 | 90 | 90 | 90 | 101.038(3) |
| Z | 4 | 8 | 2 | 4 | 2 | 1 |
| ρcalcd (mg/m3) | 2.456 | 2.440 | 2.358 | 2.112 | 2.083 | 2.060 |
| μcalcd (mm−1) | 11.919 | 11.230 | 11.392 | 8.101 | 7.665 | 7.877 |
| Volume (Å3) | 1177.20(11) | 2500.49(16) | 615.85(6) | 1740.82(15) | 920.48(3) | 447.61(4) |
| Conventional R | 0.0192 | 0.0326 | 0.0264 | 0.0243 | 0.0305 | 0.0350 |
| wR2 | 0.0478 | 0.0825 | 0.0690 | 0.0582 | 0.0690 | 0.0913 |
| Independent reflections |
1453 | 3057 | 1429 | 2307 | 2454 | 2121 |
| No. reflections measured |
6926 | 9019 | 2838 | 21880 | 14648 | 5315 |
| R int | 0.03 | 0.036 | 0.045 | 0.0377 | 0.027 | 0.041 |
| Data/Restraints/Pa rameters |
1452/0/87 | 3057/0/190 | 1423/13/71 | 2307/0/124 | 2453/20/142 | 2121/0/125 |
Each of the PtIV complexes (4–6) crystallised in a different crystal system and space group, however they all contain a two-fold axis of symmetry (Figure 2) and adopt approximate octahedral geometry; the O–Pt–O angles are 179.18(14)° (4), 165.12(19)° (5) and 179.994° (6). The Pt–Nα distances are 2.022(3) Å, 2.032(4) Å and 2.049(6) Å respectively, and the Pt–N(ring) distances 2.033(3) Å, 2.052(4) Å and 2.028(6) Å, respectively. The angles at the coordinated azide groups are 115.0(3)°, 118.5(4)° and 114.9(5)°, respectively. The crystallographic parameters are given in Table 1 and a selection of key bond lengths and angles are shown in Table 2.
Figure 2.
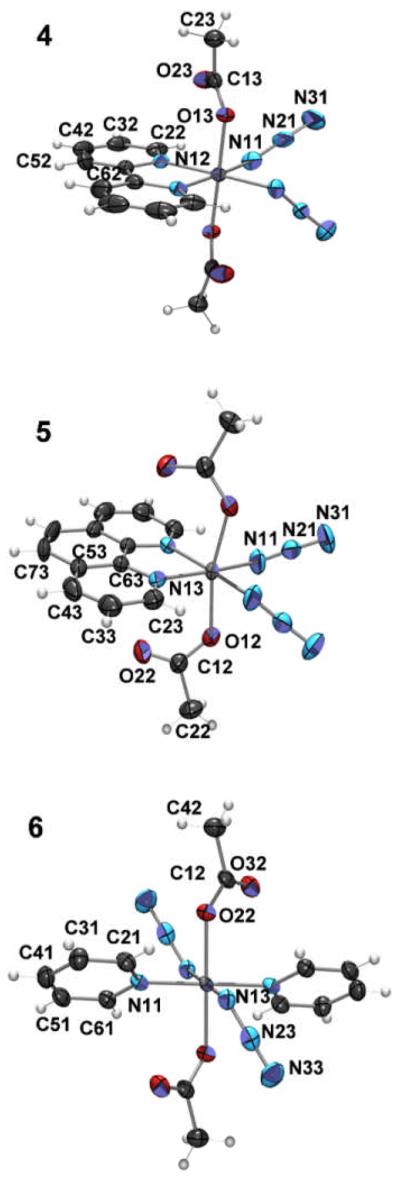
ORTEP plots of cis, trans-[Pt(bpy)(N3)2(OH)2] (4), cis, trans-[Pt(phen)(N3)2(OH)2] (5) and (6) trans, trans, trans-[Pt(N3)2(OH)2(py)2] (6). Non-hydrogen atoms are represented by Gaussian ellipsoids at the 50% probability level. Hydrogen atoms are shown with arbitrarily small thermal parameters.
Table 2.
Selected bond lengths (Å) and angles (°) for complexes 4–6. The azide ligands are labelled Pt–Nα–Nβ–Nγ and the pyridyl nitrogens as NRing.
| Bond / Angle | Complex | ||
|---|---|---|---|
| 4 | 5 | 6 | |
| Pt–N(Ring) | 2.033(3) | 2.052(4) | 2.028(6) |
| Pt–O | 2.020(2) | 2.001(3) | 2.007(5) |
| Pt–N(α1) | 2.022(3) | 2.032(4) | 2.049(6) |
| N(α1) –N(β1) | 1.225(5) | 1.175(6) | 1.224(9) |
| N(β1) –N(γ1) | 1.147(5) | 1.154(6) | 1.142(10) |
| Pt–N(α1) –N(β1) | 115.0(3) | 118.5(4) | 114.9(5) |
| N(α1) –N(β1) –N(γ1) | 175.2(5) | 173.1(5) | 176.0(8) |
| N(Ring) –Pt–N(Ring) | 80.07(17) | 81.2(2) | 179.994 |
| O–Pt–O | 179.18(14) | 165.12(19) | 179.994 |
| N(α1) –Pt–N(α2) | 89.5(2) | 96.1(2) | 179.994 |
| N(Ring) –Pt–N(α1) | 175.25(13) | 172.40(16) | 89.3(2) |
| N(Ring) –Pt–N(α2) | 95.22(14) | 91.40(16) | 90.7(2) |
| N(Ring) –Pt–O | 93.82(11) | 95.84(13) | 95.6(2) |
| N(Ring2) –Pt–O | 85.55(11) | 95.44(14) | 84.4(2) |
| N(α1) –Pt–O | 95.50(12) | 83.17(16) | 86.4(2) |
| N(α2) –Pt–O | 85.08(12) | 86.90(16) | 93.6(2) |
In general, the structures optimized by DFT calculations are in good agreement with those obtained from X-ray crystallography. DFT tended to overestimate bond lengths of 1–6, particularly in the case of Pt–N (bpy, phen, py) where distances are up to 0.06 Å longer. In the case of complexes 1 and 2, the Pt–Na1 and Pt–Nα2 bond lengths are underestimated. Optimising the geometry in the gas phase rather than the condensed phase is a possible source of this inconsistency33 although recent results suggest that this is unlikely to significantly affect the values34. DFT-calculated angles are within a few degrees of the X-ray values; the only exception being the angles relating to the azide ligands; for all complexes, X-ray and DFT gave differences in the orientation of the N3 groups although this is unlikely to be energetically significant. Data are summarized in the Supplementary Information (Tables S1–4).
Orbital Analysis
The PtIV complexes 4 (bpy) and 5 (phen) have similar frontier orbitals, those of 4 are shown in Figure 3. The HOMOs of 4 and 5 are mainly azide-based, with a small contribution from Pt. The HOMO shows a bonding interaction between the two azide Nα atoms, while it is antibonding with respect to the Nα-Nβ and Pt-Nα bonds. The HOMO–1 is similar in shape to the HOMO, while the HOMO–2 is more diffuse, having contributions also from the acetate groups and the chelating ligand. The LUMO and LUMO+1 of 4 and 5 are strongly antibonding orbitals; the LUMO is antibonding towards the Pt–N(azide) and Pt–N(bpy, phen) bonds, LUMO+1 is antibonding towards Pt–N(azide) and Pt–O(Ac) bonds. Higher in energy, the LUMO+2, LUMO+3 and LUMO+4 are all bpy- or phen-based. In complex 6 the HOMO is azide-centred. In the HOMO–1 and HOMO–2 the acetate character progressively increases, and the HOMO–3 is mainly acetate-based. In 6, the LUMO has antibonding character for the Pt–N(azide) bonds, while the LUMO+1 is antibonding towards both acetate and azide ligands. Both the LUMO+2 and LUMO+3 are py-centred.
Figure 3.

Selected orbitals for complexes [Pt(bpy)(N3)2] (1) and [Pt(OAc)2(N3)2(bpy)] (4).
The PtII complexes show similar features in the frontier orbitals which play a significant role in the UV-vis spectra. However, there is a major and fundamental difference in the unoccupied orbitals. The LUMO and LUMO+1 of 1 and 2 are bpy- or phen-based π* orbitals, those of 1 are shown in Figure 3. The lowest-energy antibonding orbital is LUMO+2 which lies ca.1 eV above LUMO+1. Complex 3 has orbitals that resemble those of 6, differing in that the LUMO has lost its antibonding character and the other empty orbitals are of higher energy.
UV-vis spectroscopy and TD-DFT singlet transitions
Computed UV-vis spectra of complexes 1 and 2 were in agreement with previously published data.18 Theoretical UV-vis spectra of 4–6 were calculated by TD-DFT with the CPCM solvent model and then overlaid with the experimental spectra which were recorded in water (Figure 4). The most representative electron density difference maps for the singlet transitions of 4–6 are also reported in Figure 4. Experimental and theoretical data are summarized in Tables 3-5.
Figure 4.

Calculated (blue line) and experimental (black line) absorption spectra of 4–6 in H2O. The excited states are shown as vertical bars with heights equal to the extinction coefficients. EDDMs of selected singlet transitions, are reported on the right (electron density migrates from light pink areas to purple ones). Theoretical curves and EDDMs were obtained using the program GaussSum 1.05.68
Table 3.
Experimental and calculated absorption properties of 4.
| λmax, nm ε(M−1 cm−1) |
Tra | Composition | Energy, eV (nm) |
Oscillator Strength |
Assignment |
|---|---|---|---|---|---|
| ~350 | 6 | HOMO–2→LUMO+1 (62%) HOMO–1→LUMO+1 (17%) |
3.43 (361) | 0.0105 | LMCT |
| 304(11402) and 315(9756) |
16 | HOMO–7→LUMO (17%) HOMO–6→L+1 (67%) |
3.98 (312) | 0.0127 | LMCT |
| 18 | HOMO–7→LUMO+1 (47%) HOMO–6→LUMO (40%) |
4.04 (307) | 0.0161 | LMCT | |
| 19 | HOMO–7→LUMO (11%) HOMO–4→LUMO+2 (54%) HOMO–3→LUMO+2 (17%) |
4.07 (304) | 0.0525 | LLCT | |
| 250 (19102) |
20 | HOMO–9→LUMO (15%) HOMO–8→LUMO (28%) HOMO–7→LUMO+1 (30%) HOMO–6→LUMO (16%) |
4.31(287) | 0.0969 | LMCT |
| 21 | HOMO–7→LUMO (37%) HOMO–6→LUMO+1 (10%) HOMO–5→LUMO+2 (35%) |
4.32 (287) | 0.0496 | LMCT/LLCT | |
| 22 | HOMO–9→LUMO+1 (21%) HOMO–8→LUMO+1 (25%) HOMO–5→LUMO+2 (22%) |
4.42 (280) | 0.1933 | LLCT/MLCT | |
| 23 | HOMO–9→LUMO+1 (22%) HOMO–8→LUMO+1 (27%) HOMO–5→LUMO+2 (20%) |
4.44 (279) | 0.1581 | LLCT/MLCT | |
| 25 | HOMO–9→LUMO (19%) HOMO–8→LUMO (65%) |
4.48 (277) | 0.0556 | LLCT/MLCT | |
| 26 | HOMO→LUMO+3 (91%) | 4.56 (272) | 0.0314 | LLCT | |
| 28 | HOMO–9→LUMO (57%) | 4.61 (268) | 0.1447 | LMCT | |
| 29 | HOMO→LUMO+4 (83%) | 4.65 (267) | 0.047 | LLCT | |
| 30 | HOMO–2→LUMO+3 (80%) HOMO–1→LUMO+3 (14%) |
4.76 (260) | 0.0119 | LLCT |
Tr = transition number as obtained in the TDDFT calculation output.
Table 5.
Experimental and calculated absorption properties of 6.
| λmax, nm ε(M−1 cm−1) |
Tra | Composition | Energy, eV (nm) |
Oscillator Strength |
Assignment |
|---|---|---|---|---|---|
| 307 (16900) |
9 | HOMO–4→LUMO (11%) HOMO–4→ LUMO+1 (65%) HOMO–2→LUMO+1 (20%) |
3.75 (330) | 0.0424 | LMCT |
| 10 | HOMO–4→LUMO+1 (18%) HOMO–2→LUMO+1 (76%) |
3.79 (327) | 0.0116 | LMCT | |
| 12 | HOMO–6→LUMO (64%) HOMO–4→LUMO (16%) |
4.01 (309) | 0.1278 | LMCT | |
| 13 | HOMO–6→LUMO (28%) HOMO–4→LUMO (40%) HOMO–4→LUMO+1 (10%) |
4.06 (305) | 0.3458 | LMCT | |
| 259 (12500) |
19 | HOMO–10→LUMO (72%) HOMO–6→LUMO+1 (16%) |
4.46 (277) | 0.0208 | LMCT |
| 21 | HOMO–10→LUMO (12%) HOMO–6→LUMO+1 (72%) |
4.51 (275) | 0.011 | LMCT | |
| 25 | HOMO–3→ LUMO+2 (75%) HOMO–2→LUMO+3 (10%) |
4.67 (265) | 0.0357 | LLCT | |
| 29 | HOMO–1→LUMO+3 (87%) | 4.82 (257) | 0.0155 | LLCT |
Tr = transition number as obtained in the TDDFT calculation output.
Complex 4 shows a maximum at 250 nm and a pronounced shoulder around 310 nm; a weak absorbance is also present at ca. 350 nm. Similarly, the main absorbance of 5 is at 272 nm and a shoulder, although significantly less pronounced, is visible at 300 nm. In addition, 5 has two weak bands in the range 330–360 nm. TD-DFT calculations performed without relativistic correction give satisfactory results in the case of 5, despite a small red shift in the absorbance energies. On the contrary, less satisfactory is the determination of the 310-nm band intensity for 4. However, the general character of the electronic transitions of 4 was confirmed by performing calculations with different software (see Computational Details and Supplementary Information). For both complexes, the lowest-energy absorbance is due to ligand-to-metal charge transfer (LMCT)23 states (state S6 for both 4 and 5), where electron density migrates from the acetate groups to the metal. At higher energy, the transitions composing the shoulder are a mixture of LMCT (N3 → Pt) and ligand-to-ligand charge-transfer states (LLCT, N3 → bpy, phen). LMCTs are predominant in 4, while LLCTs are predominant in 5. The highest-energy bands are of mixed character in both complexes, and bpy and phen have marginal roles.
For complex 6, the band centred at 304 nm has an almost pure LMCT (py → Pt,N3) character.35 TDDFT assigns the band at 260 nm to LLCT transitions (OAc → py), but underestimates their oscillator strength values. The λmax of complex 6 is shifted towards the red region compared to complexes 4 and 5 which is due to the trans geometry of the azide ligands.36
Photochemistry of 4–6
Aqueous solutions of platinum (IV) complexes 4–6 (ca. 50 μM) were irradiated with UVA light (power ca. 1.5 mW/cm2), and UV-visible spectra were recorded after 0, 1, 5, 15, 30 and 60 min of irradiation (see Figure 5). Marked changes in the spectra occurred following irradiation. For 4 and 5, a large and weak band appeared at ca. 370 nm corresponding to formation of PtII species,18 and a decrease in the UV band at ca. 250 nm was observed. Complex 6 behaved slightly differently since no absorption appeared at wavelengths longer than 350 nm upon irradiation, with a new band appearing at 250 nm while at 300 nm there was a significant decrease in absorbance. The pH of the non-buffered aqueous solutions of 4, 5 and 6 after 2h UVA irradiation did not increase. Following irradiation of 6, ESI-MS analysis (positive ion mode) showed peaks corresponding to [Pt(OH)2(py)2 + H]+ (obs: 388.4 m/z, calc: 388.1 m/z) and its fragment [Pt(OH)(py)2]+ (obs: 370.4 m/z, calc: 371.1 m/z) (see Figure S2).
Figure 5.

UV-visible spectra of cis, trans-[Pt(bpy)(N3)2(OH)2] (4), cis, trans-[Pt(phen)(N3)2(OH)2] (5) and trans, trans, trans-[Pt(N3)2(OH)2(py)2] (6) after UVA irradiation for 0 (–), 1 ( ), 5 (
), 5 ( ), 15 (
), 15 ( ), 30 (
), 30 ( ), 60 (
), 60 ( ) and 120 (
) and 120 ( ) min. The arrows denote an increase or decrease of absorbance with increasing irradiation time.
) min. The arrows denote an increase or decrease of absorbance with increasing irradiation time.
Photoactivation studies of complexes 4–6 using visible (green) light (514 nm, 60 mW/cm2) were carried out on 1 mM 90% D2O / 10% d6-acetone solutions, acetone being used to aid dissolution. 1H NMR spectra were recorded after 0, 15, 30 and 60 min irradiation. Changes in the spectra for 4–6 were observed following irradiation with the acetate region providing the clearest picture of speciation; as judged by the free and bound acetate resonances (Figure 6). Complex 6 gave rise to free acetate the fastest, with ca. 85% reacting after only 15 min of irradiation, and after 30 min only a very small peak corresponding to bound acetate was visible. In contrast, only half of complexes 4 and 5 had reacted after 30 min, and after 60 min ca. 40% of each of the original compounds remained.
Figure 6.

1H NMR spectra showing loss of bound acetate on irradiation for complexes 4–6. The appearance of free acetate was confirmed by spiking with acetic acid; the very slight change in chemical shift of the free acetate is due to variation in the pH of the solutions.
Discussion
The standard protocol of using an aqueous solution of silver nitrate to remove chlorides from a PtII centre to produce reactive aqua adducts reportedly produces oxygen-bridged dimers when ligands such as bipyridine or phenanthroline are present.37 For this reason we employed the method of direct substitution of chloride by azide in DMF. Problems were encountered while attempting to oxidise the PtII complexes 1–3 by the method previously used to generate PtIV azides (H2O2 added to an aqueous suspension of the PtII diazide species)21,36,38 since complexes 1–3 required harsher conditions to undergo oxidation. The use of acetic acid followed by acetic anhydride allowed us to oxidise these complexes and to synthesise the diacetato PtIV products 4–6 in good yield.
Although there are many examples of PtIV bipyridine and phenanthroline compounds with halide or alkyl ligands39 complexes 4–6 appear to be the first reported PtIV pyridine, phenanthroline and bis-pyridine azido complexes. The oxidation of the PtII-azido precursors is hindered by their sensitivity to both heat and light. In contrast, compounds such as [Pt(L)R2] (L = bpy, phen; R = alkyl, aryl) are easily oxidised by H2O2,39g,h indeed such reactions are reported to proceed rapidly at room temperature. Ligands such as methyl form strong σ-bonds to PtII, raising the energy of the HOMO and thereby making the PtII species more susceptible to oxidation.39h Oxidation of halogen-containing complexes [Pt(L)X2] (X = Cl, Br, I) with H2O2 is much more difficult due to the greater ionic component in Pt–X bonds and the resultant reduced donation of charge to the metal centre. Halido PtIV α-diimine complexes have been produced by using oxidants such as Cl2, Br2 and/or more forcing conditions (e.g. high temperature or photocatalysis).39d-f,i None of these synthetic routes are suitable for oxidising [PtII(L)(N3)2] (L = bpy, phen) diazido complexes to a PtIV complex with oxygen bound in axial positions. The preference for groups such as OH or OCOCH3 in the axial positions arises from their stabilising effect on PtIV, which is a desirable feature for potential photoactivatable prodrugs since compounds containing axial chloride ligands are often readily reduced in vivo without irradiation.10
The oxidation route we employed to synthesize the diacetato complexes, which proceeds via initial formation of the monohydroxido monoacetato species [Pt(L)(N3)2(OAc)(OH)] (L = bpy, phen, (py)2) in H2O/acetic acid, is anticipated to occur with retention of the OH oxygen in the second step, in accordance with the literature.40 The oxidation step may proceed directly to the mixed OH/OAc complex with participation from the acetate anion41 or via a dihydroxido species, followed by nucleophilic substitution of the acetate. If the oxidation occurs via the latter mechanism, the first step in the substitution is anticipated to be protonation of OH. Since only the monosubsituted species [Pt(L)(N3)2(OAc)(OH)] is isolated, this would imply that the pKaH of the second axial hydroxido is lower than the pKa of acetic acid (4.75).42
Crystal structures and DFT electrostatic potentials
The bond lengths and angles of complexes 1 and 2 (Tables S1 and S2) are comparable to similar reported structures.43,44 For a discussion of the crystal packing of 1 see the supporting information.
Complex 3 shows a significant distortion from square-planar geometry which can be attributed to steric interactions between the azide ligands and the pyridine rings. Deviations from square planar towards tetrahedral geometry are common for PtII complexes containing bulky or rigid ligands.45,46 Complex 3 is symmetrical with regard to the azido ligands, the bond lengths within the azido groups are consistent with those found in other azido compounds.47,48
The azide bond lengths and angles for complexes 4–6 (Table 2) compare well with other platinum azide structures.36 The Pt–N(ring) bond lengths of 4–6 are ca. 0.02–0.04 Å longer than those of the corresponding PtII compounds (1–3); a difference which has also been reported for [PtIV(bpy)Cl4]9d and [PtII(bpy)Cl2].44
Distortion from ideal octahedral geometry occurs in all three PtIV structures (4–6). The axial Pt–O(acetate) bonds are bent with respect to the plane formed by the equatorial ligands in 4 and 5, possibly to minimise intramolecular repulsions; the bending of the axial acetate bond in similar PtIV complexes has been attributed to intramolecular hydrogen bonding.49,50 The restricted bites of the chelating bipyridine and phenanthroline ligands (ca. 80°) in 4 and 5 also cause distortion. Complexes 4–6 contain intermolecular hydrogen-bonded networks, and some weak π-π stacking exists between molecules of complex 5. Interestingly, complex 4 shows a weak AcO⋯H–C(bpy) interaction which is not present in 5, where acetate groups are interacting with the π electrons of the phen ligand. Figure 7 shows the changes in the electrostatic potential surfaces of 2 and 5 due to such an interaction.
Figure 7.

Electrostatic potential (ESP) surface of complex 2 and 5. ESP surfaces are shown both in space (with positive and negative regions shown in blue and red, respectively) and mapped on electron densities (isovalue = 0.004) of the molecule (ESP colour scale is such that δ+ → δ− in the direction blue → green → yellow → orange → red).
Photochemistry and TD-DFT
The photoreactions of PtIV azide complexes are of interest due to the potential use of such complexes as photoactivated anticancer agents.36
Absence of isosbestic points indicates that more than one photoproduct is obtained by irradiation. According to calculations and experimental work done on similar systems,51 it is reasonable to assume that both azide and acetate ligands can easily be displaced by solvent molecules upon irradiation. In fact, the strongly antibonding character of LUMO and LUMO+1 suggests that all the transitions having contributions from such orbitals are dissociative. As shown in Figure 6, formation of free acetate in irradiated samples can be easily monitored by 1H NMR. Furthermore, ligand dissociation can be confidently associated with reduction of PtIV to PtII, consistent with the appearance of the MLCT-like absorption at 370 nm (for 4 and 5) and the reported behaviour of [PtIV(bpy)Cl4].39c
The UV-vis spectra of complexes 4 and 5 after 2 h UVA irradiation were remarkably similar to those reported52 for [Pt(L)(OH)2] and [{Pt(L)(μ-OH)}2] (L = bpy, phen) suggesting that these were significant photoproducts. ESI-MS of irradiated solutions of 6 revealed that the py ligands can be retained as the acetate is lost, with species such as [Pt(OH)2(py)2 + H]+ detected (Figure S2); such aqua adducts would be effective for DNA binding if photoactivation is carried out in vivo. No dimers were observed, perhaps hindered by the trans geometry.
The lack of increase in pH of the solutions of 4–6 after 2 h UVA irradiation is in contrast to solutions of analogous PtIV-azido compounds containing NH3 ligands for which a sharp increase in pH to >10 was seen under similar conditions.51 We suggest that this is because continued irradiation of the NH3-containing complexes results in release of the coordinated ammine; it is not clear whether bpy, phen and py are released on continued irradiation, but they are significantly less basic thanNH3.
Complexes 4–6 underwent photoreaction with green light (514 nm) giving rise to free acetate. Although the 1H NMR spectra of 4 clearly show an NMR peak for the released acetate, irradiation of 5 and 6 produced more complicated spectra, possibly due to formation of significant mono-acetato Pt intermediates or through weak electrostatic interactions of the free acetate with the photoproducts. Although the absorbance in the UV-vis spectrum at 514 nm for all three complexes is very low, the photodissociative behaviour may be explained by the computed singlet transitions in Table 6. Low-energy transitions of a highly dissociative nature exist for complexes 4–6 (all involving LUMO and LUMO+1) despite the very small oscillator strengths; excitation of these transitions with green light can evidently still produce ligand dissociation.
Table 6.
Calculated low-energy absorption properties of 4–6.
| Tra | Composition | Energy, eV (nm) |
Oscillator Strength |
|---|---|---|---|
| 4 | |||
| 1 | HOMO→LUMO (91%) | 2.60 (477) | 0.0007 |
| 2 | HOMO–1→LUMO (25%), HOMO→LUMO+1 (66%) |
2.80 (442) | 0.0001 |
| 3 | HOMO–1→LUMO (62%) HOMO→LUMO +1 (28%) |
3.10 (400) | 0.0027 |
| 4 | HOMO–2→LUMO+1 (17%), HOMO–1→LUMO+1 (76%) |
3.25 (381) | 0.0019 |
| 5 | |||
| 1 | HOMO→LUMO (91%) | 2.55 (485) | 0.0006 |
| 2 | HOMO–2→LUMO (17%), HOMO–1→LUMO (−12%), HOMO→LUMO+1 (64%) |
2.77 (447) | 0.0001 |
| 3 | HOMO–2→LUMO (−29%), HOMO–1→LUMO (36%), HOMO→LUMO+1 (30%) |
3.06 (405) | 0.0032 |
| 4 | HOMO–2→LUMO+1 (49%), HOMO–1→LUMO+1 (−44%) |
3.22 (384) | 0.0032 |
| 6 | |||
| 1 | HOMO→LUMO (93%) | 2.38 (520) | 0.0 |
| 2 | HOMO–2→LUMO (−10%), HOMO–1→LUMO (85%) |
2.91 (427) | 0.0021 |
| 3 | HOMO→LUMO+1 (96%) | 3.09 (401) | 0.0 |
| 4 | HOMO–5→LUMO (45%), HOMO–3→LUMO (−41%) |
3.27 (379) | 0.0 |
| 5 | HOMO–2→LUMO (82%), HOMO–1→LUMO (10%) |
3.29 (377) | 0.0068 |
Tr = transition number as obtained in the TDDFT calculation output.
Experimental
Methods and Materials
Materials
Silver nitrate, pyridine (py), d6-acetone, d6-DMSO, D2O and 1,4-dioxane were purchased from Aldrich, NaN3, HCl (37%), acetic anhydride and acetic acid (> 99%) from Fisher, H2O2 and 1,10-phenanthroline (phen) from Sigma, K2[PtCl4] from Alfa Aesar, and 2,2′-bipyridine (bpy) from Acros.
NMR Spectroscopy
NMR spectra were recorded at 298 K on a Bruker DMX500 (1H: 500.13 MHz) magnet. Samples were prepared in d6-acetone, d6-DMSO, D2O or 90% H2O / 10% D2O with 1H chemical shifts referenced internally to dioxane (δ 3.75 ppm in D2O; δ 3.764 ppm for 90% H2O / 10% D2O). All data were processed with XWIN-NMR software (Version 3.6, Bruker, UK Ltd.). All J values are quoted in Hz.
Mass Spectrometry
Positive ion electrospray mass spectrometry (ESI-MS) was performed on a Platform II Mass Spectrometer (Micromass, Manchester, UK). The capillary voltage was 3.5 V and the cone voltage typically varied between 5 and 30 V. The source temperature was adjusted depending on the solvent. Data were acquired and processed with Mass Lynx (Version 2.0) software.
UV-visible Spectroscopy
UV-visible electronic absorption spectra were recorded on a Varian Cary 300 UV-visible spectrophotometer in 1-cm path-length cuvettes. Data were processed with Microcal Origin 5.0.
Light Sources
The ultraviolet light source used for photochemical studies was a broadband UVA lamp (2 × 15 W tubes, model VL-215L; Merck Eurolab, Poole, UK) which operated with a maximum output at 365 nm. Samples were irradiated at a distance of 10 cm from the lamp, where the power was ca. 1.5 mW/cm2, delivering a dose of 10 J/cm2 over 2 h. The laser (Coherent Innova 70C Spectrum) used for irradiation at 514 nm was equipped with a fibre optic (FT-600-UMT, Ø = 600 μm; Elliot Scientific) to enable delivery of light to a sample in the NMR probe. The fibre optic was placed 2 mm above the solution in the NMR tube at which distance the power (ca. 60 mW/cm2) was measured with a Coherent Fieldmate power meter (OP2-VIS head).
pH Measurements
pH values were measured with an Orion 710A pH meter equipped with a chloride-free micro-combination electrode (Aldrich) calibrated with Aldrich standard buffers (pH 4, 7 and 10).
X-ray Crystallography
Diffraction data were collected with Mo-Kα radiation (λ = 0.71073 Å) on a Bruker Smart Apex CCD diffractometer equipped with an Oxford Cryosystems low-temperature device operating at 150 K. Data were corrected for absorption using the program SADABS.53 The crystal structures of 1, 4 and 5 were solved by Patterson methods (SHELXS54 or DIRDIF55) The structures of 2, 3 and 6 were solved by direct methods (DIRDIF55 or SIR9256). All structures were refined against F2 using SHELXL54 or CRYSTALS.57 The crystal structures of 1 – 6 have been deposited in the Cambridge Crystallographic Data Centre under the accession numbers CCDC 709183,709182, 709184, 709185, 709186 and 709187 respectively, and the cif files are available in the Supplementary Information. ORTEP diagrams were generated using ORTEP 3v258 and POV-Ray3.6.59
Computational details
The calculations were generally performed with the Gaussian 03 (G03) program60 employing the DFT method, Becke three-parameter hybrid exchange functional61 and Lee-Yang-Parr's gradient corrected correlation functional62 (B3LYP). The LanL2DZ basis set63 and effective core potential were used for the Pt atom and the 6-31G** basis set64 was used for all other atoms. Geometry optimizations of 1–6 in the ground state were performed in the gas phase from the x-ray crystallographic structure and the nature of all stationary points was confirmed by normal mode analysis. The conductor-like polarizable continuum model method (CPCM)65 with water as solvent was used to calculate the electronic structure and the excited states of 1–6 in solution. Thirty-two singlet excited states and the corresponding oscillator strengths were determined with a Time-dependent Density Functional Theory (TD-DFT)66 calculation. The computational results are summarized in Tables 3-5 and in the Supplementary Information, where only electronic transitions with an oscillator strength value (f) higher than 0.01 are reported. The electronic distribution and the localization of the singlet excited states were visualized using the electron density difference maps (EDDMs).67 GaussSum 1.0568 was used for EDDMs calculations and for the electronic spectrum simulation.
Additional geometry optimizations and energy calculations on 4 were performed using the Amsterdam Density Functional 2007 program (ADF) at the gradient-corrected density functional theory (DFT) level using BP86 functional in combination with the TZP basis sets.69 A small frozen core was used for efficient treatment of the inner atomic shells. Uncontracted Slater-type orbitals (STOs) were used as basis functions. Relativistic effects were considered by the zeroth-order regular approximation (ZORA). Electronic excitation energies were computed with the asymptotically correct XC potential obtained with the statistical average of (model) orbital potentials (SAOP)70 using scalar relativistic Time-Dependent Density Functional Theory (TD-DFT) in the ADF program.
Syntheses
Caution! No problems were encountered during this work, however heavy metal azides are known to be shock sensitive detonators, therefore it is essential that any platinum azide compound is handled with care.
[Pt(bpy)(N3)2] (1)
[Pt(bpy)Cl2] (98.5 mg) was prepared from K2[PtCl4] and 2,2′-bipyridine in an 89% yield by the literature method.29 The isolated product was suspended in DMF (15 mL) and NaN3 (0.152 g, 2.14 mmol) added. After stirring at 298 K in the dark for 3 d, the volume was reduced to 1–2 mL and H2O added to precipitate the product. [Pt(bpy)(N3)2] was collected by filtration, washed with water, ethanol and diethyl ether, then dried under vacuum (93.1 mg, 91.7 %). δH (500 MHz; d6-DMSO; dioxane) 8.91 (d, H6,6′, 1J5,6 5.7, 2H), 8.57 (d, H3,3′, 1J3,4 8.0, 2H), 8.40 (t, H4,4′, 1J4, 5 7.7, 2H), 7.82 (t, H5,5′, 2H). Crystals suitable for X-ray structure determination were grown from DMF at 277 K.
[Pt(phen)(N3)2] (2)
The synthesis of [Pt(phen)Cl2] and [Pt(phen)(N3)2] was carried out by a similar method as described for the corresponding 2,2′-bipyridine complexes. From [Pt(phen)Cl2] (100 mg) obtained 2 (85.2 mg, 82.8 %). δH (500 MHz; d6-DMSO, dioxane): 9.20 (d, H2,9, 1J 5.3, 2H), 9.02 (d, H4,7, 1J 8.3, 2H), 8.28 (s, H5,6, 2H), 8.14 (d, H3,8, 2H). Crystals suitable for X-ray structure determination were grown from DMF at 277 K.
Trans-[Pt(N3)2(py)2] (3)
Trans-[PtCl2(py)2] (166.5 mg) was synthesised by the literature method30 and converted to the diazido complex 3 (162.6 mg, 94.7 %) by the same method used for preparation of 1 and 2. δH (500 MHz; d6-acetone, dioxane): 8.86 (dd, Ho, 1Jo,m 6.6, 2Jo,p 1.7, 4H), 8.12 (tt, Hp, 1Jp,m 7.7, 2H), 7.67 (dd, Hm, 4H). λmax (DMF)/nm: 273 (ε/M−1 cm−1 14 500) and 328sh (3456). Crystals suitable for X-ray structure determination were grown from pyridine at 277 K.
Trans, cis-[Pt(bpy)(OAc)2(N3)2] (4)
[Pt(bpy)(N3)2] (10.0 mg, 0.023 mmol) was suspended in acetic acid (> 99 %, 4 mL) and H2O2 (30 %, 0.23 mmol, 17 μL) added. After stirring at 298 K for 2 h, all the solvent was removed from the bright orange solution and acetic anhydride (3 mL) added. After stirring at 298 K for 3 d the solution was again reduced to dryness and ice-cold H2O added. The yellow solid was collected by filtration, washed with water, ethanol and ether and dried under vacuum. Crystallisation from a 50:50 acetone/water solution at 277 K gave 4 (7.3 mg, 57.4 %). λmax (H2O)/nm: 250 (ε/M −1 cm−1 19100), 304 (11400) and 315 (9760). δH (500 MHz; d6-acetone, dioxane): 9.30 (d, H6,6′, 1J5,6 5.9, 2H), 8.69 (d, H3,3′, 1J3,4 7.9, 2H), 8.47 (t, H4,4′, 1J4,5 7.9, 2H), 8.04 (t, H5,5′, 2H), 1.65 (s, CH3, 6H). ESI-MS m/z: 554.1 [M + H]+; 576.0 [M + Na]+. Crystals suitable for X-ray structure determination were grown from water at 277 K.
Trans, cis-[Pt(phen)(OAc)2(N3)2] (5)
Synthesised from 2 by the oxidation method outlined for synthesis of 4. Crystals suitable for structure determination were grown from H2O at 277 K. Yield of 5: 4.7 mg, 42.4 %. UV-vis (H2O)/nm: 272 (ε/M−1 cm−1 17900), 300sh (5840), 337 (1060) and 352 (796). δH (500 MHz; d6-acetone, dioxane): 9.56 (d, H2,9, 1J 5.5, 2H), 9.06 (d, H4,7, 1J 8.1, 2H), 8.39 (s, H5,6, 2H), 8.36 (d, H3,8, 2H), 1.56 (s, CH3, 3H). ESI-MS m/z: 578.3 [M + H]+, 600.3 [M + Na]+, 616.4 [M + K]+.
Trans, trans, trans-[Pt(OAc)2(N3)2(py)2] (6)
Synthesised from 3 by the oxidation method outlined for synthesis of 4. Yield of 6: 6.7 mg, 56.9 %. Crystals suitable for structure determination were grown from H2O at 277 K. UV-vis (H2O)/nm: 259 (ε/M−1 cm−1 12500) and 307 (16900). δH (500 MHz; d6-acetone, dioxane): 8.95 (d, Ho, 1J 5.9, 4H), 8.30 (t, Hp, 1J 7.5, 2H), 7.83 (t, Hm, 4H), 1.92 (s, CH3, 6H). ESI-MS m/z: 556.4 [M + H]+, 578.3 [M + Na]+, 594.5 [M + K]+.
Conclusions
Complexes 4–6 are the first reported examples of PtIV-azido complexes which contain phenanthroline, bipyridine and bispyridine ligands. The x-ray crystal structures of these complexes show near-octahedral geometry for 4 and 6, with a significant distortion of the axial groups to give a O–Pt–O angle of 165.12° for complex 5. Irradiation of complexes 4–6 with both UVA (365 nm) and green (514 nm) light results in reduction of the complexes to PtII species, with release of one or both of the axial acetate ligands. Generation of reactive aqua species (through azide release) provides promising novel mechanisms for cytotoxic activity against cancer cells. Irradiation of complexes 4–6 in water is not accompanied by a large increase in pH as was detected for the previously-reported ammine complex cis, trans, cis-[Pt(N3)2(OH)2(NH3)2],51 probably due to the reduced lability of the α-diimine ligands on photoactivation, compared to NH3, in agreement with the mass spectrometric data. TDDFT calculations enabled us to show the presence of strongly dissociative low energy transitions for complexes 4–6 since we could identify key orbitals and excited states, their relative positions and dependence on the nature of the ligands. In future work we hope to predict the electronic properties of complexes prior to synthesis. This will allow us to focus our efforts on compounds which exhibit the most appropriate absorption bands.
Supplementary Material
Table 4.
Experimental and calculated absorption properties of 5.
| λmax, nm ε(M−1 cm−1) |
Tra | Composition | Energy, eV (nm) |
Oscillator Strength |
Assignment |
|---|---|---|---|---|---|
| 337 (1060) and 352 (796) |
6 | HOMO–3→LUMO +1 (10%) HOMO–2→LUMO +1 (35%) HOMO–1→LUMO +1 (43%) |
3.38 (367) | 0.0103 | LMCT |
| 300 (5840) |
16 | HOMO–2→LUMO +2 (55%) HOMO–1→LUMO +2 (15%) HOMO→LUMO +3 (20%) |
3.89 (318) | 0.0249 | LLCT |
| 17 | HOMO–2→LUMO +3 (18%) HOMO–1→LUMO +3 (76%) |
3.90 (317) | 0.0125 | LLCT | |
| 19 | HOMO–3→LUMO +2 (70%) HOMO–2→LUMO +2 (11%) |
3.96 (313) | 0.0288 | LLCT/IL | |
| 272 (17900) |
27 | HOMO–6→LUMO +2 (20%) HOMO–3→LUMO +3 (52%) |
4.27 (290) | 0.0131 | LLCT |
| 28 | HOMO–10→LUMO (23%) HOMO–9→LUMO (24%) HOMO–8→LUMO +1 (26%) HOMO–7→LUMO (14%) |
4.28 (290) | 0.1048 | LMCT | |
| 29 | HOMO–8→LUMO (29%) HOMO–7→LUMO +1 (16%) HOMO–6→LUMO +2 (19%) |
4.33 (287) | 0.1087 | LMCT/d-d | |
| 30 | HOMO–9→LUMO (51%) HOMO–5→LUMO +3 (34%) |
4.40 (282) | 0.1474 | Mixed | |
| 31 | HOMO–10→L+1 (34%) HOMO–9→LUMO +1 (50%) |
4.42 (281) | 0.0225 | LMCT | |
| 32 | HOMO–10→LUMO (24%) HOMO–9→LUMO (22%) HOMO–5→LUMO +3 (39%) |
4.45 (279) | 0.0242 | Mixed |
Tr = transition number as obtained in the TDDFT calculation output.
Acknowledgement
This work was supported by a Scottish Enterprise Proof of Concept Award, by the MRC (G0701062) for N.F., the EPSRC (EP/E000945X) for H-C. T. and the EU (EU FP7 Marie Curie Fellowship Action IEF 220281 PHOTORUACD) for L.S. We thank members of the EU COST Action D39 for stimulating discussions.
Footnotes
Electronic Supplementary Information (ESI) is available: Crystallographic data for compounds 1–6, Ortep plots of 1 and 2, mass spectrometric data and TDDFT singlet excited-state data. CCDC numbers 709182 (2), 709183 (1), 709184 (3), 709185 (4), 709186 (5), 709187 (6) relate to the crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
References
- 1.Muro ML, Diring S, Wang X, Ziessel R, Castellano FN. Inorg. Chem. 2008 doi: 10.1021/ic800316x. DOI:10.1021/ic800316x. [DOI] [PubMed] [Google Scholar]
- 2.Tzeng B-C, Fu W-F, Che C-M, Chao H-Y, Cheung K-K, Peng S-M. Dalton Trans. 1999;6:1017–1023. [Google Scholar]
- 3.Wong KM-C, Tang W-S, Chu BW-K, Zhu N, Yam VW-W. Organometallics. 2004;23:3459–3465. [Google Scholar]
- 4.Cocchi M, Virgili D, Sabatini C, Fattori V, Di Marco P, Maestri M, Kalinowski J. Synth. Mater. 2004;147:253–256. [Google Scholar]
- 5.Geary EAM, Yellowlees LJ, Jack LA, Oswald IDH, Parsons S, Hirata N, Durrant JR, Robertson N. Inorg. Chem. 2005;44:242–250. doi: 10.1021/ic048799t. [DOI] [PubMed] [Google Scholar]
- 6.a) Wong E, Giandomenico CM. Chem. Rev. 1999;99:2451–2466. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]; b) Jain N, Mittal R, Ray KS, Srivastava TS, Bhattacharya RK. J. Inorg. Biochem. 1987;31:57–64. doi: 10.1016/0162-0134(87)85005-5. [DOI] [PubMed] [Google Scholar]; c) Kumar L, Kandasamy NR, Srivastava TS, Amonkar AJ, Adwankar MK, Chitnis MP. J. Inorg. Biochem. 1985;23:1–11. doi: 10.1016/0162-0134(84)85001-1. [DOI] [PubMed] [Google Scholar]; d) Jain N, Mittal R, Srivastava TS, Satyamoorthy K, Chitnis MP. J. Inorg. Biochem. 1994;53:79–94. doi: 10.1016/0162-0134(88)80029-1. [DOI] [PubMed] [Google Scholar]; e) Mansuri-Torshizi H, Srivastava TS, Parekh HK, Chitnis MP. J. Inorg Biochem. 1992;45:135–148. doi: 10.1016/0162-0134(92)80008-j. [DOI] [PubMed] [Google Scholar]; f) Gund A, Keppler BK. Angew. Chem. Int. Ed. 1994;33:186–188. [Google Scholar]; g) Mansuri-Torshizi H, Ghadimy S, Akbarzadeh N. Chem. Pharm. Bull. 2001;49:1517–1520. doi: 10.1248/cpb.49.1517. [DOI] [PubMed] [Google Scholar]
- 7.Dhar S, Liu Z, Thomale J, Dai H, Lippard SJ. J. Am. Chem. Soc. 2008;130:11467–11476. doi: 10.1021/ja803036e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall MD, Mellor HR, Callaghan R, Hambley TW. J. Med. Chem. 2007;50:3403–3411. doi: 10.1021/jm070280u. [DOI] [PubMed] [Google Scholar]
- 9.a) Hill MG, Bailey JA, Miskowski VM, Gray HB. Inorg. Chem. 1996;35:4585–4590. [Google Scholar]; b) Canty AJ, Hoare JL, Patel J, Pfeffer M, Skelton BW, White AH. Organometallics. 1999;18:2660–2667. [Google Scholar]; c) Kapteijn GM, Meijer MD, Grove DM, Veldman N, Spek AL, van Koten G. Inorg. Chim. Acta. 1997;264:211–217. [Google Scholar]; d) Hambley TW. ActaCryst. 1986;C42:49–51. [Google Scholar]
- 10.Nakabayashi Y, Erxleben A, Létinois U, Pratviel G, Meunier B, Holland L, Lippert B. Chem. Eur. J. 2007;13:3980–3988. doi: 10.1002/chem.200601271. [DOI] [PubMed] [Google Scholar]
- 11.Zayat L, Calero C, Albores P, Baraldo L, Etchenique R. J. Am. Chem. Soc. 2003;125:882–883. doi: 10.1021/ja0278943. [DOI] [PubMed] [Google Scholar]
- 12.Pinnick DV, Durham B. Inorg. Chem. 1984;23:1440–1445. [Google Scholar]
- 13.Anbalagan V, Srivastava TS. J. Photochem. Photobiol. 1992;66A:345–353. [Google Scholar]
- 14.Anbalagan V, Srivastava TS. J. Photochem. Photobiol. 1994;77A:141–148. [Google Scholar]
- 15.Anbalagan V, Srivastava TS. J. Photochem. Photobiol. 1995;89A:113–119. [Google Scholar]
- 16.Anbalagan V. J. Coord. Chem. 2003;56:161–172. [Google Scholar]
- 17.Cuny GD, Landgrebe KD, Smith TP. Bioorg Med. Chem. Lett. 1999;9:237–240. doi: 10.1016/s0960-894x(98)00714-8. [DOI] [PubMed] [Google Scholar]
- 18.Shukla S, Kamath SS, Srivastava TS. J. Photochem. Photobiol. 1988;44A:143–152. [Google Scholar]
- 19.a) Salassa L, Garino C, Salassa G, Gobetto R, Nervi C. J. Am. Chem. Soc. 2008;130:9590–9597. doi: 10.1021/ja8025906. [DOI] [PubMed] [Google Scholar]; b) Vlček A. Coord. Chem. Rev. 1998;177:219–256. doi: 10.1016/j.ccr.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gabrielsson A, Záliš S, Matousek P, Towrie M, Vlček A. Inorg. Chem. 2004;43:7380–7388. doi: 10.1021/ic049548n. [DOI] [PubMed] [Google Scholar]; d) Karidi K, Garoufis A, Tsipis A, Hadjiliadis N, den Dulk H, Reedijk J. Dalton Trans. 2005;7:1176–1187. doi: 10.1039/b418838a. [DOI] [PubMed] [Google Scholar]; e) Dunietz BD, Dreuw A, Head-Gordon M. J. Phys. Chem. B. 2003;107:5623–5629. [Google Scholar]; f) De Angelis F, Car R, Spiro TG. J. Am. Chem. Soc. 2003;125:15710–15711. doi: 10.1021/ja037373v. [DOI] [PubMed] [Google Scholar]
- 20.a) Vlček A, Záliš S. Coord. Chem. Rev. 2007;251:258–287. [Google Scholar]; b) Salassa L, Garino C, Albertino A, Volpi G, Nervi C, Gobetto R, Hardcastle KI. Organometallics. 2008;27:1427–1435. [Google Scholar]; c) Garino C, Ruiu T, Salassa L, Albertino A, Volpi G, Nervi C, Gobetto R, Hardcastle KI. Eur. J. Inorg. Chem. 2008;23:3587–3591. [Google Scholar]
- 21.Mackay FS, Moggach SA, Collins A, Parsons S, Sadler PJ. Inorg. Chim. Acta. 2008 DOI:10.1016/j.ica.2008.02.039. [Google Scholar]
- 22.Mackay FS, Woods JA, Heringová P, Kašparková J, Pizzaro AM, Moggach SA, Parsons S, Brabec V, Sadler PJ. Proc. Natl. Acad. Sci. USA. 2007;104:20743–20748. doi: 10.1073/pnas.0707742105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bednarski PJ, Mackay FS, Sadler PJ. Anti-Cancer Agents Med. Chem. 2007;7:75–93. doi: 10.2174/187152007779314053. [DOI] [PubMed] [Google Scholar]
- 24.Farrer NJ, Sadler PJ. Aust. J. Chem. 2008;61:669–674. [Google Scholar]
- 25.a) Bonnett R. Metal Complexes for Photodynamic Therapy. In: McCleverty JA, Meyer TJ, editors. Comprehensive Coord. Chem. II. Vol. 9. Elsevier; Oxford: 2004. pp. 945–1003. [Google Scholar]; b) Dougherty TJ, Marcus SL. Eur. J. Cancer. 1992;28A:1734̃1742. doi: 10.1016/0959-8049(92)90080-l. [DOI] [PubMed] [Google Scholar]; c) Brancaleon L, Moseley H. Lasers Med. Sci. 2002;17:173–186. doi: 10.1007/s101030200027. [DOI] [PubMed] [Google Scholar]
- 26.Nikolenko V, Yuste R, Zayat L, Baraldo LM, Etchenique R. Chem. Commun. 2005;13:1752–1754. doi: 10.1039/b418572b. [DOI] [PubMed] [Google Scholar]
- 27.Dougherty TJ. Adv. Photochem. 1992;17:275. [Google Scholar]
- 28.Bowman K, Dori Z. Inorg. Chem. 1970;9:395–397. [Google Scholar]
- 29.Newkome GR, Theriot KJ, Fronczek FR, Villar B. Organometallics. 1989;8:2513–2523. [Google Scholar]
- 30.Kauffman GB. Inorg. Synth. 1963;7:249–253. [Google Scholar]
- 31.Lee Y-A, Yoo KH, Jung O-S. Bull. Chem. Soc. Jpn. 2003;76:107–110. [Google Scholar]
- 32.Ren S, Cai L, Segal BM. Dalton Trans. 1999;9:1413–1422. [Google Scholar]
- 33.Hocking RK, Deeth RJ, Hambley TW. Inorg. Chem. 2007;46:8238–8244. doi: 10.1021/ic701166p. [DOI] [PubMed] [Google Scholar]
- 34.Deeth RJ, Tai H-C. unpublished work. [Google Scholar]
- 35.Vogler A, Kern A, Hüttermann J. Angew. Chem. Int. Ed. Engl. 1978;17:524–525. [Google Scholar]
- 36.Mackay FS, Woods JA, Moseley H, Ferguson J, Dawson A, Parsons S, Sadler PJ. Chem. Eur. J. 2006;12:3155–3161. doi: 10.1002/chem.200501601. [DOI] [PubMed] [Google Scholar]
- 37.Wimmer S, Castan P. Inorg. Chim. Acta. 1988;142:13–15. [Google Scholar]
- 38.Müller P, Schröder B, Kratochwil NA, Coxall RA, Parkin A, Parsons S, Sadler PJ. Angew. Chem. Int. Ed. 2003;42:335–339. doi: 10.1002/anie.200390110. [DOI] [PubMed] [Google Scholar]
- 39.a) Hodges KD, Rund JV. Inorg. Chem. 1975;14:525–528. [Google Scholar]; b) Peloso A. Dalton Trans. 1976;11:984–988. [Google Scholar]; c) Vogler A, Kunkely H. Angew. Chem. Int. Ed. Engl. 1982;21:209–210. [Google Scholar]; d) Aye K-T, Vittal JJ, Puddephatt RJ. Dalton Trans. 1993:1835–1839. [Google Scholar]; e) Rostovtsev VV, Henling LM, Labinger JA, Bercaw JE. Inorg. Chem. 2002;41:3608–3619. doi: 10.1021/ic0112903. [DOI] [PubMed] [Google Scholar]; f) Fanizzi FP, Natile G, Lanfranchi M, Tiripicchio A, Laschi F, Zanello P. Inorg. Chem. 1996;35:3173–3182. doi: 10.1021/ic960125y. [DOI] [PubMed] [Google Scholar]; g) Rashidi M, Nabavizadeh M, Hakimelahi R, Jamali S. Dalton Trans. 2001;23:3430–3434. [Google Scholar]; h) Thorshaug K, Fjeldahl I, Romming C, Tilset M. Dalton Trans. 2003;21:4051–4056. [Google Scholar]; i) Whang S, Estrada T, Hoggard PE. Photochem. Photobiol. 2004;79:356–359. doi: 10.1562/2003-12-05-ra.1. [DOI] [PubMed] [Google Scholar]
- 40.Giandomenico CM, Abrams MJ, Murrer BA, Vollano JF, Rheinheimer MI, Wyer SB, Bossard GE, Higgins JD. Inorg. Chem. 1995;34:1015–1021. doi: 10.1021/ic00109a004. [DOI] [PubMed] [Google Scholar]
- 41.Tamasi G, Cini R, Intini FP, Sivo MF, Natile G. Angew. Chem. Int. Ed. 2004;43:5081–5084. doi: 10.1002/anie.200454111. [DOI] [PubMed] [Google Scholar]
- 42.Weast RC, Astle MK, editors. CRC Handbook of Chemistry and Physics. (60th Ed) 1979:D165. [Google Scholar]
- 43.Connick WB, Henling LM, Marsh RE, Gray HB. Inorg. Chem. 1996;35:6261–6265. [Google Scholar]
- 44.Osborn RS, Rogers D. Dalton Trans. 1974;9:1002–1004. [Google Scholar]
- 45.Darensbourg DJ, Decuir TJ, Stafford NW, Robertson JB, Draper JD, Reibenspies JH, Kathó A, Joó F. Inorg. Chem. 1997;36:4218–4226. [Google Scholar]
- 46.Miedaner A, Raebiger JW, Curtis CJ, Millar SM, DuBois DL. Organometallics. 2004;23:2670–2679. [Google Scholar]
- 47.Maag H, Rydzewski RM. J. Org. Chem. 1992;57:5823–5831. [Google Scholar]
- 48.Ribas J, Monfort M, Diaz C, Bastos C, Solans X. Inorg. Chem. 1994;33:484–489. [Google Scholar]
- 49.Neidle S, Snook CF, Murrer BA, Barnard CFJ. Acta Cryst. 1995;C51:882–884. doi: 10.1107/s0108270194013600. [DOI] [PubMed] [Google Scholar]
- 50.Kim KM, Lee Y-A, Lee SS, Sohn YS. Inorg. Chim. Acta. 1999;292:52–56. [Google Scholar]
- 51.Ronconi L, Sadler PJ. Chem. Commun. 2008;2:235–237. doi: 10.1039/b714216a. [DOI] [PubMed] [Google Scholar]
- 52.Wimmer S, Castan P, Wimmer FL, Johnson NP. Dalton Trans. 1989;3:403–412. [Google Scholar]
- 53.Sheldrick GM. SADABS. University of Göttingen; Göttingen (Germany): 2004. [Google Scholar]
- 54.Sheldrick GM. Acta Cryst. 2008;A64:112–122. [Google Scholar]
- 55.Beurskens PT, Beurskens G, Bosman WP, de Gelder R, Garcia Granda S, Gould RO, Israel R, Smits JMM. University of Nijmegen; Toernooiveld 1, 6525 ED Nijmegen, The Netherlands: 1996. [Google Scholar]
- 56.Altomare A, Cascarano G, Giacovazzo G, Guagliardi A, Burla MC, Polidori G, Camalli M. J. Appl. Cryst. 1994;27:435–436. [Google Scholar]
- 57.Betteridge PW, Carruthers JR, Cooper RI, Prout K, Watkin J. J. Appl. Cryst. 2003;36:1487. [Google Scholar]
- 58.Farrugia LJ. J. Appl. Crystallogr. 1997;30:565. [Google Scholar]
- 59. Available to download from http://www.povray.org/download/
- 60.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr., Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski J, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson BG, Chen W, Wong MW, Gonzalez C, Pople JA. GAUSSIAN 03 (Revision D.01) Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 61.Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 62.Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 63.Hay PJ, Wadt WR. J. Chem. Phys. 1985;82:270–283. [Google Scholar]
- 64.McLean AD, Chandler GS. J. Chem. Phys. 1980;72:5639–5648. [Google Scholar]
- 65.a) Cossi M, Rega N, Scalmani G, Barone V. J. Comput. Chem. 2003;24:669–681. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]; b) Cossi M, Barone V. J. Chem. Phys. 2001;115:4708–4717. [Google Scholar]; c) Barone V, Cossi M. J. Phys.Chem. A. 1998;102:1995–2001. [Google Scholar]
- 66.a) Casida ME, Jamorski C, Casida KC, Salahub DR. J. Chem. Phys. 1998;108:4439–4449. [Google Scholar]; b) Stratmann RE, Scuseria GE, Frisch MJ. J. Chem. Phys. 1998;109:8218–8224. [Google Scholar]
- 67.Browne WR, O'Boyle NM, McGarvey JJ, Vos JG. Chem. Soc. Rev. 2005;34:641–663. doi: 10.1039/b400513a. [DOI] [PubMed] [Google Scholar]
- 68.O'Boyle NM, Vos JG. GaussSum. Dublin City University; 2005. Available at http://gausssum.sourceforge.net. [Google Scholar]
- 69.Te Velde G, Bickelhaupt FM, Baerends EJ, Fonseca Guerra C, Van Gisbergen SJA, Snijders JG, Ziegler T. J. Comput. Chem. 2001;22:931–967. [Google Scholar]
- 70.Schipper PRT, Gritsenko OV, van Gisbergen SJA, Baerends EJ. J. Chem. Phys. 2000;112:1344–1352. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.