

Abstract
Obtaining high stereoselectivity in glycosylation reactions is often challenging in the absence of neighboring group participation. In this study, we demonstrate that activation of glycosyl trichloroacetimidate donors with immobilized perchloric acid on silica (HClO4–SiO2) provides higher α-selectivity than trimethylsilyl triflate (TMSOTf) for reactions that do not involve neighboring group participation.
Keywords: Glycosylation, Trichloroacetimidates, Stereoselectivity, Perchloric acid on silica
Carbohydrates play an important role in a variety of biological processes and are important constituents of many natural products and drugs.1 Studies on this class of biopolymers require access to structurally-defined, homogeneous compounds; however, carbohydrates are notoriously difficult to isolate from natural sources and, when available, are typically only available in small quantities. Chemical and chemo-enzymatic methods provide a useful alternative for obtaining carbohydrates.2–4 Both natural and unnatural carbohydrates can be obtained, and much larger quantities of pure material can be prepared.
The key step in the synthesis of carbohydrates is formation of the glycosidic linkages between monosaccharide units.2–4 Both α and β linkages are possible, and control of stereochemistry is critical. In some cases, stereochemistry can be controlled by use of protecting groups that are capable of neighboring group participation. For example, esters at the C-2 position of a glycosyl donor typically provide high selectivity for the 1,2-trans glycoside product. Stereocontrol in the absence of neighboring group participation is considerably more challenging.5 Many factors, such as steric hindrance of protecting groups, reaction solvent, and temperature, can affect the stereochemical outcome of a glycosylation reaction, and these effects are typically difficult to predict for any given donor-acceptor pair. Better methods to control stereoselectivity in the absence of neighboring group participation are needed.
Perchloric acid on silica (HClO4–SiO2) has recently been introduced as a user-friendly acid catalyst for a variety of organic transformations, such as protection–deprotection reactions,6–8 rearrangements9, 10 and esterifications.11 HClO4–SiO2 is an air stable powder that is easily prepared, is conveniently handled, and can readily be removed from reaction mixtures by filtration. HClO4–SiO2 has been examined previously as a catalyst for glycosylation reactions.12, 13 In particular, it was used as an alternative to the highly moisture sensitive trimethylsilyl triflate (TMSOTf) for the activation of trichloroacetimidate glycosyl donors. HClO4–SiO2 performed in all cases as well as TMSOTf for the activation of C-2 acylated donors, leading to the desired β-products for the Glc/Gal-series and α-products for Man-series compounds with high chemical yields. Much less was known about the effects of HClO4–SiO2 activation on the stereochemical outcome in the absence of neighboring group participation. In this study, we demonstrate that activation of glycosyl trichloroacetimidate donors with HClO4–SiO2 provides enhanced α-selectivity relative to TMSOTf in the absence of neighboring group participation.
Our initial studies focused on the synthesis of glycopeptides. Glycopeptides are typically prepared via solid phase peptide synthesis with incorporation of suitably protected glyco-amino acids at the appropriate position(s). For most O-linked glycans, the central connection between the glycan portion and peptide involves an α linkage between a GalNAc residue and a serine or threonine. Therefore, stereoselective formation of the requisite α-linkage between protected GalNAc derivatives and either serine or threonine is critical for the synthesis of this family of carbohydrates.
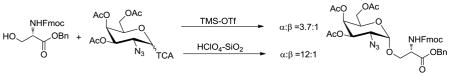
The synthesis of the key glycosidic linkage between GalNAc and serine/threonine has been studied in detail previously, and a variety of anomeric leaving groups have been investigated. Most frequently, glycosyl bromides,14–17 trichloroacetimidates (TCA),18–20 phenylsulfides,21, 22 sulfoxides,23 and phenylselenides24 have been used as glycosyl donors with adequately protected acceptor amino acids. In general, many glycosylation methods and conditions will give good α-selectivity when the secondary alcohol of threonine is used as the acceptor. With primary alcohols, such as serine derivatives, stereoselectivities are typically much lower. For example, glycosylation of Fmoc-protected serine acceptor 1 with 2-azido-2-deoxygalactose imidate 225 provided the corresponding glycoside 3 with an α to β ratio of only 1.4.20 Therefore, better methods for preparing these compounds were needed.
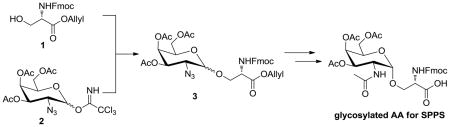
To improve the α-selectivity when glycosylating serine derivatives, we varied a number of reaction parameters. We started by optimizing the reaction conditions for the standard TMSOTf activated glycosylation of the TCA-donor 2 and serine acceptor 4 with respect to α-selectivity and overall chemical yield. Because the reaction solvent and temperature can have a dramatic influence, both factors were tested systematically for their ability to drive the reaction to form predominantly the desired α-product. The reaction media chosen in this study were based on solvents and solvent systems that were reported to successfully lead to α-glycosylation, regardless of the anomeric leaving group.26, 27 The results of these experiments are summarized in Table 1. The product ratios were determined by 1H NMR spectroscopy (400 MHz, CDCl3).
Table 1.
Effects of solvent and temperature on stereoselectivity
 | |||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst | Temperature [°C] | Yield[a] | Ratio[b] (α/β) |
| 1 | DCM | TMSOTf | −30 | 98% | 1.4 |
| 2 | ether | TMSOTf | −30 | 71% | 1.9 |
| 3 | DCM–ether (1:1) | TMSOTf | −30 | 97% | 1.9 |
| 4 | THF | TMSOTf | −30 | 76% | 1.0 |
| 5 | DCM–THF (1:1) | TMSOTf | −30 | 90% | 1.0 |
| 6 | nitromethane–ether (1:1) | TMSOTf | −30 | 77% | 1.6 |
| 7 | toluene | TMSOTf | −30 | 78% | 0.8 |
| 8 | toluene–dioxane (1:1) | TMSOTf | −30 | 94% | 2.0 |
| 9 | DCM–dioxane (1:1) | TMSOTf | −30 | 97% | 2.0 |
| 10 | DCM–dioxane (1:1) | TMSOTf | 0 | 96% | 3.8 |
| 11 | DCM–dioxane (1:1) | TMSOTf | rt | 86% | 5.3 |
Combined yield of both anomers after purification
Determined by 1H NMR spectroscopy
Variation of the solvent had a minor influence on the stereoselectivity of the glycosylation reaction at −30°C (entries 1–9). α:β ratios ranged from slightly favoring the β-product (toluene, entry 7) to modest α-selectivity (toluene–dioxane, entry 8 and DCM–dioxane, entry 9). In accordance with the literature, reaction media containing the participating solvents ether or dioxane showed an increased α-selectivity, compared to the nonparticipating solvents DCM and toluene. However, no selectivity was observed for the participating solvent THF. Interestingly, when the participating solvents ether and THF were paired with the nonparticipating DCM to form binary solutions, a marked increase in chemical yield was observed, without affecting the stereoselectivity (entries 2–5). Due to the high melting point of dioxane, no data could be obtained for the pure solvent at −30 °C. However, when paired with toluene or DCM (entries 8 and 9), the highest α-selectivities (α:β = 2.0) and chemical yields (94% and 97%, respectively) within this set of experiments were reached. Although both solvent systems behaved quite similar in these experiments, we continued our study with the mixture of DCM–dioxane, due to the low solubility of certain acceptors and donors in toluene based solvents, allowing a more general approach. Raising the reaction temperature from −30°C to 0 °C (entry 10) and room temperature (entry 11) resulted in additional increases in α-selectivity from 2.0 to 3.8 and 5.3, respectively, without a significant loss in overall chemical yield.
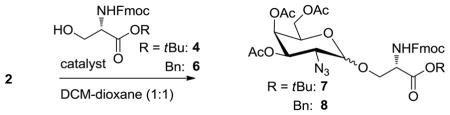
Next, we turned our attention to the influence of the catalyst on the α/β-product distribution of this reaction. Glycosylations were performed at 0 °C and room temperature with the same concentrations of donor and acceptor and employing an equal mol% of catalyst as compared to the TMSOTf-catalyzed reaction (Table 2).
Table 2.
Influence of catalyst and additives on stereoselectivity
 | |||||
|---|---|---|---|---|---|
| Entry | Acceptor | Catalyst | Temperature [°C] | Yield[a] | Ratio[b] (α/β) |
| 1 | 4 | TMSOTf | 0 | 96% | 3.8 |
| 2 | 4 | HClO4–SiO2 | 0 | 93% | 8.0 |
| 3 | 4 | TMSOTf + SiO2 | 0 | 91% | 3.8 |
| 4 | 4 | TMSOTf + LiClO4 | 0 | 96% | 3.9 |
| 5 | 4 | TMSOTf | rt | 86% | 5.3 |
| 6 | 4 | HClO4–SiO2 | rt | 42% | 24.0 |
| 7 | 6 | TMSOTf | rt | 98% | 3.7 |
| 8 | 6 | HClO4–SiO2 | rt | 95% | 12.0 |
Combined yield of both anomers after purification
Determined by 1H NMR spectroscopy
Under the same reaction conditions, the use of HClO4–SiO2 consistently resulted in higher ratios of α/β-selectivity for the serine acceptor 4, compared to the standard TMSOTf catalyzed reactions (entries 1,2: 3.8 to 8.0 and entries 5,6: 5.3 to 24.0). To determine whether this effect is related solely to the presence of solid SiO2 in the reaction mixture, a control experiment with TMSOTf as catalyst and additional silica was performed under the optimized conditions (entry 3). No change of selectivity was observed compared to the reaction without the additive, ruling out an exclusive effect of the silica. Additionally, we performed a control experiment with TMSOTf as catalyst and 1.5 equivalents of LiClO4 present in the reaction mixture (entry 4). Again, no change in selectivity was observed, indicating that the enhanced α-selectivity is not simply due to the presence of the perchlorate ion. When performed at room temperature, a significant decrease in overall yield was observed for the HClO4–SiO2 activated glycosylation (entry 6). This was due to partial cleavage of the acid labile tert-butyl ester moiety on the amino acid 4. Therefore, this set of experiment was repeated with an acid stable benzyl ester (6), resulting again in a significant increase in α-selectivity for the HClO4–SiO2 activated reaction (entries 7,8: 3.7 to 12.0), while maintaining very good chemical yields.
These results encouraged us to look into a more general application of the HClO4–SiO2 catalyst for α-selective glycosylations. Therefore, a set of representative and easily accessible trichloroacetimidate glycosyl donors, bearing non-participating protecting groups at C-2, were selected. Besides the previously introduced disarmed galactosyl donor 2, we also performed glycosylations with glucose (15)28 and mannose (18)29 donors. The hydroxyl groups on these donors are protected with benzyl ethers, giving rise to more reactive, or “armed”,30 donors relative to ester-protected donors. To cover a variety of representative acceptors, glycosylations were performed with the commercially-available 1,2:3,4-di-O-isopropylidene-α-D-galactopyranose (11) and 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose (13). To complete our investigations on the Tn-antigen, the benzyl protected threonine 9 was also studied as acceptor. Reactions were performed under optimized conditions at either 0 °C or room temperature. The results from these experiments are listed in Table 3.
Table 3.
Comparisons of α selectivities for various donor-acceptor pairs
| Entry | Donor | Acceptor | Product | Conditions | Yield[a] | Ratio[b] (α/β) |
|---|---|---|---|---|---|---|
| 1 | 2 |
 9 |
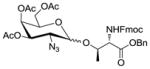 1031 |
TMSOTf rt | 79% | 8.2 |
| 2 | 2 | 9 | 10 | HClO4-SiO2 rt | 81% | 19 |
| 3 | 2 |
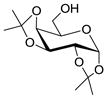 11 |
 1232 |
TMSOTf 0°C | 96% | 6.1 |
| 4 | 2 | 11 | 12 | HClO4-SiO2 0°C | 94% | 8.1 |
| 5 | 2 |
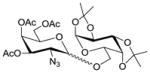
 13 |
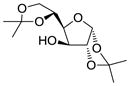
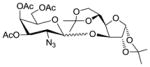 14 |
TMSOTf 0°C | 84% | 13 |
| 6 | 2 | 13 | 14 | HClO4-SiO2 0°C | 86% | 32 |
| 7 |
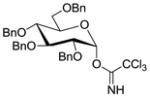 15 |
11 |
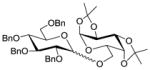 1633 |
TMSOTf 0°C | 93% | 3.0 |
| 8 | 15 | 11 | 16 | HClO4-SiO2 0°C | 90% | 3.0 |
| 9 | 15 | 11 | 16 | HClO4-SiO2 rt | 85% | 4.0 |
| 10 | 15 | 13 |
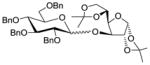 1734 |
TMSOTf 0°C | 84% | 4.0 |
| 11 | 15 | 13 | 17 | HClO4-SiO2 0°C | 88% | 9.3 |
| 12 |
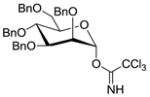 18 |
11 |
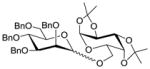 1935 |
TMSOTf 0°C | 85% | 3.0 |
| 13 | 18 | 11 | 19 | HClO4-SiO2 0°C | 88% | 10 |
Combined yield of both anomers after purification
Determined by 1H NMR spectroscopy
In all of the studied glycosylation reactions with representative donors and acceptors, the HClO4–SiO2 performed better, or at least as well as, the standard activator, TMSOTf, under the same reaction conditions. As noted before, the less reactive secondary acceptors tended to show a higher selectivity for the α-product, as compared to the more reactive primary alcohols. Therefore, using threonine based acceptors (9) for constructing glycosylated amino acids resulted in better α/β-ratios, as compared to acceptors of the serine type (entries 1, 2). Nonetheless, the α-selectivities achieved with serine 4 under the presented reaction conditions (vide supra) are sufficiently high for efficient syntheses and were readily applied by us for the multi-gram scale preparation of the Tn-antigen.
In the case of the armed glucose donor 15 and the highly reactive acceptor 11, no difference in glycosylation selectivity was observed for both catalysts (α/β = 3.0, entries 7,8). Raising the temperature from 0 °C to room temperature led to a slight increase in α-selectivity to 4.0 (entry 9), as already observed for donor–acceptor pair 2 and 4. The epimeric mannose donor 18, on the other hand, showed a marked increase in α-selectivity in the glycosylation with acceptor 11 after changing the catalyst from TMSOTf to HClO4–SiO2. The enhanced α-selectivity is especially interesting in light of the fact that activation of mannosyl bromides with silver silicate, a heterogeneous activator, produces primarily β-mannose products.36
In summary, we have demonstrated that the heterogeneous catalyst, HClO4–SiO2, provides enhanced α-selectivity in glycosylations involving trichloroacetimidate donors with non-participating protecting groups. The chemical yields are comparable to those achieved by TMSOTf activation, and the catalyst and conditions are applicable to a range of donor and acceptor pairs. Given the improved selectivity and ease of handling, HClO4–SiO2 is a useful alternative to TMSOTf for activation of trichloroacetimidates.
1. Experimental
1.1 General methods
All experiments involving water-sensitive compounds were conducted under dry conditions (positive argon pressure) using standard syringe, cannula and septa apparatus. Solvents: All solvents were purchased anhydrous (Aldrich) and stored over activated molecular sieves. Hexanes, ethyl acetate, methylene chloride, and methanol employed in chromatography were purchased HPLC-grade. Chromatography: Flash chromatography was performed with Teledyne ISCO CombiFlash Companion. TLC: analytical thin layer chromatography was performed on Analtech precoated plates (Uniplate, silica gel GHLF, 250 microns) containing a fluorescence indicator; sugar-containing compounds were visualized with the sugar spray reagent (5 mL of 4-methoxybenzaldehyde, 90 mL of ethanol, 5 mL of concentrated sulfuric acid, and 10 mL of glacial acectic acid) by heating with a heat gun. NMR spectra were recorded using a Varian Inova 400 MHz spectrometer. The coupling constants are reported in Hertz, and the peak shifts are reported in the delta (ppm) scale; abbreviations s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Mass spectra (ESI-MS) were obtained on an Agilent Technologies multimode ion source (LC/MSD SL) using the loop injection mode. Optical rotations were measured on a Jasco P-1010 polarimeter at 589 nm. Infrared spectroscopy data was obtained neat with a Jasco FT-IR/615 spectrometer. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, Georgia, 30091.
1.2 Preparation of perchloric acid immobilized on silica gel (HClO4-SiO2)
To a suspension of silica gel (20.0 g, SiliaFlash F60, 230 – 400 mesh, Silicycle, Quebec City, Canada) in Et2O (100 mL) was added HClO4 (70% aq solution, 690 μL, 8.00 mmol) and the suspension was stirred for 2h at rt. The solvent was removed under reduced pressure and the crude catalyst was dried under vacuum overnight at 80 °C to afford HClO4–SiO2 (0.4 mmol/g) as a free-flowing colourless powder. The catalyst is light sensitive and should be stored under argon in the dark to maintain activity.
1.3 General procedure for TMSOTf-activated glycosylations
Trichloroacetimidate donor (1.3 equiv.) and glycosyl acceptor (1.0 equiv.) were dissolved in anhyd. CH2Cl2 (5.0 mL/mmol donor) and anhyd. dioxane (5.0 mL/mmol donor). Activated molecular sieves (4Å, 50 mg/mL solvent) were added and the mixture was stirred for 0.5 h at rt. The reaction mixture was adjusted to the desired temperature and TMSOTf (0.075 mmol/mmol donor) was added dropwise. The reaction was stirred at the indicated temperature until TLC-analysis revealed reaction completion. All reactions with the armed donors, 15 and 18, were complete within 0.5 h. Reactions with the disarmed donor, 2, were complete within 0.5 h when carried out at r.t. and within 1 h when carried out at 0 °C. After neutralization with DIPEA, the solids were filtered off and the solvent was removed under reduced pressure. The crude was purified by silica-gel flash chromatography (EtOAc in hexanes) and product containing fractions were pooled, concentrated and subjected to 1H NMR-analysis for α/β-ratio determination. Additional chromatography provided pure compounds with spectroscopic data identical to those reported.
1.4 General procedure for HClO4-SiO2 -activated glycosylations
Trichloroacetimidate donor (1.3 equiv.) and glycosyl acceptor (1.0 equiv.) were dissolved in anhyd. CH2Cl2 (5.0 mL/mmol donor) and anhyd. dioxane (5.0 mL/mmol donor). Activated molecular sieves (4Å, 50 mg/mL solvent) were added and the mixture was stirred for 0.5 h at rt. The reaction mixture was adjusted to the desired temperature and HClO4–SiO2 (0.075 mmol/mmol donor) was added in one portion. The reaction was stirred at the indicated temperature until TLC-analysis revealed reaction completion. The solids were filtered off and the solvent was removed under reduced pressure. The crude was purified by silica-gel flash chromatography (EtOAc in hexanes) and product containing fractions were pooled, concentrated and subjected to 1H NMR-analysis for α/β-ratio determination. Additional chromatography provided pure compounds with spectroscopic data identical to those reported.
1.5 Determination of the product ratio
20 mg of pre-purified product was dissolved in 0.75 mL of CDCl3 and the 1H NMR was recorded at 400 MHz at 25 °C. The ratio was determined by integration of fully isolated peaks of each diastereomer. The diagnostic protons of each compound are listed in Table 4.
Table 4.
Diagnostic protons for determining α/β product ratios
| Diagnostic Protons | ||
|---|---|---|
| Compound | δα [ppm] | δβ [ppm] |
| 5 | 5.85 (d, 1H, J = 8.5 Hz, NHFmoc) 2.12 (s, 3H, CH3, OAc) |
5.75 (d, 1H, J = 8.5 Hz, NHFmoc) 2.10 (s, 3H, CH3, OAc) |
| 10 | 3.55 (dd, 1H, J = 11.2, 3.7 H-2) | 4.63 (dd, 1H, J = 10.8, 3.2 Hz, H-3) |
| 12 | 4.99 (d, 1H, J = 3.5 Hz, H-1) | 4.50 (d, 1H, J = 8.0 Hz, H-1) |
| 14 | 4.53 (d, 1H, J = 3.6 Hz, H-2) | 4.55 (d, 1H, J = 3.6 Hz, H-2) |
| 16 | 5.53 (d, 1H, J = 5.0 Hz, H-1) 1.53 (s, 3H, CH3, isoprop.) |
5.57 (d, 1H, J = 5.0 Hz, H-1) 1.50 (s, 3H, CH3, isoprop.) |
| 17 | 5.80 (d, 1H, J = 3.6 Hz, H-1) | 5.68 (d, 1H, J = 3.6 Hz, H-1) |
| 19 | 5.50 (d, 1H, J = 5.0 Hz, H-1) 1.48 (s, 3H, CH3, isoprop.) |
5.57 (d, 1H, J = 5.0 Hz, H-1) 1.46 (s, 3H, CH3, isoprop.) |
1.6 1,2:5,6-Di-O-isopropylidene-3-O-(3′,4′,6′-tri-O-acetyl-2′-azido-2′-deoxy-α-D-galactopyranosyl)-α-D-glucofuranose (14)
The reaction was carried out according to the general procedure for HClO4-SiO2-activated glycosylations with 3,4,6-tri-O-acetyl-2-azido-2-deoxy-α-D-galactopyranosyl trichloro-acetimidate donor 2 (100 mg, 0.210 mmol), diacetone-D-glucose acceptor 13 (42.1 mg, 0.162 mmol), activated molecular sieves (4A, 100 mg) and immobilized HClO4 on silica (40.0 mg) in CH2Cl2–dioxane (1:1, 2.0 mL) at 0 °C. The crude was purified by chromatography on silica gel (EtOAc in hexanes 20–40%) to yield the α glycosylated disaccharide 14 (79.9 mg, 86%) as a colourless foam. [α]D20 = +59.1 (c = 0.5, CHCl3); IR (neat): 2990, 2109 (N3), 1748, 1372, 1214, 1148, 1031, 847 cm−1; 1H NMR (400 MHz, CDCl3): δ 5.88 (d, 1H, J1,2 3.6 Hz, H-1); 5.42 (dd, 1H, J3′4′ 3.1 Hz, J4′5′ 1.0 Hz, H-4′); 5.32 (d, 1H, J1′2′ 3.7 Hz, H-1′); 5.20 (dd, 1H, J2′3′ 11.1 Hz, J3′4′ 3.7 Hz, H-3′); 4.53 (d, 1H, J1,2 3.6 Hz H-2); 4.39 (ddd, 1H, J4,5 8.9 Hz, J5,6a 6.0Hz, J5,6b 5.2Hz, H-5); 4.24 (d, 1H, J3,4 2.7 Hz, H-3); 4.23 – 4.19 (m, 1H, H-6′a); 4.14 (dd, 1H, J6a,6b 8.6Hz, J5,6a 6.0 Hz, H-6a); 4.10 (s, 1H, H-6′b); 4.08 (d, 1H, J4′5′ 1.0 Hz, H-5′); 4.02 (dd, 1H, J4,5 8.9 Hz, J3,4 2.7 Hz, H-4); 3.94 (dd, 1H, J6a,6b 8.6 Hz, J5,6b 5.2 Hz, H-6b); 3.82 (dd, 1H, J2′3′ 11.1 Hz, J1′2′ 3.7 Hz, H-2′); 2.12 (s, 3H, OAc); 2.04 (s, 3H, OAc); 2.02 (s, 3H, OAc); 1.47 (s, 3H, CH3, isoprop.); 1.39 (s, 3H, CH3, isoprop.); 1.32 (s, 3H, CH3, isoprop.); 1.30 (s, 3H, CH3, isoprop.) ppm; 13C NMR (100 MHz, CDCl3): 170.49, 169.94, 169.77 (3 × C=O, Ac); 112.02, 109.30 (2 × C(CH3)2, isoprop.); 83.78 (C-2); 81.41 (C-3); 80.47 (C-4); 71.28 (C-5); 68.51 (C-4′); 67.88 (C-3′); 67.43 (C-6′); 67.24 (C-5′); 61.98 (C-6); 57.93, (C-2′); 26.83, 26.77, 26.23, 25.14, 20.59 (CH3, OAc/isoporop.) ppm.; ESI-MS: 596.05 (M+Na+); Elemental Analysis for C24H35N3O13: Calculated: C, 50.26; H, 6.15; N, 7.33. Found: C, 50.10; H, 6.06; N, 7.32.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NCI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bertozzi CR, Kiessling LL. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 2.Lepenies B, Yin J, Seeberger PH. Curr Opin Chem Biol. 2010;14:404–411. doi: 10.1016/j.cbpa.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 3.Muthana S, Cao H, Chen X. Curr Opin Chem Biol. 2009;13:573–581. doi: 10.1016/j.cbpa.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boltje TJ, Buskas T, Boons GJ. Nat Chem. 2009;1:611–622. doi: 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demchenko AV. Synlett. 2003:1225–1240. [Google Scholar]
- 6.Chakraborti AK, Gulhane R. Chem Comm. 2003;9:1896–1897. doi: 10.1039/b304178f. [DOI] [PubMed] [Google Scholar]
- 7.Misra AK, Tiwari P, Madhusudan SK. Carbohydr Res. 2005;340:325–329. doi: 10.1016/j.carres.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal A, Vankar YD. Carbohydr Res. 2005;340:1661–1667. doi: 10.1016/j.carres.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Agarwal A, Rani S, Vankar YD. J Org Chem. 2004;69:6137–6140. doi: 10.1021/jo049415j. [DOI] [PubMed] [Google Scholar]
- 10.Misra AK, Tiwari P, Agnihotri G. Synthesis. 2005:260–266. [Google Scholar]
- 11.Chakraborti AK, Singh B, Chankeshwara SV, Patel AR. J Org Chem. 2009;74:5967–5974. doi: 10.1021/jo900614s. [DOI] [PubMed] [Google Scholar]
- 12.Du Y, Wei G, Cheng S, Hua Y, Linhardt RJ. Tet Lett. 2006;47:307–310. [Google Scholar]
- 13.Mukhopadhyay B, Maurer SV, Rudolph N, Van Well RM, Russell DA, Field RA. J Org Chem. 2005;70:9059–9062. doi: 10.1021/jo051390g. [DOI] [PubMed] [Google Scholar]
- 14.Liu M, Young VG, Jr, Lohani S, Live D, Barany G. Carbohydr Res. 2005;340:1273–1285. doi: 10.1016/j.carres.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 15.Liebe B, Kunz H. Helv Chim Acta. 1997;80:1473–1482. [Google Scholar]
- 16.Reipen T, Kunz H. Synthesis. 2003:2487–2502. [Google Scholar]
- 17.Kuduk SD, Schwarz JB, Chen XT, Glunz PW, Sames D, Ragupathi G, Livingston PO, Danishefsky SJ. J Am Chem Soc. 1998;120:12474–12485. [Google Scholar]
- 18.Koeller KM, Smith MEB, Wong CH. Bioorg Med Chem. 2000;8:1017–1025. doi: 10.1016/s0968-0896(00)00041-9. [DOI] [PubMed] [Google Scholar]
- 19.Gambert U, Thiem J. Carbohydr Res. 1997;299:85–89. doi: 10.1016/s0008-6215(96)00324-2. [DOI] [PubMed] [Google Scholar]
- 20.Yang YY, Ficht S, Brik A, Wong CH. J Am Chem Soc. 2007;129:7690–7701. doi: 10.1021/ja0708971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cato D, Buskas T, Boons GJ. J Carbohydr Chem. 2005;24:503–516. [Google Scholar]
- 22.Miermont A, Barnhill H, Strable E, Lu X, Wall KA, Wang Q, Finn MG, Huang X. Chem Eur J. 2008;14:4939–4947. doi: 10.1002/chem.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andreotti AH, Kahne D. J Am Chem Soc. 1993;115:3352–3353. [Google Scholar]
- 24.Karkkainen TS, Ravindranathan Kartha KP, MacMillan D, Field RA. Carbohydr Res. 2008;343:1830–1834. doi: 10.1016/j.carres.2008.03.034. [DOI] [PubMed] [Google Scholar]
- 25.Grundler G, Schmidt RR. Liebigs Ann Chem. 1984:1826–1847. [Google Scholar]
- 26.Demchenko AV, Rousson E, Boons G-J. Tetrahedron Lett. 1999;40:6523–6526. [Google Scholar]
- 27.Ishiwata A, Munemura Y, Ito Y. Tetrahedron. 2008;64:92–102. [Google Scholar]
- 28.Schmidt RR, Stumpp M. Liebigs Ann Chem. 1983:1249–1256. [Google Scholar]
- 29.Schmidt RR, Michel J, Roos M. Liebigs Ann. 1984:1343–1357. [Google Scholar]
- 30.Fraser-Reid B, Wu ZF, Udodong UE, Ottosson H. J Org Chem. 1990;55:6068–6070. [Google Scholar]
- 31.Mathieux N, Paulsen H, Meldal M, Bock K. J Chem Soc Perk. 1997;1:2359–2368. [Google Scholar]
- 32.Paulsen H, Paal M. Carbohydr Res. 1983;113:203–218. doi: 10.1016/0008-6215(83)88236-6. [DOI] [PubMed] [Google Scholar]
- 33.Adinolfi M, Barone G, Iadonisi A, Schiattarella M. Org Lett. 2003;5:987–989. doi: 10.1021/ol027353i. [DOI] [PubMed] [Google Scholar]
- 34.Grayson EJ, Ward SJ, Hall AL, Rendle PM, Gamblin DP, Batsanov AS, Davis BG. J Org Chem. 2005;70:9740–9754. doi: 10.1021/jo051374j. [DOI] [PubMed] [Google Scholar]
- 35.Singh G, Vankayalapati H. Tetrahedron Asymmetry. 2000;11:125–138. [Google Scholar]
- 36.Paulsen H, Lockhoff O. Chem Ber. 1981;114:3102–14. [Google Scholar]