

Abstract
Vigabatrin was a major drug in the treatment of epilepsy until the discovery that it was associated with an irreversible constriction of the visual field. Nevertheless, the drug is still prescribed for infantile spasms and refractory epilepsy. Disorganization of the photoreceptor nuclear layer and cone photoreceptor damage have been described in albino rats. To investigate the vigabatrin-elicited retinal toxicity further, we examined the retinal tissue of albino mice treated with two vigabatrin doses. The higher dose did not always cause the photoreceptor layer disorganization after 1 month of treatment. However, it triggered a massive synaptic plasticity in retinal areas showing a normal layering of the retina. This plasticity was shown by the withdrawal of rod but not cone photoreceptor terminals from the outer plexiform layers towards their cell bodies. Furthermore, both rod bipolar cells and horizontal cells exhibited dendritic sprouting into the photoreceptor nuclear layer. Withdrawing rod photoreceptors appeared to form ectopic contacts with growing postsynaptic dendrites. Indeed, contacts between rods and bipolar cells, and between bipolar cells and horizontal cells were observed deep inside the outer nuclear layer. This neuronal plasticity is highly suggestive of an impaired glutamate release by photoreceptors because similar observations have been reported in different genetically modified mice with deficient synaptic transmission. Such a synaptic deficit is consistent with the decrease in glutamate concentration induced by vigabatrin. This description of the neuronal plasticity associated with vigabatrin provides new insights into its retinal toxicity in epileptic patients.
Keywords: bipolar cells, horizontal cells, photoreceptors, toxicity, vision
Introduction
Vigabatrin, gamma-vinyl GABA (VGB), is a drug that used to be widely used in the treatment of partial epilepsy, pharmacoresistant epilepsy and infantile spasm (French et al., 1996; Lortie et al., 1997; Curatolo et al., 2006). VGB treatment greatly reduced the seizure frequency and made many patients seizure-free (Livingston et al., 1989). VGB is an irreversible inhibitor of GABA-transaminase and its antiepileptic action is a result of it increasing the concentration of GABA in the brain. Unfortunately, peripheral visual field loss in patients treated with VGB was reported during the late 1990s (Krauss et al., 1998; Ruether et al., 1998; Daneshvar et al., 1999). This functional impairment of vision was associated with altered electro-retinogram (ERG) measurements, suggesting damage to the retina in the visual system. The most consistently altered measurements include increased latency and reduced amplitude of the photopic ERG b-wave and abnormalities of flicker responses (Krauss et al., 1998; Ruether et al., 1998; Daneshvar et al., 1999; Miller et al., 1999; Harding et al., 2000; Coupland et al., 2001; Westall et al., 2002). All of these electrophysiological changes are indicative of a dysfunction in the cone photoreceptor pathway. However, other studies also reported decreases in amplitude of the scotopic ERG b-wave consistent with these patients also suffering an alteration in the rod phototransduction pathway (Daneshvar et al., 1999). The reversible decrease in electro-oculogram amplitude suggested that the cellular interactions between photoreceptors and the retinal pigment epithelium were affected (Arndt et al., 1999; Lawden et al., 1999). Finally, atrophy of the retinal fibre layer was observed in both infants (Buncic et al., 2004) and adults (Wild et al., 2006) taking vigabatrin.
In the retina, GABA activates GABAC receptors that are more sensitive than GABAA receptors and do not desensitize (Feigenspan & Bormann, 1998; Lukasiewicz et al., 2004). Furthermore, GABA can remain excitatory in the adult retina (Vardi et al., 2000; Varela et al., 2005; Duebel et al., 2006). VGB was shown to increase the retinal GABA concentration by up to sevenfold (Cubells et al., 1987; Neal et al., 1989) and therefore to a level that could trigger GABA excitotoxicity. Retinal examination identified major disorganization of the outer nuclear layer (ONL) in VGB-treated animals (Butler et al., 1987), long before visual field constriction in VGB-treated patients was reported. Retinal damage was further documented recently by descriptions of major glial reactions in rats and rabbits (Duboc et al., 2004; Ponjavic et al., 2004). In addition, cone photoreceptors showed disorganized or lost inner/outer segments in central areas with a normal retinal morphology (Duboc et al., 2004). However, these findings did not explain the loss of transmission from rod photoreceptors to bipolar cells indicated by scotopic ERG measurements (Coupland et al., 2001).
We report evidence of an expected plasticity at photoreceptor synapses during the VGB treatment in mice, which could explain these electrophysiological results in patients under scotopic conditions.
Materials and methods
Animal treatment
All animal experiments were performed in compliance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Visual Research and with the guidelines of the INSERM ethics committee. BALB/c mice (16 treated, 10 controls) were purchased from Janvier (Le Genest-St-Isle, France) at 6 weeks of age. VGB was dissolved in 0.9% NaCl at 100 mg/mL and injected intraperitoneally daily for 30 days at doses of 5 and 3 mg/day as described for treatment of animal epilepsy (Andre et al., 2001). These doses (250 or 150 mg/kg) are in the same range or lower than those prescribed to children (100 mg/kg, Bialer et al., 2001) or infants (250 mg/kg, Bialer et al., 2001; 400 mg/kg, Aicardi et al., 1996).
Histology
Animals were killed by CO2 inhalation. Eye cups were fixed overnight at 4 °C in 4% (w/v) paraformaldehyde in 0.01 M phosphate-buffered saline, pH 7.4. The tissue was cryoprotected by immersion in phosphate-buffered saline containing, successively, 10, 20 and 30% sucrose at 4 °C and then embedded in OCT (Labonord, Villeneuve d’Ascq, France). Vertical sections (8–10 μm thickness) were permeabilized for 5 min in phosphate-buffered saline containing 0.1% Triton X-100 (Sigma, St Louis, MO, USA), rinsed and incubated in phosphate-buffered saline containing 1% bovine serum albumin (Eurobio, Les-Ulis, France), 0.1% Tween 20 (Sigma) and 0.1% sodium azide (Merck, Fontenay-Sous-Bois, France) for 2 h at room temperature (20°C). The primary antibody was added to the solution and the samples incubated for 2 h at room temperature. The polyclonal antibodies used were directed against protein kinase C alpha (PKCα) (1: 2000, Sigma), vesicular glutamate transporter 1 (VGLUT1) (1: 2000, Millipore, Billerica, MA, USA), Goα (K-20; 1 : 100, Santa Cruz Biotechnology, Santa Cruz, CA, USA), calbindin D-28K (1 : 500, Millipore), mouse cone arrestin (1 : 20 000; Zhu et al., 2002) or glial fibrillary acidic protein (GFAP) (1 : 200, Dako, Carpinteria, CA USA). The monoclonal antibodies were directed against Goα (1 : 2000, Millipore), bassoon (1 : 100, StressGen, Ann Arbour, MI, USA) or calbindin D-28K (1 : 500, Sigma). After rinses, sections were incubated with the secondary antibody, goat anti-rabbit IgG or rabbit anti-mouse IgG conjugated to either Alexa TM594 or Alexa TM488 (1 : 500, Molecular Probes, Eugene, OR, USA), for 2 h. The dye, diamidiphenyl-indole (Sigma), was added to the solutions for the last incubations. Sections were rinsed, mounted with Fluorsave reagent (Calbiochem, San Diego, CA, USA) and viewed with a DM 5000B microscope (Leica Microsystems SAS, Rueil Malmaison, France) equipped with a Photometrics cool SNAP TM FX camera (Ropper Scientific, Tuscon, AZ, USA). For higher resolutions, preparations were observed with a confocal inverted microscope [Leica TCS Spectral (SP2) instrument] using 63 and 100 oil immersion objectives both with a numerical aperture of 1.4. A krypton/argon mixed-gas laser and two helium/neon mixed-gas lasers were used to generate the bands at 488, 543 and 633 nm, respectively.
Results
In mice treated daily with 5 mg/day, there was disorganization of the retinal architecture with photoreceptor nuclei moving into the layers of inner/outer segments toward the retinal pigment epithelium (see Fig. 1). However, this disorganization of the ONL was observed in only five of the 16 treated mice. Therefore, the photoreceptor layer can become disorganized in VGB-treated mice but this effect was not observed in all VGB-treated animals.
Fig. 1.

Disorganization of the photoreceptor ONL in VGB-treated mice. Diamidiphenyl-indole-stained vertical sections of the retina showing the linear ONL in a control mouse (A) and the photoreceptor nuclei migrating toward the retinal pigment epithelium (RPE) in a VGB-treated animal (B). Scale bar, 25 μm. INL, inner nuclear layer.
As retinal damage is often associated with retinal gliosis and an increase in GFAP expression, we stained retinal sections of VGB-treated and control animals with a GFAP antibody. In control albino mice, some glial Müller cells were intensely GFAP immunopositive (Fig. 2A); VGB treatment did not increase the intensity of this GFAP immunolabelling (Fig. 2B) except in highly disorganized areas (see below).
Fig. 2.

Cone cell damage without glial reaction in VGB-treated mice. Vertical retinal sections in control (A, C and E) and VGB-treated animals (B, D and F) stained with diamidiphenyl-indole to visualize cell bodies (blue in A–F) and immunolabelled with antibodies directed against the GFAP protein (red in A and B) and against cone arrestin (red in C–F). The GFAP immunostaining shows that glial Müller cells are immunopositive in an untreated albino mouse (A) with no enhancement in a VGB-treated animal (B). (C–F) Cone arrestin immunolabelling (CARRES in red) reveals that there are fewer cone outer segments (OSs) in the VGB-treated mouse (D and F) than in the control animal (C and E). Note the disorganization of the ONL close to the area showing cone damage in D. Scale bars, 25 μm. INL, inner nuclear layer.
Cone photoreceptors in VGB-treated mice were then examined following their identification with the mouse cone arrestin antibody (Zhu et al., 2002). Cone outer segments and their terminals were intensely labelled with the cone arrestin antibody (Fig. 2C and E). In VGB-treated mice, cone outer/inner segments were altered in particular areas, whereas their terminals remained intensely labelled in the outer plexiform layer (OPL) (Fig. 2D and F). These observations indicate that the VGB treatment led to cone damage.
To examine the reaction of inner retinal neurones to the VGB treatment, ON rod bipolar cells were stained with the antibody directed against the protein PKCα. In control animals, ON bipolar cells exhibited cell bodies with very short dendrites extending to the OPL and axons reaching the inner part of the inner plexiform layer (Fig. 3A and B). By contrast, in VGB-treated animals, rod ON bipolar cells extended their dendrites deep into the ONL (Fig. 3C and D). Note that on the diamidiphenyl-indole-stained section (Fig. 3C) this rod bipolar cell plasticity was observed in areas with no disorganization of the ONL. Indeed, growing dendrites of postsynaptic neurones were present throughout the retinal sections, suggesting that these features of plasticity occurred in postsynaptic neurones independently of the ONL disorganization. To determine whether this plasticity of postsynaptic neurones was restricted to bipolar cells, we used a calbindin antibody to label horizontal cells. Horizontal cell bodies in controls were located at the distal part of the inner nuclear layer with their dendritic processes extending into the OPL (Fig. 3E and F). In VGB-treated animals, horizontal cell processes were not limited to the OPL but they extended deep into the ONL (Fig. 3G and H). Therefore, the VGB treatment induced plasticity in both bipolar and horizontal cells.
Fig. 3.

Morphological changes in the outer retinal cells of VGB-treated animals. Vertical retinal sections from control (A, B, E, F, I, J, M and N) and VGB-treated (C, D, G, H, K, L, O and P) animals both stained with diamidiphenyl-indole (A, C, E, G, I, K, M and O) and immunolabelled with antibodies directed against one of the following: PKCα (B and D), calbindin (F and H), VGLUT1 (J and L) or bassoon (N and P). In the control animal, PKCα-positive ON bipolar cells (B) and calbindin-positive horizontal cells (F) have their dendritic tips in the OPL. In contrast, in the VGB-treated mouse, rod bipolar cells (D) and horizontal cells (H) have dendritic processes extending deep into the ONL. On the photoreceptor side, VGLUT1-positive structures (J) and bassoon-positive synaptic ribbons (N) are only found in the plexiform layers (F) of control animals, whereas they are observed deep within the ONL of the VGB-treated animals (H and P). Scale bar, 25 μm. IPL, inner plexiform layer; INL, inner nuclear layer.
To examine if this plasticity of postsynaptic neurones to photoreceptors was related to changes in synaptic terminals of photoreceptors themselves, we used an antibody directed against VGLUT1, a protein of photoreceptor synaptic vesicles. On retinal sections of control animals, the VGLUT1 immunolabelling was restricted to the OPL (Fig. 3I and J), whereas it was observed deep within the ONL in VGB-treated animals (Fig. 3K and L). We then used an antibody directed against the protein bassoon to study the synaptic specialization of photoreceptors, the synaptic ribbon. In the control retina, bassoon-positive structures were observed in both the OPL and inner plexiform layer (Fig. 3M and N), whereas in VGB-treated animals some synaptic ribbons were clearly identified in the ONL (Fig. 3O and P). These observations indicate that the synaptic plasticity of postsynaptic neurones was associated with a withdrawal of photoreceptor synapses.
Labelling with the cone arrestin antibody surprisingly suggested that cone photoreceptor terminals remained in the OPL even in highly disorganized areas (see Fig. 2D). To confirm that the neuronal morphology of cone terminals was not affected by VGB treatment in areas with sprouting of postsynaptic neurones, retinal sections were immunolabelled with the cone arrestin antibody and ON bipolar cells were identified with the Goα antibody. Although the OPL structure in VGB-treated mice was not as linear as in control animals, cone terminals remained in the OPL, whereas Goα-positive dendrites of ON bipolar cells were detected very deep inside the ONL (Fig. 4A–C). These observations suggested that rod but not cone photoreceptors were withdrawing their terminals toward their cell bodies.
Fig. 4.
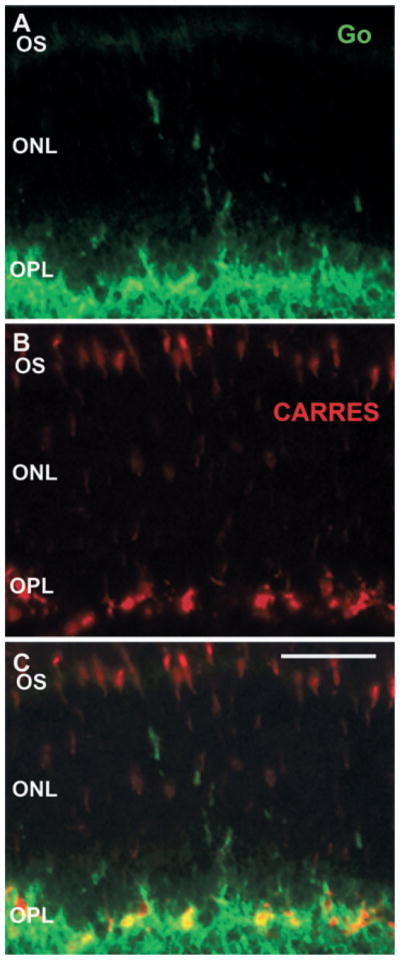
No detectable withdrawal of cone photoreceptor terminals in VGB-treated animals. Vertical retinal sections in a VGB-treated mouse stained with antibodies directed against the protein Goα (green in A and C) and against cone arrestin (CARRES in red in B and C). Note the presence of Goα-positive ON bipolar cell dendrites in the ONL of VGB-treated animals (green in A and C), whereas cone arrestin-immunopositive terminals remain localized in the OPL (red in B and C) as in the control animals. Scale bar, 25 μm. OS, outer segment.
To determine if the plasticity of photoreceptor postsynaptic neurones also involved neurones participating in cone terminals, cone ON bipolar cells were visualized by double immunolabelling with the protein Goα antibody to stain both cone and rod ON bipolar cells, and the protein PKCα antibody specifically to identify rod ON bipolar cells. In the inner nuclear layer, rod bipolar cells were labelled by both the anti-Goα antibody and the anti-PKCα antibody (green in Fig. 5A and D and yellow in Fig. 5C and F), whereas cone ON bipolar cells were only labelled by the Goα antibody (red in Fig. 5B, C, E and F). The sections were examined at high resolution with the confocal microscope; only rod ON bipolar cells exhibited dendrites extending into the ONL. Indeed, all Goα-positive dendrites seen to extend into the ONL were labelled with the PKCα antibody (yellow in Fig. 5F); this suggests that cone ON bipolar cells do not extend their dendrites into the ONL. These observations demonstrated that only rod ON bipolar cells underwent plasticity following VGB treatment.
Fig. 5.

Dendritic sprouting from rod but not cone ON bipolar cells in VGB-treated mice. Vertical retinal sections in control (A–C) and VGB-treated (D–F) animals were immunolabelled with antibodies directed against the PKCα to reveal rod bipolar cells (green in A, C, D and F) and against the protein Goα to identify rod and cone bipolar cells (red in B, C, E and F). These sections were also stained with diamidiphenyl-indole to reveal the nuclear layers (blue in C and F). Rod ON bipolar cells (green in A and D) are stained by both the anti-PKCα and anti-Goα antibodies (yellow in C and F), whereas cone ON bipolar cells are labelled only by the protein Goα antibody (red in B, C, E and F). Note that in the control animal (A–C), all ON bipolar cells have their dendrites ramifying in the OPL, whereas rod ON bipolar cells extend their dendrites into the ONL of the VGB-treated mouse (yellow in F). Scale bar, 20 μm.
We then examined if the withdrawing rod terminals and the sprouting postsynaptic neurones were forming ectopic synapses. We stained photoreceptor terminals with the antibody against the synaptic protein, VGLUT1, and ON bipolar cells with the Goα antibody. Under confocal microscopy, many dendritic tips of bipolar cells that had grown into the ONL were observed to be in contact with withdrawing VGLUT1-positive photoreceptor terminals (Fig. 6A–C, inset in C). Similarly, Goα-positive bipolar cell dendrites that had grown into the ONL were often associated with calbindin-positive sprouting dendrites of horizontal cells (Fig. 6D–F, inset in F). To confirm that these contacts may be ectopic synapses, photoreceptor synaptic ribbons were immunolabelled with the bassoon antibody. As described above, these ribbon synapses were all distributed within the OPL in the control animals (Fig. 7A) but they were found deep within the ONL among photoreceptor nuclei in VGB-treated animals (Fig. 7B). In this location, although few synaptic ribbons appeared to be isolated on the optical section observed by confocal microscopy (arrowhead in the upper part of Fig. 7B), many were seen to be in contact with PKCα-immunopositive sprouting dendrites of rod bipolar cells (Fig. 7B, inset). These observations were consistent with the formation of ectopic synapses by rod photoreceptors in the ONL.
Fig. 6.

Maintenance of cell contacts during cellular plasticity, induced by the VGB treatment, at the photoreceptor synapses. (A–C) Vertical retinal sections in VGB-treated animals stained with antibodies directed against the protein Goα (green in A, C, D and F), calbindin (red in B and C) or VGLUT1 (E and F). Both Goα-positive ON bipolar cells (A) and calbindin-positive horizontal cells (B) have dendrites growing into the ONL. The dendritic tips of these two cell populations are often seen in close apposition in the ONL (C, inset). Similarly, when photoreceptor terminals are labelled with VGLUT1, numerous tips of ON bipolar cell dendrites growing into the ONL can be seen in close contact with a VGLUT1-positive withdrawn photoreceptor terminal (inset in F). INL, inner nuclear layer. Scale bar, 25 μm.
Fig. 7.

Formation of ectopic synapses by rod photoreceptors. Vertical retinal sections in a control (A) and a VGB-treated (B) animal immunolabelled with antibodies directed against the protein Goα (green) and against bassoon (red) and stained with diamidiphenyl-indole (blue). In control animals (A), bassoon-immunopositive synaptic ribbons (red) are located in the OPL below the layer of photoreceptor nuclei and above the inner nuclear layer containing the Goα-positive bipolar cell bodies (green). In the VGB-treated mouse, some bassoon-positive structures are found among photoreceptor nuclei in the ONL (arrowheads) where they are often in contact with sprouting bipolar cell dendrites, thereby forming ectopic synapses (B). The inset illustrates such ectopic synapses between bassoon-positive structures and growing bipolar cell dendrites. Scale bar, 20 μm.
To investigate the dose dependence of this neuronal plasticity and of the photoreceptor layer disorganization, mice were treated with a lower VGB dose (3 mg/day). Under these conditions, retinal areas with photoreceptor layer disorganization were observed in all animals (n = 7) (Fig. 8); GFAP expression was abnormally high in all of these disorganized retinal areas (Fig. 8A and D). Surprisingly, alterations of cell morphology with sprouting of rod bipolar cells in the ONL were mainly restricted to the areas of photoreceptor layer disorganization (Fig. 8E). These observations indicate that the different histological changes (photoreceptor layer disorganization and neuronal plasticity) may be associated with different VGB doses.
Fig. 8.

Glial cell reaction and bipolar cell plasticity in disorganized retinal areas of VGB-treated mice. Vertical retinal sections in a control (A–C) and a VGB-treated (D–F) animal immunolabelled with antibodies directed against GFAP (green in A, C, D and F) and against PKCα (red in B, C, E and F) and stained with the diamidiphenyl-indole nuclear dye (blue in A–F). Note that the ONL is disorganized in the VGB-treated animal (D–F) with reference to the control animal (A–C). In this disorganized area, more GFAP-positive glial cell processes are observed (D). The dendrites of PKCα-positive bipolar cell dendrites, which normally contact photoreceptor terminals in the OPL (B), are found to have grown into the ONL at the site of the disorganization in the retina (E). Scale bar, 25 μm.
Discussion
The first report of VGB retinal toxicity described a disorganization of the ONL at the periphery in treated albino rats (Butler et al., 1987). This disorganization of the ONL with photoreceptor nuclei abutting the retinal pigment epithelium was subsequently confirmed (Duboc et al., 2004). Retinal damage was further substantiated by descriptions of cone photoreceptor damage (Duboc et al., 2004) and major glial reactions revealed as an increase in GFAP immunolabelling intensity (Ponjavic & Andreasson, 2001; Duboc et al., 2004). In the present study, we confirmed the disorganization of the photoreceptor layer and cone damage in VGB-treated albino mice with a higher incidence at the lowest VGB dose. In mice treated with the higher dose of VGB (5 mg/day) the most drastic change was the plasticity of postsynaptic bipolar and horizontal cells, involving withdrawal of presynaptic rod photoreceptor synapses. This plasticity of postsynaptic neurones was not observed in all animals and, as was the case in mice treated with a low dose of VGB, was restricted to highly damaged retinal areas in VGB-treated rats (data not shown). By contrast, mice treated with the higher VGB dose displayed neuronal plasticity in all retinal areas prior to any disorganization of the ONL. This study therefore suggests that VGB treatment may cause various different phenomena including neuronal plasticity, cone damage and ONL dysplasia.
Plasticity of retinal neurones has been reported in different animal models including ageing mice (Liets et al., 2006). In retinitis pigmentosa, photoreceptor degeneration is associated with photoreceptor axon sprouting into the inner nuclear layer, whereas postsynaptic neurones show only minor plasticity (Li et al., 1995). By contrast, plasticity of postsynaptic neurones has been reported in animal models of retinal detachment that involve photoreceptor apoptosis (Cook et al., 1995). This plasticity of the postsynaptic neurones is associated with deep withdrawal of photoreceptor synaptic terminals into the ONL (Lewis et al., 1998; Liets et al., 2006). Such plasticity has also been observed in a number of genetically modified animals in which synaptic transmission from photoreceptor to postsynaptic neurones is impaired. Examples of this observed plasticity include: (i) the Nob2 mouse with a mutation on the Cacna1f gene encoding the alpha1F subunit of photoreceptor voltage-dependent calcium channels (Chang et al., 2006); (ii) the knockout mice for the Ca2+-binding protein, Cabp4, specific to photoreceptors (Haeseleer et al., 2004); and (iii) the knockout mice for bassoon, the photoreceptor synaptic protein (Dick et al., 2003). In contrast, no synaptic plasticity was observed when the defect originated in the postsynaptic cells as, for example, in ON bipolar cells in either mGluR, metabotropic glutamate receptor 6 (mGluR6) (Masu et al., 1995) or Goα knockout mice (Dhingra et al., 2000). These various findings are consistent with the idea that a defect in photoreceptor synaptic transmission triggers plasticity of postsynaptic neurones, whereas dysfunction of postsynaptic neurones concerning reception of the presynaptic signal does not alter their synaptic organization. The VGB-induced photoreceptor impairment is further indicated by presynaptic plasticity in rod photoreceptors withdrawing their terminals as observed in both retinal detachment (Lewis et al., 1998) and in bassoon knockout mice with ectopic synapse formation in the ONL (Dick et al., 2003). Therefore, the plasticity of retinal neurones at rod photoreceptor synapses provides evidence for a defect in synaptic transmission from these photoreceptors.
Photoreceptors are glutamatergic neurones continuously releasing glutamate in the dark; this release is modulated by light stimulation such that the glutamate concentration in the synaptic cleft is a linear function of light intensity. We propose that the observed defect in photoreceptor synaptic transmission is related to the reduction in glutamate and glutamine concentrations previously reported in various brain structures of VGB-treated animals (Bernasconi et al., 1988; Halonen et al., 1991; Loscher & Horstermann, 1994). This decrease in glutamate concentration is related to a decrease in glutamine synthetase activity in VGB-treated animals (Waniewski & Martin, 1995). This would explain the formation of ectopic synapses in the ONL as seen in animal models with deficient photoreceptor synapses. It could also explain the degeneration of cone photoreceptors, because cone degeneration has also been described in bassoon knockout mice (Specht et al., 2007). However, it cannot explain the disorganization of the ONL seen in both VGB-treated mice and rats because such a disorganization of the photoreceptor layers has never been reported in any of the genetically modified animals described above. This decrease in glutamate concentration could therefore contribute significantly to the formation of ectopic synapses and cone degeneration but photoreceptor layer disorganization must presumably involve additional mechanisms. In particular, it is unclear if the VGB-induced GABA increase (Cubells et al., 1987; Neal et al., 1989) contributes to these events.
In humans, the measurements most consistently altered by VGB treatment include an increase in latency and a decrease in amplitude of the photopic ERG b-wave, and abnormalities of flicker responses (Krauss et al., 1998; Ruether et al., 1998; Daneshvar et al., 1999; Miller et al., 1999; Harding et al., 2000; Coupland et al., 2001; Westall et al., 2002). All of these electrophysiological changes are indicative of a dysfunction in the cone photoreceptor pathway. Alterations in the cone pathway have been confirmed by analysis of colour vision, visual acuity and other visual tasks depending on cone visual perception (Miller et al., 1999; Hilton et al., 2002). The absence of a significant difference in the ERG a-wave amplitude does not mean that cone photoreceptors themselves were not affected because the photopic ERG a-wave is attributed to neurones postsynaptic to cone photoreceptors in the primate retina (Bush & Sieving, 1994). The cone damage observed at all VGB doses in mice is therefore consistent with the decrease in the photopic ERG in VGB-treated epileptic patients.
Considering the rod pathway, the scotopic ERG a-wave is directly related to the kinetics of the rod photoreceptor response whereas the scotopic ERG b-wave is representative of ON rod bipolar cell function. A significant number of VGB-treated epileptic patients also display a scotopic ERG b-wave reduction (Coupland et al., 2001; Hardus et al., 2003). The observed plasticity of neurones postsynaptic to rod photoreceptors and the correlated impairment of synaptic transmission could readily explain these ERG changes. An impairment of the synaptic transmission from rod to ON bipolar cells should not affect the scotopic ERG a-wave but may decrease the amplitude of the ERG b-wave as in mGluR6 knockout mice (Masu et al., 1995). The withdrawal of rod photoreceptor terminals associated with the growth of postsynaptic dendrites may allow contacts to be maintained as in the bassoon knockout mice, thereby limiting the reduction in the scotopic ERG b-wave amplitude. Impairment of rod synaptic transmission and the correlated retinal cell plasticity could therefore explain the scotopic ERG b-wave amplitude reduction without change in the scotopic ERG a-wave amplitude. Observation of the retinal plasticity only at higher doses in VGB-treated mice may explain the inconsistency of the reduction in the scotopic ERG b-wave amplitude in VGB-treated epileptic patients.
Recent estimates of VGB-associated visual field loss suggest that approximately 30% of epileptic patients receiving this anticonvulsant treatment at normal therapeutic levels suffer visual abnormalities (Daneshvar et al., 1999; Lawden et al., 1999). Although neuronal plasticity can explain the change in the scotopic ERG-b wave, the reversible change in electro-oculogram in VGB-treated epileptic patients (Arndt et al., 1999; Lawden et al., 1999) cannot be a consequence of synaptic plasticity. The electro-oculogram provides an indication of a dysfunction at the interface between photoreceptors and the retinal pigment epithelium. This interface dysfunction may in fact trigger the neuronal plasticity. Indeed, in models of retinal detachment where photoreceptors are physically disconnected from the retinal pigment epithelium, a similar neuronal plasticity has been observed with rod photoreceptors withdrawing their terminals, whereas ON bipolar cells and horizontal cells exhibit dendrites sprouting into the ONL (Lewis et al., 1998). Cones, in contrast to rods, were not retracting their terminals. Cone photoreceptor damage in VGB-treated rats and the observed reactive retinal glial cells crossing the outer limiting membrane have already been suggested as showing similarities with retinal detachment (Duboc et al., 2004). The plasticity of postsynaptic neurones that we describe is further consistent with features of retinal detachment (Lewis et al., 1998). The VGB treatment may therefore cause reversible changes at the interface between photoreceptors and the retinal pigment epithelium, and these events may trigger the irreversible damage to the neuronal network like that observed following retinal detachment.
Conclusion
We report VGB-elicited neuronal plasticity that has not previously been described and gives insight into the observed electrophysiological alterations in VGB-treated epileptic patients. Treating mice with VGB should therefore provide an excellent animal model for investigating the molecular mechanisms of VGB toxicity in different strains of genetically modified mice.
Acknowledgments
We thank Philippe Fontange for his help with confocal microscopy and the referees for very helpful comments. This work was supported by INSERM, the University Pierre and Marie Curie (Paris 6), Fondation Ophtalmologique Adolphe de Rothschild (Paris), Assistance Publique-Hopitaux de Paris, Fédération des Aveugles de France, Agence Nationale pour la recherche (GABARET), European Economic Community (EVI-GENORET-512036) and Ovation Pharmaceuticals, Agence Nationale pour la Recherche (GAB-ARET). F.J. and Q.-P.W. received fellowships from the University of Tichrine (Syria) and the city of Paris (France), respectively. C.M.C. is the Mary D. Allen Chair in Vision Research, Doheny Eye Institute and thanks Mary D. Allen for her generous endowment and the NIH (EY015851) for their support. VGB was provided by Dr Malouvier (Sanofi-Aventis, Paris, France).
Abbreviations
- ERG
electroretinogram
- GFAP
glial fibrillary acidic protein
- mGluR
metabotropic glutamate receptor
- ONL
outer nuclear layer
- OPL
outer plexiform layer
- PKCα
protein kinase C alpha
- VGB
vigabatrin
- VGLUT1
vesicular glutamate transporter 1
References
- Aicardi J, Mumford JP, Dumas C, Wood S. Vigabatrin as initial therapy for infantile spasms: a European retrospective survey. Sabril IS Investigator and Peer Review Groups. Epilepsia. 1996;37:638–642. doi: 10.1111/j.1528-1157.1996.tb00627.x. [DOI] [PubMed] [Google Scholar]
- Andre V, Ferrandon A, Marescaux C, Nehlig A. Vigabatrin protects against hippocampal damage but is not antiepileptogenic in the lithium-pilocarpine model of temporal lobe epilepsy. Epilepsy Res. 2001;47:99–117. doi: 10.1016/s0920-1211(01)00299-6. [DOI] [PubMed] [Google Scholar]
- Arndt CF, Derambure P, Defoort-Dhellemmes S, Hache JC. Outer retinal dysfunction in patients treated with vigabatrin. Neurology. 1999;52:1201–1205. doi: 10.1212/wnl.52.6.1201. [DOI] [PubMed] [Google Scholar]
- Bernasconi R, Klein M, Martin P, Christen P, Hafner T, Portet C, Schmutz M. Gamma-vinyl GABA: comparison of neurochemical and anticonvulsant effects in mice. J Neural Transm. 1988;72:213–233. doi: 10.1007/BF01243421. [DOI] [PubMed] [Google Scholar]
- Bialer M, Johannessen SI, Kupferberg HJ, Levy RH, Loiseau P, Perucca E. Progress report on new antiepileptic drugs: a summary of the Fifth Eilat Conference (EILAT V) Epilepsy Res. 2001;43:11–58. doi: 10.1016/s0920-1211(00)00171-6. [DOI] [PubMed] [Google Scholar]
- Buncic JR, Westall CA, Panton CM, Munn JR, MacKeen LD, Logan WJ. Characteristic retinal atrophy with secondary ‘inverse’ optic atrophy identifies vigabatrin toxicity in children. Ophthalmology. 2004;111:1935–1942. doi: 10.1016/j.ophtha.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush RA, Sieving PA. A proximal retinal component in the primate photopic ERG a-wave. Invest Ophthalmol Vis Sci. 1994;35:635–645. [PubMed] [Google Scholar]
- Butler WH, Ford GP, Newberne JW. A study of the effects of vigabatrin on the central nervous system and retina of Sprague Dawley and Lister-Hooded rats. Toxicol Pathol. 1987;15:143–148. doi: 10.1177/019262338701500203. [DOI] [PubMed] [Google Scholar]
- Chang B, Heckenlively JR, Bayley PR, Brecha NC, Davisson MT, Hawes NL, Hirano AA, Hurd RE, Ikeda A, Johnson BA, McCall MA, Morgans CW, Nusinowitz S, Peachey NS, Rice DS, Vessey KA, Gregg RG. The nob2 mouse, a null mutation in Cacna1f: anatomical and functional abnormalities in the outer retina and their consequences on ganglion cell visual responses. Vis Neurosci. 2006;23:11–24. doi: 10.1017/S095252380623102X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook B, Lewis GP, Fisher SK, Adler R. Apoptotic photoreceptor degeneration in experimental retinal detachment. Invest Ophthalmol Vis Sci. 1995;36:990–996. [PubMed] [Google Scholar]
- Coupland SG, Zackon DH, Leonard BC, Ross TM. Vigabatrin effect on inner retinal function. Ophthalmology. 2001;108:1493–1496. doi: 10.1016/s0161-6420(01)00638-8. discussion 1497–1498. [DOI] [PubMed] [Google Scholar]
- Cubells JF, Blanchard JS, Makman MH. The effects of in vivo inactivation of GABA-transaminase and glutamic acid decarboxylase on levels of GABA in the rat retina. Brain Res. 1987;419:208–215. doi: 10.1016/0006-8993(87)90585-3. [DOI] [PubMed] [Google Scholar]
- Curatolo P, Bombardieri R, Cerminara C. Current management for epilepsy in tuberous sclerosis complex. Curr Opin Neurol. 2006;19:119–123. doi: 10.1097/01.wco.0000218225.50807.12. [DOI] [PubMed] [Google Scholar]
- Daneshvar H, Racette L, Coupland SG, Kertes PJ, Guberman A, Zackon D. Symptomatic and asymptomatic visual loss in patients taking vigabatrin. Ophthalmology. 1999;106:1792–1798. doi: 10.1016/S0161-6420(99)90345-7. [DOI] [PubMed] [Google Scholar]
- Dhingra A, Lyubarsky A, Jiang M, Pugh EN, Jr, Birnbaumer L, Sterling P, Vardi N. The light response of ON bipolar neurons requires G[alpha]o. J Neurosci. 2000;20:9053–9058. doi: 10.1523/JNEUROSCI.20-24-09053.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick O, tom Dieck S, Altrock WD, Ammermuller J, Weiler R, Garner CC, Gundelfinger ED, Brandstatter JH. The presynaptic active zone protein bassoon is essential for photoreceptor ribbon synapse formation in the retina. Neuron. 2003;37:775–786. doi: 10.1016/s0896-6273(03)00086-2. [DOI] [PubMed] [Google Scholar]
- Duboc A, Hanoteau N, Simonutti M, Rudolf G, Nehlig A, Sahel JA, Picaud S. Vigabatrin, the GABA-transaminase inhibitor, damages cone photoreceptors in rats. Ann Neurol. 2004;55:695–705. doi: 10.1002/ana.20081. [DOI] [PubMed] [Google Scholar]
- Duebel J, Haverkamp S, Schleich W, Feng G, Augustine GJ, Kuner T, Euler T. Two-photon imaging reveals somatodendritic chloride gradient in retinal ON-type bipolar cells expressing the biosensor Clomeleon. Neuron. 2006;49:81–94. doi: 10.1016/j.neuron.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Feigenspan A, Bormann J. GABA-gated Cl- channels in the rat retina. Prog Retin Eye Res. 1998;17:99–126. doi: 10.1016/s1350-9462(97)00008-6. [DOI] [PubMed] [Google Scholar]
- French JA, Mosier M, Walker S, Sommerville K, Sussman N. A double-blind, placebo-controlled study of vigabatrin three g/day in patients with uncontrolled complex partial seizures. Vigabatrin Protocol 024 Investigative Cohort. Neurology. 1996;46:54–61. doi: 10.1212/wnl.46.1.54. [DOI] [PubMed] [Google Scholar]
- Haeseleer F, Imanishi Y, Maeda T, Possin DE, Maeda A, Lee A, Rieke F, Palczewski K. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat Neurosci. 2004;7:1079–1087. doi: 10.1038/nn1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halonen T, Pitkanen A, Saano V, Riekkinen PJ. Effects of vigabatrin (gamma-vinyl GABA) on neurotransmission-related amino acids and on GABA and benzodiazepine receptor binding in rats. Epilepsia. 1991;32:242–249. doi: 10.1111/j.1528-1157.1991.tb05251.x. [DOI] [PubMed] [Google Scholar]
- Harding GF, Wild JM, Robertson KA, Rietbrock S, Martinez C. Separating the retinal electrophysiologic effects of vigabatrin: treatment versus field loss. Neurology. 2000;55:347–352. doi: 10.1212/wnl.55.3.347. [DOI] [PubMed] [Google Scholar]
- Hardus P, Verduin W, Berendschot T, Postma G, Stilma J, van Veelen C. Vigabatrin: longterm follow-up of electrophysiology and visual field examinations. Acta Ophthalmol Scand. 2003;81:459–465. doi: 10.1034/j.1600-0420.2003.00085.x. [DOI] [PubMed] [Google Scholar]
- Hilton EJ, Cubbidge RP, Hosking SL, Betts T, Comaish IF. Patients treated with vigabatrin exhibit central visual function loss. Epilepsia. 2002;43:1351–1359. doi: 10.1046/j.1528-1157.2002.00502.x. [DOI] [PubMed] [Google Scholar]
- Krauss GL, Johnson MA, Miller NR. Vigabatrin-associated retinal cone system dysfunction: electroretinogram and ophthalmologic findings. Neurology. 1998;50:614–618. doi: 10.1212/wnl.50.3.614. [DOI] [PubMed] [Google Scholar]
- Lawden MC, Eke T, Degg C, Harding GF, Wild JM. Visual field defects associated with vigabatrin therapy. J Neurol Neurosurg Psychiat. 1999;67:716–722. doi: 10.1136/jnnp.67.6.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GP, Linberg KA, Fisher SK. Neurite outgrowth from bipolar and horizontal cells after experimental retinal detachment. Invest Ophthalmol Vis Sci. 1998;39:424–434. [PubMed] [Google Scholar]
- Li ZY, Kljavin IJ, Milam AH. Rod photoreceptor neurite sprouting in retinitis pigmentosa. J Neurosci. 1995;15:5429–5438. doi: 10.1523/JNEUROSCI.15-08-05429.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liets LC, Eliasieh K, van der List DA, Chalupa LM. Dendrites of rod bipolar cells sprout in normal aging retina. Proc Natl Acad Sci USA. 2006;103:12 156–12 160. doi: 10.1073/pnas.0605211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston JH, Beaumont D, Arzimanoglou A, Aicardi J. Vigabatrin in the treatment of epilepsy in children. Br J Clin Pharmacol. 1989;27(Suppl 1):109S–112S. doi: 10.1111/j.1365-2125.1989.tb03470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lortie A, Chiron C, Dumas C, Mumford JP, Dulac O. Optimizing the indication of vigabatrin in children with refractory epilepsy. J Child Neurol. 1997;12:253–259. doi: 10.1177/088307389701200407. [DOI] [PubMed] [Google Scholar]
- Loscher W, Horstermann D. Differential effects of vigabatrin, gamma-acetylenic GABA, aminooxyacetic acid, and valproate on levels of various amino acids in rat brain regions and plasma. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:270–278. doi: 10.1007/BF00169293. [DOI] [PubMed] [Google Scholar]
- Lukasiewicz PD, Eggers ED, Sagdullaev BT, McCall MA. GABAC receptor-mediated inhibition in the retina. Vis Res. 2004;44:3289–3296. doi: 10.1016/j.visres.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Masu M, Iwakabe H, Tagawa Y, Miyoshi T, Yamashita M, Fukuda Y, Sasaki H, Hiroi K, Nakamura Y, Shigemoto R. Specific deficit of the ON response in visual transmission by targeted disruption of the mGluR6 gene. Cell. 1995;80:757–765. doi: 10.1016/0092-8674(95)90354-2. [DOI] [PubMed] [Google Scholar]
- Miller NR, Johnson MA, Paul SR, Girkin CA, Perry JD, Endres M, Krauss GL. Visual dysfunction in patients receiving vigabatrin: clinical and electrophysiologic findings. Neurology. 1999;53:2082–2087. doi: 10.1212/wnl.53.9.2082. [DOI] [PubMed] [Google Scholar]
- Neal MJ, Cunningham JR, Shah MA, Yazulla S. Immunocytochemical evidence that vigabatrin in rats causes GABA accumulation in glial cells of the retina. Neurosci Lett. 1989;98:29–32. doi: 10.1016/0304-3940(89)90368-6. [DOI] [PubMed] [Google Scholar]
- Ponjavic V, Andreasson S. Multifocal ERG and full-field ERG in patients on long-term vigabatrin medication. Doc Ophthalmol. 2001;102:63–72. doi: 10.1023/a:1017589301855. [DOI] [PubMed] [Google Scholar]
- Ponjavic V, Granse L, Kjellstrom S, Andreasson S, Bruun A. Alterations in electroretinograms and retinal morphology in rabbits treated with vigabatrin. Doc Ophthalmol. 2004;108:125–133. doi: 10.1023/b:doop.0000036780.96560.74. [DOI] [PubMed] [Google Scholar]
- Ruether K, Pung T, Kellner U, Schmitz B, Hartmann C, Seeliger M. Electrophysiologic evaluation of a patient with peripheral visual field contraction associated with vigabatrin. Arch Ophthalmol. 1998;116:817–819. [PubMed] [Google Scholar]
- Specht D, Tom Dieck S, Ammermuller J, Regus-Leidig H, Gundelfinger ED, Brandstatter JH. Structural and functional remodeling in the retina of a mouse with a photoreceptor synaptopathy: plasticity in the rod and degeneration in the cone system. Eur J Neurosci. 2007;26:2506–2515. doi: 10.1111/j.1460-9568.2007.05886.x. [DOI] [PubMed] [Google Scholar]
- Vardi N, Zhang LL, Payne JA, Sterling P. Evidence that different cation chloride cotransporters in retinal neurons allow opposite responses to GABA. J Neurosci. 2000;20:7657–7663. doi: 10.1523/JNEUROSCI.20-20-07657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela C, Rivera L, Blanco R, De la Villa P. Depolarizing effect of GABA in horizontal cells of the rabbit retina. Neurosci Res. 2005;53:257–264. doi: 10.1016/j.neures.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Waniewski RA, Martin DL. Repeated administration of gamma-vinylGABA reduces rat brain glutamine synthetase activity. J Neurochem. 1995;65:355–362. doi: 10.1046/j.1471-4159.1995.65010355.x. [DOI] [PubMed] [Google Scholar]
- Westall CA, Logan WJ, Smith K, Buncic JR, Panton CM, Abdolell M. The Hospital for Sick Children, Toronto. Longitudinal ERG study of children on vigabatrin. Doc Ophthalmol. 2002;104:133–149. doi: 10.1023/a:1014656626174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild JM, Robson CR, Jones AL, Cunliffe IA, Smith PE. Detecting vigabatrin toxicity by imaging of the retinal nerve fiber layer. Invest Ophthalmol Vis Sci. 2006;47:917–924. doi: 10.1167/iovs.05-0854. [DOI] [PubMed] [Google Scholar]
- Zhu X, Li A, Brown B, Weiss ER, Osawa S, Craft CM. Mouse cone arrestin expression pattern: light induced translocation in cone photoreceptors. Mol Vis. 2002;8:462–471. [PubMed] [Google Scholar]