

Abstract
Objective
We evaluated different dose effects of rIL-25 on acute ulcerative colitis.
Materials and methods
Mice were fed 2.5% dextran sulfate sodium (DSS) for 5 days while infused i.p. with repeated doses of rIL-25 (0.2, 0.4 and 0.8 μg) in PBS or PBS only after every 24 h at the same time as the start of the DSS exposure. Clinical, macroscopical and microscopical assessment of colitis severity with survival study was performed. Colonic IL-25 expression and production of IFN-γ, IL-10 and IL-4 was also analyzed.
Results
At a dose of 0.2 μg, colitis was aggravated with high mortality, better improvements were observed at a dose of 0.4 μg, and colitis-induced diarrhea was reversed at a dose of 0.8 μg. The expression of IL-25 was found to decrease in severe colitis. Moreover, IL-25 inhibited production of mucosal IFN-γ, induced increase in IL-10 but not IL-4.
Conclusion
Improvements in DSS-induced colitis in response to IL-25 suggest IL-25's protective role by mechanisms including inhibition of IFN-γ with enhancement of anti-inflammatory release.
Keywords: Acute colitis, IL-25, rIL-25, Dose, DSS
Introduction
Contrary to other IL-17 family members (IL-17A, IL-17B, IL-17C, IL-17D, and IL-17F), IL-25/IL-17E had been shown to possess unique biological activities that promote type 2 immune response [1–4]. Currently, evidence has also been growing on the involvement of IL-25 in allergic [5, 6], anti-tumor [7] and anti-inflammatory activities [8, 9] in various human and animal models. As extensive studies on the biological functions of the IL-17 family are still underway, biological activities typical of type 1 inflammatory responses have been suggested for IL-17A, IL-17B, and IL-17C [10]. Otherwise, a number of cytokines have been shown to play an important role in the progression of IBD with a complex and dynamic network that regulates mucosal immune responses via cytokines, chemokines, and growth factors [11]. On the basis of their roles, cytokines have been grouped into pro-inflammatory, regulatory, healing and repair and those which are predominantly chemokine [12].
The DSS model is a well-characterized model of colitis which has been shown to be ideal for studying the colitis immune mechanism, epithelial repairing process and drug therapy with cytokine profile resembling human IBD [12, 13]. Clinically, mice administered with DSS tend to develop colon inflammation with symptoms such as weight loss, diarrhea, and rectal bleeding [14, 15]. Microscopically epithelial erosion, mucosal infiltration and ulcers with subsequent destruction of crypt architecture of the colonic mucosa can be revealed [12, 14, 15]. In our previous study, we hinted out inhibitory effects of IL-25 on the development and progression of acute DSS-induced colitis in mice [16]. In the present investigation, we further used different concentrations of rIL-25 to perform differential dose studies, expression of IL-25 in colitis was determined and levels of colonic INF-γ, IL-10 and IL-4 production were measured in order to trace mechanisms by which IL-25 tend to regulate the development and progression of acute DSS-induced colitis in mice.
Materials and methods
Animals
Female C57BL/6 mice aged 6–8 weeks were purchased from Shanghai Experimental Center of Chinese Academy of Science (Shanghai, China). Throughout the experiments they were maintained at 22°C under a 12 h day/night cycle. The study protocol was approved by the Ethical and Research Committee of China Medical University and materials relating to animal experiments conform to the standards currently applied in China.
Induction of acute colitis and rIL-25 i.p. treatment
Acute colitis was induced by oral administration of 2.5% (w/v) DSS (molecular weight 36,000–50,000, MP Biomedical ICN, France) in drinking water ad libitum for 5 days. Three groups of DSS + rIL-25 treatment mice (n = 6 mice/group) were given i.p. different doses of rIL-25 (R&D System, MN, USA) (0.2, 0.4 and 0.8 μg in 0.1 ml PBS) at the same time as the start of the DSS exposure. The administration of the same doses of rIL-25 was repeated after every 24 h for the duration of 4 days. Mice in DSS + PBS group (n = 6) underwent identical procedures but were only injected i.p. with 0.1 ml PBS. All mice were sacrificed at day 6.
General assessment of colitis
General assessment of the development and progression of colitis was performed as previously described in our paper [16]. A modified scoring system was adopted based on change in body weight, stool consistency and bloody stool (Table 1). Disease activity index (DAI) in each group was determined as the sum total of scores divided by three. At day 6 mice from all groups were sacrificed by cervical dislocation.
Table 1.
Scoring system for disease activity index
| Score | Weight loss | Stool consistency | Blood stool |
|---|---|---|---|
| 0 | None | Normal | No blood |
| 1 | 1 ~ 5% | Slightly loose | Slightly bloody |
| 2 | 6 ~ 10% | Loose stools | Bloody |
| 3 | 11 ~ 20% | ||
| 4 | >20% | Diarrhea | Gross bleeding |
The disease activity index (DAI) is a mean of individual scores of weight loss, stool consistency, and bleeding
Stool consistency; A normal stool = stool with an appearance of well formed pellets; a slightly loose stool = a stool with any absence of well formed pellets; loose stool = stool with pasty, semi formed, soft material that not adhere to anal fur; diarrhea = liquid stool that adheres to anal fur
Gross bleeding = an appearance of visible blood that adheres to anal fur
The mice were blindly scored for the DAI
For macroscopic colitis assessment, the colon from each mouse was dissected, photographed and the length from the ileocecal junction to the anal verge was measured and recorded. Colonic tissue samples from each mouse were fixed in 4% paraformaldehyde, embedded in paraffin wax, sectioned and then were stained with hematoxylin and eosin. Histological evaluation was performed in a blinded fashion using a detailed and validating scoring system by Cooper et al. [15]. Attributed scoring systems were given as follows; normal colonic mucosa defined as grade 0; shortening and loss of the basal one-thirds of crypts with mild inflammation and edema in the mucosa defined as grade 1; loss of the basal two-third of the crypts with moderate inflammation in the mucosa defined as grade 2; loss of entire crypts with severe inflammation in the mucosa with remaining surface epithelium defined as grade 3; and the loss of entire crypts and surface epithelium with massive inflammation in the mucosa, muscularis propria and sub mucosa defined as grade 4.
Survival study
For survival study, acute colitis was induced by oral administration of 2.5% (w/v) DSS (molecular weight 36,000–50,000, MP Biomedical ICN, France) in drinking water ad libitum for the duration of 7 days. At that time, the mice were returned to normal drinking water while assessed for their survival. Three groups of DSS + rIL-25 treatment mice (n = 10 mice/group) were given i.p. different doses of rIL-25 (R&D System, MN, USA) (0.2, 0.4 and 0.8 μg in 0.1 ml PBS) at the same time as the start of the DSS exposure. The administration of the same doses of rIL-25 was repeated after every 24 h for the duration of 4 days. Mice in DSS + PBS group (n = 10) underwent identical procedures but were only injected i.p. with 0.1 ml PBS.
Western blot analysis
Frozen samples of distal colonic tissues from each mouse group were homogenized with RIPA Lysis Buffer (100 mg/ml) (Applygen Technologies Inc. China) supplemented with an aprotinin cocktail of inhibitors (Roche, Mannheim, Germany) and incubated at 4°C for 10 min. The homogenized solutions were centrifuged at 12,000g at 4°C for 10 min. Supernatants were separated and protein concentration in the lysates quantified with a Bradford quantitative protein assay kit (Applygen Technologies Inc., China). Total protein lysates 20 μg per lane were loaded and electrophoresed on a 10% SDS-PAGE before being electro-transferred to Hybond polyvinylidene difluoride membranes at 60 V for 2 h. The membrane was blocked with 5% nonfat milk in Tris-buffered saline supplemented with 0.05% Tween 20 (pH 7.6) at 4°C overnight and incubated with goat anti-mouse IL-25 polyclonal antibody (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at room temperature for 2 h. After washing, membrane was incubated with horseradish peroxidase-conjugated anti-mouse secondary antibody (1:10,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at room temperature for 2 h. After extensive washing, the blot was developed by using the ECL chemiluminescent detection kit (TransGen Biotech Co. Ltd., China) according to the manufacturer’s instructions. Images of target protein were obtained by the use of MF–ChemiBis 3.2 (DNR–Bio-Imaging System Ltd., Jerusalem, Israel).
Immunohistofluorescent staining
To evaluate expressions of IL-25 protein in the colonic mucosa, tissue sections (5 μm) from each mouse group were pretreated by boiling in citrate buffer 10 mM (pH 6.1) in a microwave oven. After cooling, non-specific binding was blocked with 5% BSA blocking reagent followed by incubation with primary antibody goat polyclonal anti-mouse IL-25 or isotype-matched IgG control antibodies (1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C in a humidified chamber. After incubation with the primary antibody, sections were washed with three changes of PBS and treated with fluorescein isothiocyanate (FITC) conjugated donkey anti-mouse secondary antibody (1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 30 min at 37°C in a dark chamber. Thereafter, the sections were extensively washed, mounted with aqueous UltraCruz Mounting Medium (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and then were examined with a fluorescence microscope (BX 61; Olympus, Japan) and images were taken by a digital CCD Image system (DP 71; Olympus, Japan) attached to the fluorescence microscope.
Cytokines assay
Murine IFN-γ, IL-10 and IL-4 cytokines were analyzed by ELISA kit (eBioscience Inc. San Diego, USA) according to the manufacturer’s instructions. Colonic tissue samples from each group were collected and homogenized with PBS homogenizing buffer (100 mg/ml) containing 1% Triton X-100 (AMRESCO Inc., USA) supplemented with a cocktail of protease inhibitors (AMRESCO Inc., USA). The homogenized solutions were centrifuged at 12,000g for 10 min, and the supernatants were separated into aliquots and stored at −70°C.
Statistics
Results are presented as mean ±SD using SAS Software (version 9.13; SAS Institute Inc., NC, USA). The statistic difference between means was analyzed using analysis of variance (ANOVA) for overall comparison and the Student–Newman–Keuls (SNK) test as post-test for individual comparisons. The mortality data were analyzed by Kaplan–Meier Survival curves. P < 0.05 was considered as statistically significant.
Results
General observations
In studying the development and progression of DSS-induced colitis, mice were orally fed 2.5% DSS for five consecutive days in order to establish acute colitis while minimizing mortality. In our study, we realized that decrease in body weight in mice treated with DSS + PBS was not as dramatic as those that received DSS + 0.2 μg rIL-25 and DSS + 0.8 μg rIL-25 (Fig. 1). Mice that received DSS exhibited marked variations in clinical symptoms during the development of colitis (Table 2). The disease index activity (DIA) in mice treated with 0.4 μg rIL-25 was significantly reduced compared to mice treated with DSS + PBS and those treated with DSS + 0.2 μg rIL-25 (P < 0.05). Marked variations in stool consistency were also observed between all groups. Mice treated with 0.2 μg rIL-25 showed no improvement. However, mice treated with 0.4 μg rIL-25 and 0.8 μg rIL-25 exhibited reduced diarrhea and gross bleeding (Table 2). As shown in Fig. 2, at necropsy, macroscopic inspection of colons revealed signs of severe colitis in mice treated with DSS + PBS compared to mild colitis in all mice treated with rIL-25. Since shortening of colon length is an indication of colonic inflammation [17], our study revealed significant difference between groups. The shortening of colons in mice treated with DSS + PBS was significant compared to the mice treated with 0.2 μg rIL-25, 0.4 μg rIL-25, and 0.8 μg rIL-25, respectively (P < 0.05) (Table 2). Our findings reveal that treatment with rIL-25 at all doses can result in certain forms of reduction of colon inflammation. Histological analysis was blindly carried out on sections from the distal colon stained by hematoxylin and eosin (Fig. 3a–e). We revealed variations in colonic damage between groups. Mice treated with 0.4 μg rIL-25 displayed less severe colonic damage (grade 2) compared to the mice treated with 0.2 μg rIL-25 and 0.8 μg rIL-25 (grade 3). Histological score was lower in mice treated with 0.4 μg rIL-25 compared to mice treated with 0.2 μg rIL-25 and 0.8 μg rIL-25 (Fig. 3f) (P < 0.05). Our histological findings correlated with clinical and macroscopic findings on dose variation effects of rIL-25 in the development and progression of acute DSS-induced colitis.
Fig. 1.

Changes in body weight of mice with DSS-induced colitis. The mice were fed DSS for 5 days and were repeatedly infused into the peritoneal cavity with rIL-25 (at indicated doses) or PBS after every 24 h for the duration of 4 days. The values represent means ±SD (n = 6 mice/group)
Table 2.
Clinical variables in different experimental groups at day 6
| Group (n = 6) | Mean DAIa | Diarrheab | Gross bleedingb | Mean colon length (cm)a |
|---|---|---|---|---|
| DSS + PBS | 2.6 ± 0.25 | 6/6 | 4/6 | 4.3 ± 0.33# |
| DSS + 0.2 μg rIL-25 | 2.7 ± 0.08 | 6/6 | 5/6 | 5.0 ± 0.21 |
| DSS + 0.4 μg rIL-25 | 2.2 ± 0.15* | 3/6 | 2/6 | 6.0 ± 0.18 |
| DSS + 0.8 μg rIL-25 | 2.5 ± 0.22 | 2/6 | 3/6 | 5.4 ± 0.27 |
aData are the mean ± SD
bNumber of mice with diarrhea or gross bleeding/total number of mice in each group
* P < 0.05 versus DSS + PBS and DSS + 0.2 μg rIL-25
# P < 0.05 versus DSS + 0.2 μg rIL-25, DSS + 0.4 μg rIL-25 and DSS + 0.8 μg rIL-25
Fig. 2.

Pictures of colons from normal mice (a), DSS + PBS treated mice (b) and DSS + rIL-25 treated mice at the dose of 0.2 μg (c), 0.4 μg (d) and 0.8 μg (e). Mice were killed at day 6. Treatments of rIL-25 were found to improve DSS-induced colitis macroscopically
Fig. 3.

Representative histological changes in H & E stained colonic tissues from normal mice (a), DSS + PBS treated mice (b) and DSS + rIL-25 treated mice at the dose of 0.2 μg (c), 0.4 μg (d) and 0.8 μg/0.1 ml PBS (e) with their histological scores (f). Treatments of rIL-25 at a dose 0.4 μg was found to improve DSS-induced colitis compared to the DSS + PBS treated mice and mice received rIL-25 at a dose of 0.2 and 0.8 μg. Mice were killed at day 6. The scores represent means ±SD (n = 6 mice/group). *P < 0.05 when compared to DSS + PBS and mice treated with rIL-25 at a dose of 0.2 μg and 0.8 μg
Survival study
Acute colitis was successively induced by administration of drinking water supplemented with 2.5% DSS for 7 days. There was marked variations in survival of mice between all groups (Fig. 4). In comparison to other groups, mice treated with 0.4 μg rIL-25 showed improvement and prolonged survival. The mice treated with 0.2 μg rIL-25 were fatally affected with severe bleeding and dramatic loss of body weight, 100% of mice in this group were dead as early as at day 8. Mice treated with DSS + PBS and mice treated with 0.8 μg rIL-25 revealed no difference in survival rate while mice treated with 0.4 μg rIL-25 had prolonged survival. Our study demonstrated efficiency of IL-25 at a dose of 0.4 μg.
Fig. 4.
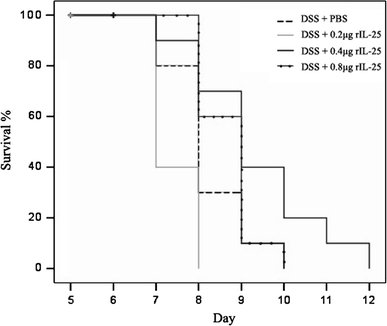
Kaplan–Meier survival curve. Mice were fed DSS to day 7 then returned to normal drinking water while assessed for their survival. rIL-25, at indicated doses, or PBS was given i.p. as repeated injections after every 24 h for the duration of 4 days. DSS + PBS (n = 10) and DSS + rIL-25 at a dose of 0.2 μg (n = 10), 0.4 μg (n = 10) and 0.8 μg (n = 10). Mice treated with rIL-25 at a dose of 0.4 μg exhibited improved and prolonged survival
Expression of IL-25 in colonic tissues
The western blot analysis was applied to assess the expression of IL-25 in colonic tissues. A band with an approximate molecular weight of 37 kd protein was detected, showing a considerable decrease in protein band intensity with increase in colitis as evident in the DSS + PBS treated group, DSS + 0.2 μg rIL-25 treated group and DSS + 0.8 μg rIL-25 treated group (Fig. 5). These findings reveal decrease in IL-25 expression in mice with severe colitis.
Fig. 5.
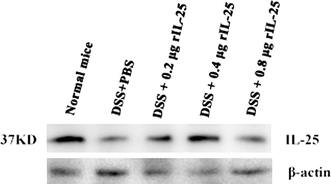
Western blots analysis. IL-25 expression was visually diminished in mice with severe colitis. rIL-25, at indicated doses, or PBS were injected i.p. repeatedly after every 24 h for the duration of 4 days. Mice were killed at day 6
Cellular source of IL-25 in colonic mucosa
To confirm the expression of IL-25 and to identify its cellular source, we further performed immunohistofluorescence study. IL-25 was expressed on the epithelium surface of normal colonic mucosa and colonic mucosa with mild inflammation (mice treated with 0.4 μg rIL-25) (Fig. 6a, d). However, no IL-25 expression was noted on entirely damaged mucosa. For this reason, we confirmed decrease in IL-25 expression in mice with severe colitis while assuming colonic epithelial cells as IL-25 cellular sources.
Fig. 6.
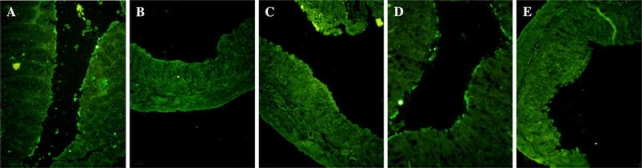
Immunofluorescent stained distal colonic tissues sections in normal mice (a), DSS + PBS treated mice (b) and DSS + rIL-25 treated mice at the dose of 0.2 μg (c), 0.4 μg (d) and 0.8 μg (e), respectively. IL-25 was expressed on the colonic mucosal epithelium surface in normal mice (a) and DSS + 0.4 μg rIL-25 treated mice (d). rIL-25, at indicated doses, or PBS were injected i.p. repeatedly after every 24 h for the duration of 4 days. Mice were killed at day 6
Cytokines assay
Previously, we reported production of IL-17A, IL-23p19/p40 and TGF-β1 in the development of acute DSS-induced colitis [16]. Herein, investigations of IFN-γ, L-10 and IL-4 secretion in colonic tissue homogenates revealed significant decrease in IFN-γ levels (P < 0.05) in mice treated with rIL-25 at different doses (Fig. 7a). In comparison with DSS + PBS treated mice, the level of IL-10 contents was significantly elevated in mice treated with 0.2 μg rIL-25 (P < 0.05). However, elevation of IL-10 levels in mice treated with 0.4 μg rIL-25 and 0.8 μg rIL-25 was statistically very close though not at significant levels (Fig. 7b). IL-4 levels were undetectable in all groups. Thus, treatment with rIL-25 resulted in decrease in IFN-γ colonic production with subsequent increase in IL-10 in this acute colitis model.
Fig. 7.

a Exogenous treatment of rIL-25 inhibited production of colonic IFN-γ at all dose, *P < 0.05 versus DSS + PBS. b The level of IL-10 was significantly elevated in DSS + rIL-25 at a dose of 0.2 μg treated mice (*P < 0.05 versus DSS + PBS) and close to the significant level at a dose of 0.4 and 0.8 μg. rIL-25 at indicated doses, or PBS were injected i.p. repeatedly after every 24 h for the duration of 4 days. Mice were killed at day 6. The values represent means ±SD (n = 6 mice/group)
Discussion
On the basis of clinical symptoms and histopathological changes, development and progression of colitis has been classified into acute, chronic and recovery phases [12]. Thus, the present study was only focused on the acute phase. It has been reported that differences in gender and genetic background are major determinants for susceptibility and inflammatory response in DSS-induced colitis models, in which case male mice were more severely affected than females [18]. Nonetheless, C57BL/6 mice were found to be more susceptible to DSS infections than BALB/c mice [12]. IL-25 has been implicated in the initiation of type 2 immunity [1] which involves coordinated regulation of innate and adaptive immune responses necessary for controlling pathogen invasion at mucosal sites and resistance to infections. In our recent study [16], we reported inhibitory effects of IL-25 on the development of acute DSS-induced colitis, in which clinical symptoms and histopathological changes were significantly reduced. In the present investigation, treatment of mice with different doses of rIL-25, resulted distinct dose related variations in the development of the clinical symptoms, histopathological changes and severity of DSS-induced colitis. Previously, treatment of different strains of mice with different doses of purified IL-25 protein at different time points resulted in Th2-type inflammatory responses with massive pathological changes [1]. Remarkably, our clinical findings showed significant reversal of colitis-induced diarrhea with resistance to the development of colitis-induced bleeding. On the contrary, at a small dose rIL-25 aggravated DSS-colitis. In addition, mild macroscopic morphological changes with less significant colonic shortening were noted in all mice treated with rIL-25. Consistent with these findings, histological observations revealed improvements of colitis with decline in mice mortality in a dose dependency manner. Thus, these findings reveal distinct IL-25 dose effects in protection of acute DSS-induced colitis.
Following western blot analysis, we detected expression of IL-25 in colonic tissues of normal mice. However, the expression of IL-25 was observed to decrease with increase in severity of colitis suggesting a protective role for IL-25 as evident in the mice treated with rIL-25 at a dose of 0.4 μg. In identifying colonic IL-25 cellular sources, we performed immunofluorescence study. We have observed the presence of IL-25 fluorescence on the colonic epithelium surface. Despite earlier reports on expression of IL-25 by Th2 cells [1] and mast cells [19], recently multiple cellular sources have also been identified in several tissues including alveolar macrophage with particle inhalation [20], activated eosinophils and basophils [21], brain microglia [9], some subpopulation of gut-associated T cell [8] and lung epithelial cells [5]. Thus, with the presence of IL-25 on mucosal epithelial surfaces of normal mice in our study, we may have found colonic epithelial mucosal cells as another IL-25 producer.
In attempting to trace the IL-25 inhibitory mechanism in this acute model, we choose to determine IFN-γ contents in colonic tissue homogenate as a pro-inflammatory (Th1) cytokine which has been widely reported to play a major role in the development DSS-induced colitis compared to TNF-α [22–24]. Studies on DSS-induced colitis revealed a robust production of IFN-γ not only in colonic tissue homogenate [12, 22] but also at the mRNA level with significant expression in colonic tissues [13]. In the present investigation, colonic IFN-γ contents were significantly lowered at all rIL-25 doses. This is in line with a report by Owyang et al. [9] in which infected IL-25 deficient mice exhibited severe inflammation and exacerbated pathology associated with increased production of IFN-γ. In addition, Owyang et al. also indicated that IL-25 can either directly inhibit or limit the expression and production of IFN-γ or IL-17. Our previous study [16] showed no inhibition of IL-17 by IL-25, however, observations from our present study showed inhibition of IFN-γ suggesting anti-inflammatory activity of IL-25 via inhibition or suppression of IFN-γ rather than IL-17 pathway in acute DSS-induced colitis. On the other hand, the above reports also seemed to support the hypothesis that IFN-γ is among essential mediators in the initiation of DSS-induced colitis.
Moreover, in our previous study [16], we revealed significant increase of TGF-β1 in mice administered with rIL-25. Since TGF-β1 was suggested to play an interrelated role with IL-10 in the prevention of colitis [25], herein we also wished to determine the impact of IL-10 in the regulation of DSS-induced colitis by IL-25. IL-10 is an anti-inflammatory cytokine shown to attenuate mucosal inflammation by inhibiting both antigen presentation and subsequent release of pro-inflammatory cytokines [26]. It was also reported that, in down-regulating the level of Th1 cytokine production, IL-10 whenever it remains unchecked, would otherwise inhibit TGF-β response [27]. As TGF-β production is necessary in the regulation of Th1 T cell-mediated mucosal inflammation, IL-10 has been reported to be a secondary factor required in facilitating TGF-β production [25]. However, some studies have also suggested that an increase in IL-10 in acute DSS-colitis might be likely due to an attempt to counteract and dampen the increased Th1 response [13]. Consistent with our data, study of acute DSS-induced colitis was also revealed that expression of IL-10 is up-regulated with an increase in severity of colonic mucosal injury [13]. On top of that, despite elevation of IL-10 production in the DSS + PBS treated mice, there was much increase of IL-10 production in DSS + rIL-25 treated mice. Hence association of IL-25 with IL-10 still remains unknown. Evidently, IL-25 deficient mice have been shown to exhibit severe destructive inflammation with an increased level of IL-10 though in conclusion it was not found to act in IL-25-dependent manner [8]. Moreover, studies by Fort et al. [1] also found no change in the gene expression of IL-10 in the mice treated with IL-25. Since we could not confirm the association of increased IL-10 with IL-25 we suggested further studies on the relationship of IL-25 with IL-10 and the environment which determine the biological consequences of IL-10 elevation in DSS-induced colitis.
Finally, we analyzed colonic production of IL-4 based on reported induction of IL-4 by IL-25[1]. Unlike IL-10, we have been unable to detect the level of IL-4 in this acute model. Meanwhile, only a very low level of IL-4 was reported in colonic tissue homogenates from C57BL/6 and BALB/c with acute DSS-induced colitis [12]. Similarly, at the mRNA level, no significant expression of IL-4 mRNA transcript was observed on colonic tissues from mice with either acute DSS-induced colitis [12, 13] or rIL-25 infusion [1]. IL-4 still remains among the anti-inflammatory cytokines whose roles are less well characterized in IBD [26]. IL-4 is believed to be a key cytokine in controlling the efficiency of the type 2 response [28] and to influence the balance of Th1 and Th2 cells towards an anti-inflammatory one [13]. Since IL-25 has been reported to induce IL-4 and Th2 cytokine response [1], it is obvious that there was no early induction of IL-4 by exogenous IL-25.
In conclusion, our study demonstrated IL-25 dose dependence improvements in colitis. A possible mechanism is either through direct inhibition or suppression of the expression and production of IFN-γ with subsequent enhancement of anti-inflammatory cytokine release. In addition to that, we revealed a decrease in mucosa IL-25 expression in acute colitis. These results suggest further study to explore the possible IL-25 protective role in the pathophysiology of acute ulcerative colitis.
Acknowledgments
The present work was supported by a grant from the Natural Science Foundation of Liaoning province, China (No. 20072101).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- rIL-25
Recombinant interleukin-25
- DSS
Dextran sulfate sodium
- IL
Interleukin
- i.p.
Intraperitoneal
- DAI
Disease activity index
- IBD
Inflammatory bowel disease
Contributor Information
S. S. Salum Mchenga, Phone: +86-24-23256666, Email: salummchenga@yahoo.com
D. Wang, Phone: +86-24-23256666, Email: wangdanan@mail.cmu.edu.cn
F. M. Janneh, Email: janefmusa@yahoo.com
Y. Feng, Phone: +86-24-23256666, Email: yonghui_feng@hotmail.com
P. Zhang, Phone: +86-24-23256666, Email: jyzhangpei@163.com
Z. Li, Phone: +86-24-23256666, Email: lizongxi@yahoo.com
C. Lu, Phone: +86-24-23256666, FAX: +86-24-23255291, Email: chllu@hotmail.com
References
- 1.Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–995. doi: 10.1016/S1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 2.Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, et al. New IL-17 family members promote Th1 or Th2 responses in the lung: In vivo function of the novel cytokine IL-25. J Immunol. 2002;169:443–453. doi: 10.4049/jimmunol.169.1.443. [DOI] [PubMed] [Google Scholar]
- 3.Pan G, French D, Mao W, Maruoka M, Risser P, Lee J, et al. Forced expression of murine IL-17E induces growth retardation, jaundice, a Th2-biased response, and multiorgan inflammation in mice. J Immunol. 2001;167:6559–6567. doi: 10.4049/jimmunol.167.11.6559. [DOI] [PubMed] [Google Scholar]
- 4.Kim MR, Manoukian R, Yeh R, Silbiger SM, Danilenko DM, Scully S, et al. Transgenic overexpression of human IL-17E results in eosinophilia, B-lymphocyte hyperplasia, and altered antibody production. Blood. 2002;100:2330–2340. doi: 10.1182/blood-2002-01-0012. [DOI] [PubMed] [Google Scholar]
- 5.Angkasekwinai P, Park H, Wang YH, Wang YH, Chang SH, Corry DB, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tamachi T, Maezawa Y, Ikeda K, Kagami S, Hatano M, Seto Y, et al. IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J Allergy Clin Immunol. 2006;118:606–614. doi: 10.1016/j.jaci.2006.04.051. [DOI] [PubMed] [Google Scholar]
- 7.Benatar T, Cao MY, Lee Y, Li H, Feng N, Gu X, et al. Virulizin induces production of IL-17E to enhance antitumor activity by recruitment of eosinophils into tumors. Cancer Immunol Immunother. 2008;57:1757–1769. doi: 10.1007/s00262-008-0502-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Owyang AM, Zaph C, Wilson EH, Guild KJ, McClanahan T, Miller HR, et al. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J Exp Med. 2006;203:843–849. doi: 10.1084/jem.20051496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–170. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an interleukin (IL)-25–dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. 2006;203:1105–1116. doi: 10.1084/jem.20051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS, et al. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melgar S, Karlsson A, Michaëlsson E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1328–G1338. doi: 10.1152/ajpgi.00467.2004. [DOI] [PubMed] [Google Scholar]
- 13.Egger B, Bajaj-Elliott M, MacDonald TT, Inglin R, Eysselein VE, Büchler MW. Characterisation of acute murine dextran sodium sulphate colitis: cytokine profile and dose dependency. Digestion. 2000;62:240–248. doi: 10.1159/000007822. [DOI] [PubMed] [Google Scholar]
- 14.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 15.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 16.Mchenga SS, Wang D, Li C, Shan F, Lu C. Inhibitory effect of recombinant IL-25 on the development of dextran sulfate sodium-induced experimental colitis in mice. Cell Mol Immunol. 2008;5:425–431. doi: 10.1038/cmi.2008.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Axelsson LG, Landström E, Bylund-Fellenius AC. Experimental colitis induced by dextran sulphate sodium in mice: beneficial effects of sulphasalazine and olsalazine. Aliment Pharmacol Ther. 1998;12:925–934. doi: 10.1046/j.1365-2036.1998.00357.x. [DOI] [PubMed] [Google Scholar]
- 18.Mähler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO, et al. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol. 1998;274:G544–G551. doi: 10.1152/ajpgi.1998.274.3.G544. [DOI] [PubMed] [Google Scholar]
- 19.Ikeda K, Nakajima H, Suzuki K, Kagami S, Hirose K, Suto A, et al. Mast cells produce interleukin-25 upon Fc epsilon RI-mediated activation. Blood. 2003;101:3594–3596. doi: 10.1182/blood-2002-09-2817. [DOI] [PubMed] [Google Scholar]
- 20.Kang CM, Jang AS, Ahn MH, Shin JA, Kim JH, Choi YS, et al. Interleukin-25 and interleukin-13 production by alveolar macrophages in response to particles. Am J Respir Cell Mol Biol. 2005;33:290–296. doi: 10.1165/rcmb.2005-0003OC. [DOI] [PubMed] [Google Scholar]
- 21.Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, Hippe A, Corrigan CJ, Dong C, Homey B, Yao Z, Ying S, Huston DP, Liu YJ. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC activated Th2 memory cells. J Exp Med. 2007;204:1837–1847. doi: 10.1084/jem.20070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito R, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, Imanishi J, et al. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin Exp Immunol. 2006;146:330–338. doi: 10.1111/j.1365-2249.2006.03214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Y, Hunt NH, Bao S. The effect of restraint stress on experimental colitis is IFN-gamma independent. J Neuroimmunol. 2008;200:53–61. doi: 10.1016/j.jneuroim.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Horino J, Fujimoto M, Terabe F, Serada S, Takahashi T, Soma Y, et al. Suppressor of cytokine signaling-1 ameliorates dextran sulfate sodium-induced colitis in mice. Int Immunol. 2008;20:753–762. doi: 10.1093/intimm/dxn033. [DOI] [PubMed] [Google Scholar]
- 25.Fuss IJ, Boirivant M, Lacy B, Strober WT. he interrelated roles of TGF-beta and IL-10 in the regulation of experimental colitis. J Immunol. 2002;168:900–908. doi: 10.4049/jimmunol.168.2.900. [DOI] [PubMed] [Google Scholar]
- 26.Sanchez-Munoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008;14:4280–4288. doi: 10.3748/wjg.14.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melgar S, Yeung MM, Bas A, Forsberg G, Suhr O, Oberg A, et al. Over-expression of interleukin 10 in mucosal T cells of patients with active ulcerative colitis. Clin Exp Immunol. 2003;134:127–137. doi: 10.1046/j.1365-2249.2003.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voehringer D, Shinkai K, Locksley RM. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity. 2004;20:267–277. doi: 10.1016/S1074-7613(04)00026-3. [DOI] [PubMed] [Google Scholar]