

Abstract
The pregnane X receptor (PXR) is recognized as a key regulator for the induction of a large number of genes in drug metabolism and transport. The transactivation of PXR is enhanced by the glucocorticoid dexamethasone and the enhancement is linked to the induction of PXR in humans and rats. The present study was undertaken to determine the mechanism for the induction and ascertain the synergistic effect on the expression of CYP3A23, a rat PXR target. In primary hepatocytes, significant induction of PXR was detected as early as 2 h after the treatment and the maximal induction occurred at 1 μM dexamethasone. Similar induction kinetics was observed in the hepatoma line H4-II-E-C3. The induction was abolished by actinomycin D and dexamethasone efficaciously stimulated the rat PXR promoter. In addition, dexamethasone synergized esfenvalerate (an insecticide and a PXR activator) in inducing CYP3A23 and stimulating the CYP3A23 promoter. The full promoter of CYP3A23 (−1445/+74) was activated in a similar pattern as the changes in PXR mRNA in response to dexamethasone, esfenvalerate and co-treatment. In contrast, different responding patterns were detected on the stimulation of the CYP3A23 proximal promoter. Synergistic stimulation was also observed on the CYP3A4-DP-Luc reporter, the human counterpart of CYP3A23. These findings establish that transactivation is responsible for the induction of rat PXR and the induction presents potential interactions with insecticides in a species-conserved manner. The different responding patterns among CYP3A23 reporters point to an involvement of multiple transcriptional events in the regulation of CYP3A23 expression by dexamethasone, esfenvalerate and both.
1. INTRODUCTION
The pregnane X receptor (PXR), also called steroid and xenobiotic receptor, has been recognized as a key regulator for the induction of a large number of genes involved in drug disposition [1, 2]. Structurally, PXR belongs to a superfamily of the nuclear receptors and regulates target gene transcription in a ligand-dependent manner [3]. Unlike many other nuclear receptors, PXR has a rather large ligand-binding pocket, which is spherical in shape, extremely hydrophobic and expandable [4–6]. Such structural features allow PXR to interact with a wide range of chemicals. These chemicals include prescription drugs (e.g., rifampicin), herbal supplements (e.g., hyperforin) and pesticides (e.g., pyrethroids) [7–11]. In addition, many endogenous compounds are shown to activate PXR, notably steroidal compounds (e.g., corticosterone) and bile acids (e.g., lithocholic acid). Interestingly, both glucocorticoids (e.g., dexamethasone) and antiglucocorticoids (e.g., pregnenolone 16α-carbonitrile) can activate PXR [10, 12–14].
Some chemicals modulate the expression of PXR target genes through both PXR-dependent and independent mechanisms. For example, dexamethasone, a synthetic glucocorticoid, interacts directly with rodent PXR and increases the transactivation activity [9, 13]. On the other hand, this glucocorticoid enhances the binding activity of CCAAT/enhancer binding protein (CEBP), thus leading to increased transcription of the cytochrome P450 3A23 gene (CYP), a PXR target gene in rats [15]. Consistent with a role of CEBP in the dexamethasone-transactivation of PXR target genes, a natural variant of the CYP3A4 promoter responds poorly to dexamethasone, and this variant has a C→A substitution in a hepatocyte-nuclear-factor-3/CEBP site [16]. The CEBP binding site appears to be critical in the transactivation of this CYP3A gene by glucocorticoids, as this variant shows altered responsiveness to hydrocortisone as well.
In addition to individual genes, glucocorticoids likely modulate the expression of a group of genes by regulating certain cellular pathways or an event shared by these genes for their expression. Indeed, this and other laboratories have shown that dexamethasone induces the expression of PXR [17–19]. Induction of PXR has been implicated in the synergistically increased expression of many PXR target genes [20, 21]. It appears that induction of PXR by glucocorticoids is conserved cross species from humans to rodents [17, 18, 20, 21]. The induction is detected at the levels of mRNA and protein [17–19, 22]. It is well established that dexamethasone modulates gene expression through various mechanisms, particularly transactivation. However, it remains to be determined whether transactivation is involved in the induction of PXR.
The present study was undertaken to determine the molecular mechanism for the induction of rat PXR and ascertain the synergistic effect on the expression of CYP3A23 in the presence of a PXR ligand. In both primary hepatocytes and the hepatoma line H4-II-E-C3, the induction kinetics of PXR was similar in terms of concentrations- and time-dependency. The induction was abolished by actinomycin D and the rat PXR promoter was stimulated by dexamethasone. Dexamethasone at nanomolar concentrations greatly enhanced the pyrethroid esfenvalerate in inducing CYP3A23, and the enhancement was also detected on the stimulation of the CYP3A23 (rat) and CYP3A4 (human) promoters. These findings establish that dexamethasone induces rat PXR through transactivation and the induction potently enhances the expression of PXR target genes cross species.
2. MATERIALS AND METHODS
2.1. Chemicals and supplies
Dexamethasone, cycloheximide, equine serum, Hanks balanced salt solution (HBSS) and RU486 were purchased from Sigma (St. Louis, MO). Dulbecco’s Modified Eagle Medium (DMEM), Williams’ E medium and high fidelity Platinum Taq DNA polymerase were purchased from Invitrogen (Carlsbad, CA). GenJet transfection reagent was from the SignaGen Laboratories (Gaithersburg, MD). Kits for luciferase detection and P450-Glo™ were from Promega (Madison, WI). Fetal bovine serum was from HyClone laboratories (Logan, UT). Ketamine HCl was purchased from Fort Dodge Animal Health (Fort Dodge, IA). Esfenvalerate (99.5% purity) was purchased from ChemService (West Chester, PA). RNA Bee was purchased from Tel-test (Friendswood, TX). The antibodies against the glucocorticoid receptor-α or glyceradehyde-3-phosphate dehydrogenase (GAPDH) were from Abcam (Cambridge, UK). The goat anti-rabbit IgG conjugated with horseradish peroxidase was from Pierce (Rockford, IL). Nitrocellulose membranes were from Bio-Rad (Hercules, CA). Unless otherwise specified, all other reagents were purchased from Fisher Scientific (Fair Lawn, NJ).
2.2. Hepatocyte culture and treatment
Primary rat hepatocytes were isolated from adult male Sprague-Dawley rats (175–250 g) by a modified two-step collagenase digestion method, essentially as described previously [23]. Rat liver was perfused through the portal vein with calcium-free HBSS containing 0.5mM EGTA for ~1–2 min at a flow rate of 28 ml/min followed by perfusion for ~5–8 min with Williams’ E medium containing calcium and collagenase (100 U of collagenase activity/ml of media). The liver was further perfused with calcium-free HBSS containing EGTA until the liver was completely blanched (~1–2 min). Hepatocytes were dispersed from the digested liver in Williams’ E medium without collagenase and washed by low speed centrifugation (100–150 g, 5 min) 3 times. The resulting cell pellet was then suspended in Williams’ E medium containing 10% fetal bovine serum, Insulin-transferrin-sodium selenite (ITS) supplement and dexamethasone (100 nM), and the cell viability was determined by trypan blue exclusion. Hepatocytes were then plated onto collagen coated culture plates (6 or 12-well plate). The cells were allowed to attach for 3 to 4 h at 37°C in a humidified chamber with 95%/5%air/CO2. Culture plates were then gently swirled and the medium containing unattached cells was then aspirated. Fresh medium was added to each well, and the cultures were returned to the humidified chamber. Hepatocytes were cultured in the same medium for 48 h but the medium was changed at 24 h. Thereafter, hepatocytes were cultured in the medium without dexamethasone for 16 h. The hepatocytes were then cultured in DMEM containing treatment chemicals at appropriate concentrations. For RNA isolation, hepatocytes were washed in the plates and harvested in RNA Bee. For the preparation of protein lysates, hepatocytes were harvested into tubes and washed again by centrifugation. The lysates were prepared by sonication as described previously [24]. All rats were allowed free access to Purina Rodent Chow 5001 and water, and the use of animals was approved by the Institutional Animal Care and Use Committee.
2.3. Cell culture and treatment
Rat hepatoma line H4-II-E-C3 was purchased from the American Type Culture Collection (Rockville, MD). The hepatoma cells were maintained in DMEM containing 10% fetal bovine serum, 5% equine serum, penicillin (100 units per ml)/streptomycin (100 μg/ml), 1x non-essential amino acids and 1 mM sodium pyruvate. Cells were usually seeded at a density of 5 × 105 cells/well (12-well plates) in normal medium. After an overnight incubation, treatment was started with a chemical or the same volume of DMSO. The duration and concentration of treatment are specified in figure legends.
2.4. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated with RNA Bee according to the manufacturer’s manual, and the integrity of the RNA was confirmed by formaldehyde gel electrophoresis. Total RNA (1 μg) was subjected to the synthesis of the first strand cDNA in a total volume of 25 μl with random primers and M-MLV reverse transcriptase. The reactions were conducted at 25°C for 10 min, 42°C for 50 min and 70°C for 10 min. The cDNAs were then diluted 5 times and quantitative PCR was conducted with TaqMan Gene Expression Assay (Applied Biosystems, Foster City, CA). The TaqMan assay identification numbers were: rat PXR, Rn00583887_m1; tyrosine aminotransferase (Tat), Rn00562011_m1; rat α-1 acid glycoprotein, Rn00583913_m1; rat cytochrome P450 3A23 (CYP3A23), Rn01640761_gH; rat GAPDH, Rn99999916_s1; and rat polymerase II (Pol II), Rn01752026_m1. The PCR amplification was conducted in a total volume of 20 μl containing universal PCR master mixture (10 μl), gene-specific TaqMan assay mixture (1 μl), and cDNA template (6 μl). Cycling profile was 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60 °C, as recommended by the manufacturer. The mRNA levels were normalized according to the level of Pol II [25]. Amplification and quantification were done with the Applied Biosystems 7900 Real-Time PCR System.
2.5. Enzymatic assay
Hepatocytes were treated with dexamethasone, esfenvalerate or both at various concentrations for 24 h, and lysates were prepared by sonication and centrifugation as described previously [24]. The activity of CYP3A was determined with a P450-Glo™ kit, essentially as described previously [26]. Briefly, lysates (20 μg in 12.5 μl) were mixed with 12.5 μl of CYP3A4 substrate Luciferin-IPA (4X). After a 10 min pre-incubation at 37°C, the NADPH regeneration mixture (25 μl containing 400 mM KPO4) was added to initiate the enzymatic reaction. The reaction lasted from 30 min at 37°C and was terminated by adding 50 μl of Luciferin Detection Reagent. After an additional 10 min-incubation at room temperature, the luminescent signal was determined. Several controls were performed including incubation without lysates or the regeneration system. In addition, standard curves were generated with increasing amounts of recombinant CYP3A4 from BD Biosciences (San Jose, CA). While this kit is designed to measure CYP3A4 activity (human), it has been used to measure rat CYP3A activity as well [27].
2.6. Reporter constructs
Luciferase reporters harboring the rat PXR promoter at varying length were prepared by inserting the corresponding genomic fragment into the pGL3 promoterless luciferase vector at the Mlu I and Xho I sites. All genomic fragments were generated by PCR with high fidelity Platinum Taq DNA polymerase. Rat genomic DNA was used as the template for the amplification of the genomic fragment (−873 to +68), and this fragment was used to prepare the reporter rPXR873-Luc. This reporter was used as the template for all other reporters that contained a shorter sequence. The genomic fragments were numbered according to the first nucleotide of a full-length cDNA [19]. The sequences of primers are shown in Table I. To selectively disrupt the putative element of the glucocorticoid receptor, substitutions (bold) were made in the primer PXR-171MluIs-M. Several investigators have reported that substitutions of these nucleotides completely abolish the responsiveness to transactivation by the glucocorticoid receptor [28, 29]. To prepare the 3A23-198Luc reporters, primers 3A23-198MluIs and 3A23-198XhoIa were used for the PCR amplification and the CYP3A23-Luc reporter [30] was used as the template. The PCR product was inserted into the pGL3 vector through Mlu I-Xho I sites. To prepare 3A23-198mLuc reporter, primers 3A23-198ms and 3A23-198ma were first annealed through their complementary sequence (Table I) and then subjected to primer extension to fill the 3′ sequence of respective primer. The resultant double-stranded DNA was used as the template for PCR amplification with primers 3A23-198mMluIs and 3A23-198mXhoIa. Likewise, the PCR-amplified product was inserted into the pGL3 basic vector digested with Mlu I and Xho I. All constructs were confirmed by sequencing analysis. The preparation of CYP3A23-Luc and CYP3A4-DP-luc was described previously [30]. The CYP3A23-Luc reporter contains genomic fragment from −1445 to +74, whereas the CYP3A4-DP-luc reporter contains fused fragments: −362 to +53 and −7836 to −6093.
Table I.
Sequences of primers for reporter constructs
| Primer | Sequence | Reporter |
|---|---|---|
| PXR-873MluIs | 5′-gcaggacgcgttctggccaggtgccctgtct-3′ | rPXR873-Luc |
| PXR-488MluIs | 5′-gcaggacgcgttgtaaaatactgctgtccag-3′ | rPXR488-Luc |
| PXR-257MluIs | 5′-gcaggacgcgtctggtcattgctagttcagg-3′ | rPXR257-Luc |
| PXR-171MluIs | 5′-actgacgcgttgtgggcaagaacagta-3′ | rPXR171-Luc |
| PXR-171MluIs-M | 5′-actgacgcgttgtgggcaagccccgta-3′ | rPXR171m-Luc |
| PXR-91MluIs | 5′-actgacgcgttatagtttctgcgcccgt-3′ | rPXR91-Luc |
| PXR+68XhoIa | 5′-gtgctgctcgagtctcagcttaagtctttggtgt-3′ | all PXR reporters |
| 3A23-198MluIs | 5′-tacgacgcgttgttgttcactgacgccacccagaatgttaactcaaaggag-3′ | 3A23-198Luc |
| 3A23-198XhoIa | 5′-cttactcgagacactgctgcactgaggccagcctgcagtacttatatgctg-3′ | 3A23-198Luc |
| 3A23-198ms | 5′aatcaattgtcaaaggacctgaaaataggctgtagatgaacttcatgaactgtctaggggaagagagtaccaaagtccacgtga-3′ | 3A23-198mLuc |
| 3A23-198ma | 5′-aagatgggtggcaccagggatccagccagtagatggatcacctctgcctcacgtggactttggtactctcttcccctaga-3′ | 3A23-198mLuc |
| 3A23-198mMluIs | 5′-tacgacgcgttgttgttcactgacgccacccagaatcaattgtcaaaggacctgaaaataggctgtaga-3′ | 3A23-198mLuc |
| 3A23-198mXhoIa | 5′-cttactcgagacactgctgcactgaggccagcctgcagtacttatatgctggaaagatgggtggcaccagggatc-3′ | 3A23-198mLuc |
2.7. Co-transfection assays
Hepatoma cells (H4-II-E-C3) were plated in 48-well plates in DMEM supplemented with 10% delipided fetal bovine serum at a density of 1.2 × 105 cells per well. Transfection mixtures contained 50 ng of a reporter plasmid and 5 ng of TK-Renilla luciferase plasmid. Cells were transfected for 4 h and the medium was replaced with fresh medium. After cultured for 24 h, the transfected cells were treated with dexamethasone, esfenvalerate or both for 24–48 h with a change of the medium at 24 h (chemicals kept the same). The cells were washed once with phosphate buffered saline, and harvested in passive lysis buffer. After 30-min incubation at room temperature with shaking, the reporter activities were assayed with a Dual-Luciferase Reporter Assay System for the activities of two luciferases. The firefly luciferase activity, which represented the reporter activity, was initiated by mixing an aliquot of lysates (10 μl) with Luci-ferase Assay Reagent II. Then the firefly luminescence was quenched and the Renilla luminescence was simultaneously activated by adding Stop & Glo Reagent to the sample tubes. The firefly luminescence signal was normalized based on the Renilla luminescence signal.
2.8. Electrophoretic mobility shift assay (EMSA)
The EMSA experiment was performed as described previously [31]. Nuclear extracts of H4-II-E-C3 cells treated with dexamethasone (100 nM) were prepared with the nuclear and cytoplasmic extraction kit (Pierce, Rockford, IL). The sense and antisense oligonucleotides (5′-TGTGGGCAAGAACAAGTATAGTTTCTGCGCCCG-3′) were annealed by heating at 94°C for 5 min followed by gradually cooling to room temperature. The sense strand was synthesized as labeled or non-labeled form (for competition). Nuclear protein (5μg) was incubated with a double-stranded biotinylated probe (0.1 pmol) at room temperature for 20 min. In competition assays, nuclear extracts were first incubated with an unlabeled probe at a 5x or 20x excess for 5 min before addition of the labeled probe. For antibody-disruption assay, the nuclear extracts were first incubated with an antibody against glucocorticoid receptor-α on ice for 20 min and then with the labeled probe. The protein-DNA complexes were resolved by non-denaturing polyacrylamide gel electrophoresis (5%) and transferred onto a Biodyne® nylon membrane. The biotinylated probe was detected with streptavidin-conjugated horseradish peroxidase and chemiluminescent substrate (PIERCE, Rockford, IL). The chemiluminescent signal was captured by KODAK Image Station 2000, and the relative intensities were quantified by KODAK 1D Image Analysis Software (KODAK Molecular Imaging Software, Version 4.0, Rochester, NY).
2.9. Western analysis
Cell lysates (10 μg) were resolved by 7.5% SDS-PAGE in a mini-gel apparatus and transferred electrophoretically to nitrocellulose membranes. After non-specific binding sites were blocked with 5 % non-fat milk, the blots were incubated with an antibody against PXR, CYP3A23 or GAPDH. The anti-CYP3A23 antibody was purchased from Research Diagnostics (Flanders, NJ) and the preparation of the antibody against rat PXR was described elsewhere (Sachdeva et al., 2003). The primary antibodies were subsequently localized with goat anti-rabbit IgG conjugated with horseradish peroxidase. Horseradish peroxidase activity was detected with a chemiluminescent kit (SuperSignal West Pico). The chemiluminescent signal was captured by KODAK Image Station 2000 and the relative intensities were quantified by KODAK 1D Image Analysis Software. All blots were performed twice.
2.10. Other analyses
Protein concentrations were determined with BCA assay (Pierce) based on bovine serum albumin standard. Data are presented as mean ± SD of at least three separate experiments, except where results of blots are shown in which case a representative experiment is depicted in the figures. All data were reanalyzed for statistical significance with PASW Statistics 18. Significant differences were made according to One-way ANOVA followed by a DUNCAN’s multiple comparison test (p < 0.05). Lines or letters were used to indicate data-points for the comparisons. Location of DNA elements and promoter identification was performed with the Genomatix program.
3. RESULTS
3.1. Characterization of the induction of rat PXR by dexamethasone
To characterize the induction of PXR by dexamethasone, an initial effort was made to determine the induction as functions of the concentrations and the length of treatment. The characterization was made in both primary hepatocytes and the hepatoma cell line H4-IIE-C3. The induction was monitored primarily by RT-qPCR and selective samples were analyzed by Western blotting. The primary hepatocytes were maintained in full medium including dexamethasone for 48 h. However, a 16-h withdrawal of dexamethasone was performed before the treatment was initiated. As shown in Fig. 1A, significant induction of PXR in primary hepatocytes was detected even when dexamethasone was assayed at as low as 0.01 μM. The maximal induction was ~5 fold and occurred at 1 μM (Fig. 1A). It was interesting that higher concentration (i.e., 10 μM) was slightly less effective in terms of inducing PXR (Fig. 1A). Similarly, the level of PXR protein was proportionally increased with increasing levels of PXR mRNA (Fig. 1A). As expected, dexamethasone at higher concentrations (i.e., 1 and 10 μM) caused induction of CYP3A23 based on Western analysis (Fig. 1A).
Fig. 1. Induction of rat PXR in primary hepatocytes by dexamethasone.

Primary hepatocytes from Sprague-Dawley male rats (8-week old, n = 4) were isolated by a modified two-step collagenase digestion method. Hepatocytes were initially seeded and cultured in Williams’ E medium for 48 h with replacing the medium at 24 h. Thereafter, hepatocytes were cultured in dexamethasone-free medium for 16 h and then treated with dexamethasone at various concentrations (0–10 μM). Then hepatocytes were harvested for preparing cell lysates and total RNA. Total RNA was analyzed for the level of PXR mRNA by RT-qPCR and cell lysates (10 μg) were analyzed for the levels of PXR or CYP3A23 proteins by Western blotting. The RT-qPCR signals were normalized based on the abundance of Pol II mRNA. (A) Induction of PXR as a function of concentrations, and (B) Time-course study on the induction of PXR. The Ct value (threshold cycles) for DMSO controls in all time-points is around 25.65. *Statistically significant difference (p < 0.05).
We next performed a time-course study on the induction of PXR. As shown in Fig. 1B, a 2-h treatment caused significant induction, and the highest concentration (10 μM) was slightly less potent than 1 μM at all time-points tested. Next we examined whether hepatoma H4-II-E-C3 cells support similar induction kinetics primary hepatocytes. Like the induction in primary hepatocytes, the induction in H4-II-E-C3 cells occurred in a concentration-dependent manner when dexamethasone was used at 1 nM to 1 μM (Fig. 2A). No additional induction was detected in cells treated with a higher concentration (10 μM). Next we examined the induction as a function of time. As a control, the induction of tyrosine aminotransferase (Tat), a sensitive target gene of the glucocorticoid receptor [32], was monitored as well. Like the induction in the primary hepatocytes, the level of PXR mRNA was rapidly increased in response to dexamethasone (Fig. 2B). Actually, the induction of PXR reached the plateau in the hepatoma line faster than that in the primary hepatocytes (Figs. 1B and 2B). As expected, dexamethasone efficaciously induced Tat, and the maximal induction of Tat was much higher than that of PXR (17 versus 5 fold) (Fig. 2B). However, it took much longer time for Tat induction to reach the maximum than PXR induction. As a matter of fact, the increase of PXR mRNA approached the plateau after 6 h, whereas Tat mRNA exhibited continuous increases thereafter (Fig. 2B). It should be noted that H4-II-E-C3 cells supported similar induction kinetics as H4IIE [17].
Fig. 2. Induction of rat PXR in H4-II-E-C3 cells by dexamethasone (A) Concentration-response study.
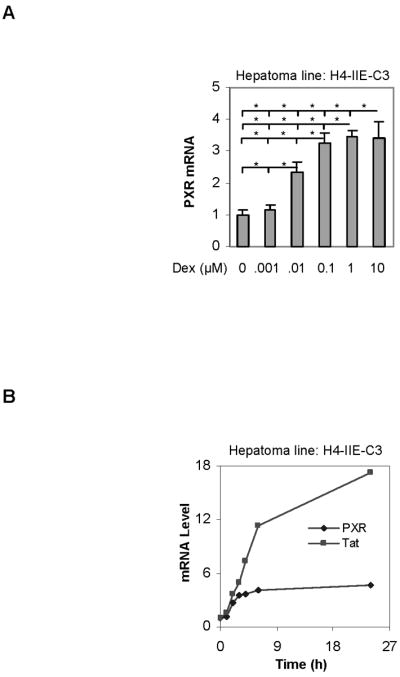
H4-II-E-C3 cells were treated for 24 h with dexamethasone at various concentrations (0–10 μM), and total RNA was analyzed for the level of PXR mRNA by RT-PCR. Similarly the mRNA level was normalized based on the level of Pol II mRNA. Data were shown from three independent experiments. *Statistically significant difference from the vehicle control (p < 0.05). *Statistically significant difference (p < 0.05). (B) Time-course study H4-II-E-C3 cells were treated with dexamethasone at 50 nM and the cells were collected at 1, 2, 3, 4, 6 and 24 h. Similarly, total RNA was analyzed by qRT-PCR for the level of PXR or Tat mRNA by qRT-PCR, and the mRNA level was normalized based on the level of Pol II mRNA. The Ct value of PXR in DMSO control in all time-points is around 26.65.
3.2. Actinomycin D and RU486 abolished PXR and Tat induction
It is well established that dexamethasone induces Tat by transactivation [32]. In this study, we have shown that PXR induction by this glucocorticoid reached the plateau faster than Tat induction, suggesting that dexamethasone induces PXR through a mechanism rather than or in addition to transactivation. To shed light on these possibilities, the induction of PXR by dexamethasone was studied in the presence of actinomycin D, an inhibitor of RNA synthesis. The treatment lasted for 6 h as this time-point approached the plateau of PXR induction and prolonged treatment with actinomycin D tended to cause cytotoxicity. As shown in Fig. 3A, actinomycin D alone caused a slight increase in PXR mRNA, although the increase was statistically insignificant. More importantly, actinomycin D abolished the increases of PXR mRNA in response to dexamethasone (Fig. 3A). As expected, the increase in Tat mRNA was completely abolished by actinomycin D. Interestingly, actinomycin D alone caused decreases in Tat mRNA by ~50% (Fig. 3A).
Fig. 3. Effect of actinomycin D, cycloheximide and RU486 on the dexamethasone-induced expression of PXR and Tat (A) Effect of actinomycin D.

H4-II-E-C3 cells were seeded at a density of 5 × 105 in 12-well plates. After an overnight incubation, the cells were treated with dexamethasone (50 nM), actinomycin D (Act D, 1 μM), or both for 6 h. The levels of PXR and Tat mRNA were determined by RT-qPCR. The signals were normalized according to the signal on Pol II mRNA. Letter “b” denotes statistical significance from letter “a”; letter “d” from letter “a” or letter “c”; and letter “a” from letter “c”. (B) Effect of RU486 H4-II-E-C3 cells were seeded as described above and subsequently treated with dexamethasone (50 nM), RU486 (0.1 μM), or both for 24 h. The levels of PXR and Tat mRNA were determined by RT-qPCR, and the signals were normalized according to the signal on Pol II mRNA. Data presented in this figure were assembled from three independent experiments. Letter “b” denotes statistical significance from letter “a”; and letter “d” from letter “c”. (C) Effect of cycloheximide on the dexamethasone-regulated expression of PXR and AGP H4-II-E-C3 cells were seeded as described above and subsequently treated with dexamethasone (50 nM), cycloheximide (CHX, 1 μM), or both for 24 h. The levels of PXR and Tat mRNA were determined by RT-qPCR, and the signals were normalized according to the signal on Pol II mRNA. Data presented in this figure were assembled from three independent experiments. Letter “b” denotes statistical significance from letter “a”; and letter “d” from letter “c”.
The glucocorticoid receptor is the major receptor transmitting the signal of glucocorticoids, particularly at low concentrations. We next examined whether induction of PXR can be antagonized by RU486, an anti-glucocorticoid [33]. H4-II-E-C3 cells were treated for 24 h with dexamethasone in the presence or absence of RU486, and the induction of PXR and Tat was monitored. As shown in Fig. 3B, this anti-glucocorticoid completely abolished dexamethasone-mediated increases in both PXR and Tat mRNA (Fig. 3B), suggesting that induction of PXR by dexamethasone is mediated by activation of the glucocorticoid receptor. To further establish this mechanism, co-transfection of the glucocorticoid receptor-β diminished the induction of PXR (data not shown). It is generally accepted that the β-form functions as a dominant negative regulator against the glucocorticoid receptor-α [34, 35]. Treatment with cycloheximide, a protein synthesis inhibitor, had no effect on the induction of PXR but completely eliminated the induction of α-1 acid glycoprotein (AGP) (Fig. 3C). It is well established that the induction of AGP by dexamethasone requires on-going protein synthesis [35], and the inability of cycloheximide to reduce PXR induction excluded an involvement of on-going protein synthesis in the induction of PXR.
3.3. Dexamethasone activates the rat PXR promoter
The studies with actinomycin D and RU486 collectively establish that dexamethasone induces PXR through transactivation, and the transactivation is supported by the glucocorticoid receptor and does not require on-going protein synthesis. We next tested whether dexamethasone directly activates the rat PXR promoter. Multiple promoter reporters were prepared to contain a genomic fragment with varying length. A computer-assisted homology search identified an element putatively responding to the glucocorticoid receptor (GR) (Left of Fig. 4A). As expected, all reporters except rPXR91-Luc were significantly activated by dexamethasone (Fig. 4A), consistent with the fact that only this reporter did not contain this GR response element. More importantly, disruption of this element (−153 to −139) led to complete loss of the responsiveness to dexamethasone (Fig. 4A). To determine whether this element binds to GR, EMSA was performed. Consistent with the reporter assay, a shifted band was detected (Fig. 4B). More importantly, the formation of the shifted band was disrupted by an antibody against GR or effectively competed by the corresponding unlabeled probe (Fig. 4B). It should be noted that the activation of PXR reporter (i.e., rPXR257-Luc) occurred in a concentration-dependent manner at low concentrations and reached the maximal activation at 50 nM (Fig. 4C).
Fig. 4. Activation of the rat PXR promoter reporters by dexamethasone (A) Identification of dexamethasone response element in the PXR promoter.

H4-II-E-C3 cells were seeded in 48-well plates at a density of 1.2 × 105. After an overnight incubation, the cells were transiently transfected by GenJet with a mixture containing a reporter (50 ng) along with 5 ng of the tk-Renilla luciferase plasmid. After incubation at 37°C for 24 h (serum-free medium), the transfected cells were treated with dexamethasone (50 nM) or the same volume of DMSO for 48 h. Luciferase activities were determined with a Dual-Luciferase Reporter Assay System and the reporter activity was normalized based on the Renilla luminescence signal. The normalized reporter activities are expressed as fold of activation. (B) EMSA analysis Nuclear extracts (5 μg) from H4-II-E-C3 cells treated with dexamethasone (100 nM) were incubated with a biotinylated probe containing the putative GR element (0.1 pmol) for 20 min. In the competition assay, nuclear extracts were pre-incubated with the unlabeled probe at 5x or 20x excess for 5 min, and then incubated with the biotinylated probe. In disruption assay, nuclear extracts were incubated first with an antibody against glucocorticoid receptor-α on ice for 20 min and then with the biotinylated probe. The protein-DNA complexes were electrophoretically resolved and transferred to a Biodyne® nylon membrane. The biotinylated probe was located with streptavidin-conjugated horseradish peroxidase and chemiluminescent substrate. (C) Concentration-dependent activation of rPXR257-Luc H4-II-E-C3 cells were seeded and transfected as above. However, only the rPXR257-Luc was used in this study. The transfected cells were treated with dexamethasone at various concentrations (0–100 nM). Data presented in this figure were assembled from three independent experiments and each experiment was performed in triplicate. Bars with a different letter indicate statistically significant differences (p < 0.05).
3.4. Synergistic induction of CYP3A23 by dexamethasone and esfenvalerate
We next examined whether increased expression of PXR by dexamethasone enhances the induction of the PXR target gene CYP3A23 in response to a PXR activator. The pyrethroid esfenvalerate was selected as the PXR activator based on its high relevance to humans. We have previously shown that esfenvalerate was comparably hydrolyzed by rat and human liver microsomes and efficaciously activated both human and rat PXR [11]. It should be noted that pyrethroids constitute a major class of insecticides and account for more than one-third of the insecticides currently marketed in the world [36, 37].
The synergistic induction study was performed in both rat primary hepatocytes and the H4-II-E-C3 cell line. However, the induction at the levels of protein and enzymatic activity was performed in hepatocytes only. Cells were treated with dexamethasone, esfenvalerate or both and harvested after a 24 h-treatment. Cell lysates and total RNA were prepared. Based on the RT-qPCR analysis, dexamethasone (50 nM) and esfenvalerate (10 μM) alone caused an induction of 2 and 8 fold, respectively (Fig. 5A). In contrast, the combined treatment caused a 20-fold induction (Fig. 5A). Similar trend of synergistic induction was detected by Western blotting (Fig. 5A), although the relative magnitude was less profound.
Fig. 5. Synergistic induction of CYP3A23 (A) Effect of dexamethasone on the induction of CYP3A23 by Esfenvalerate.

Hepatocytes from Sprague-Dawley male rats (n = 4) were treated with dexamethasone (50 nM), esfenvalerate (10 μM), or both for 24 h. Total RNA was isolated and analyzed for the level of CYP3A23 mRNA by qRT-PCR. Cell lysates (10 μg) were analyzed for the level of CYP3A23 protein by Western blotting. *Statistically significant (p < 0.05). (B) Synergistic induction of CYP3A23 as a function of concentrations of dexamethasone and esfenvalerate Hepatocytes from Sprague-Dawley male rats (n = 4) were treated with various concentrations of dexamethasone (0–100 nM), esfenvalerate (0–50 μM), or both for 24 h. Total RNA was isolated and analyzed for the level of CYP3A23 mRNA (Left) or PXR (Right) by RT-qPCR. Bars with a letter “a” indicates statistically significant difference from DMSO-control; a letter “b” from the corresponding esfenvalerate treatment; and a letter “c” from esfenvalerate alone or esfenvalerate plus 50 nM dexamethasone (p < 0.05). (C) Enzymatic activity of CYP3A23 Hepatocytes from male rats (n = 4) were treated with various concentrations of dexamethasone (0–100 nM), esfenvalerate (0–50 μM) or both for 24 h. Hepatocytes were washed four times with phosphorous-buffed saline and cell lysates (20 μg) were then prepared. Lysates were analyzed for the oxidative activity with a P450-Glo™ CYP3A4 kit and the relative oxidation activity was determined with standard curves generated from various amounts of recombinant CYP3A4. *Statistically significant (p < 0.05), and bars with a different letter indicate statistically significant differences (p < 0.05) among data-points from DMSO- but different amounts of esfenvalerate. .
The synergistic induction was studied with increasing concentrations of dexamethasone, esfenvalerate or both. Overall, an increase in either chemical caused additional induction of CYP3A23 (Left of Fig. 5B). For example, a combined treatment of dexamethasone at 50 nM and esfenvalerate at 10 μM caused a 20-fold induction. An increase of dexamethasone from 50 to 100 nM caused a 33 fold induction with the same concentration of esfenvalerate (10 μM), and an increase of esfenvalerate from 10 to 25 μM caused an induction of 82 fold with the same concentration of dexamethasone (50 nM) (Left of Fig. 5B). It should be noted that esfenvalerate alone caused little changes in the level of PXR mRNA (Right of Fig. 5B). Next, we tested whether the synergistic induction proportionally increases the activity of CYP3A23. As expected, esfenvalerate alone significantly increased CYP3A23 activity compared with the vehicle control (Fig. 5C, denoted with an alpha sign). Consistent with the synergistic induction by dexamethasone, the increase in the CYP3A23 activity was synergistically enhanced by co-treatment with this glucocorticoid (Fig. 5C, denoted with an asterisk sign).
3.5. Enhanced activation of the CYP3A23 promoter
We next examined whether dexamethasone and esfenvalerate synergistically activate the CYP3A23 promoter. We initially tested the CYP3A23-Luc promoter reporter (−1445 to +74) (Song et al., 2005). As shown in Fig. 6A (normalized reporter activity), dexamethasone at 50 nM slightly stimulated this reporter but the stimulation was statistically insignificant. In contrast, a 4-fold activation (based on normalized luciferase activity) was detected with esfenvalerate at 10 μM. More importantly, co-treatment with both chemicals increased the activation by as many as 13 fold (Left of Fig. 6A). The synergistic activation of this reporter was profound and detected with all concentrations tested. For examples, esfenvalerate at 1 μM in the present of dexamethasone caused even higher activation than esfenvalerate at 10 μM in the absence of this glucocorticoid (Right of Fig. 6A).
Fig. 6. Enhanced activation of CYP3A23 and CYP3A4 reporters.

H4-II-E-C3 cells were seeded in 48-well plates at a density of 1.2 × 105. After an overnight incubation, the cells were transiently transfected by GenJet with a mixture containing a reporter (50 ng) along with 5 ng of the tk-Renilla luciferase plasmid. After incubation at 37°C for 24 h (serum-free medium), the transfected cells were treated with dexamethasone (50 nM), esfenvalerate (10 μM), or both for 48 h. Alternatively, esfenvalerate was used at various concentrations (0–10 μM). Luciferase activities were determined with a Dual-Luciferase Reporter Assay System and the reporter activity was normalized based on the Renilla luminescence signal. (A) Activation of the CYP3A23-Luc reporter (basic + upstream regulatory sequence), (B) Activation of the CYP3A23-198Luc reporter (basic) or its mutant the CYP3A23-198mLuc reporter (basic with disrupted DexRE1), and (C) Activation of the CYP3A4-DP-Luc reporter. Data presented in this figure were assembled from three independent experiments and each experiment was performed in triplicate. *Statistical significance (Fig, 6B, Right of Figs. 6A and 6C) (p < 0.05) and bars with a different letter indicate statistically significant differences Figs. 6A and 6C (Right).
The proximal promoter of CYP3A23 has been shown to fully respond to PXR transactivation [17]. We next examined whether the synergistic activation between esfenvalerate and dexamethasone can be recaptured with the CYP3A23 proximal promoter (−193 to +5). In contrast to the CYP3A23-Luc reporter, the proximal promoter reporter 3A23-198Luc responded better to dexamethasone (50 nM) than esfenvalerate (10 μM) when assayed alone (Left of Fig. 6B). As expected, the combined treatment with both chemicals caused a synergistic activation (27 fold) (Left of Fig. 6B). The proximal promoter contains two elements with PXR binding activity: DexRE-1 and PXR (Fig. 6B), however, the PXR but not DexRE-1 supports PXR-dependent activation [17]. To determine whether the DexRE-1 element is involved in the synergistic activation, a mutant reporter (3A23-198mLuc) was prepared to have this element completely disrupted. Overall, the mutant reporter 3A23-198mLuc showed a similar responding pattern as the wild-type reporter 3A23-198Luc to dexamethasone, esfenvalerate and co-treatment (Fig. 6B). However, the magnitude of the response was much lower. For example, the co-treatment led to a 7-fold activation of the mutant reporter 3A23-198mLuc but a 27-fold activation of the corresponding wild type reporter 3A23-198Luc (Fig. 6B). Another difference was that the 3A23-198Luc reporter but not its mutant was significantly activated when esfenvalerate was assayed alone. Finally, we tested whether the CYP3A4-DP-Luc reporter, the human counterpart of CYP3A23, was synergistically activated by the co-treatment. The overall activation pattern of the CYP3A4-DP-Luc reporter was highly similar to that of the CYP3A23-Luc promoter reporter (Figs. 6A and 6C). However, the CYP3A4-DP-Luc reporter showed much greater responsiveness regardless of treatment alone or in combination (Figs. 6A and 6C).
4. DISCUSSION
Glucocorticoids are known to enhance the induction of multiple drug-metabolizing enzymes in various species including humans and rats [8, 17, 38, 39]. PXR is recognized as a major regulator in the induced expression of these enzymes [1, 2]. It is generally accepted that increased expression of PXR by glucocorticoids contributes significantly to their enhanced induction of drug-metabolizing enzymes [17, 18, 20, 21]. In this study, we have shown that dexamethasone, a synthetic glucocorticoid, increases the expression of rat PXR through trans-activation. Multiple experimental approaches further establish that the transactivation results from the action of ligand-activated glucocorticoid receptor-α (the classic glucocorticoid pathway). Nanomolar dexamethasone profoundly synergizes esfenvalerate (a PXR ligand) in inducing the expression of CYP3A23, presenting a PXR-based ligand-inducer interaction with toxicological significance.
The induction of rat PXR, like the induction of Tat, occurred rapidly and was statistically significant even by nanomolar dexamethasone (Fig. 2B). However, there were notable differences in the induction of these two genes. Based on the magnitude of the induction, the PXR gene was less inducible. For example, dexamethasone at 50 nM caused a 17-fold induction of Tat. In contrast, this concentration at the same time-point induced PXR by only ~4 fold (Fig. 2B). The greater inducibility of Tat was not due to a lower basal expression. Based on the Ct values (threshold cycles) for both genes, the PXR gene had a lower basal expression than the Tat gene (~26 versus 23). In addition, it appears that the PXR mRNA had a longer half life than the Tat mRNA. While actinomycin D abolished dexamethasone-mediated increases in PXR and Tat mRNA, this inhibitor alone actually had opposing effect on the levels of PXR and Tat mRNA. Actinomycin D slightly increased PXR mRNA but markedly decreased Tat mRNA (Fig. 3A). Assumed that actinomycin D inhibited the transcription of all cellular genes equally, the relative increase suggests that PXR mRNA has a longer half life than the average half life of all cellular mRNA but the opposite is true with Tat mRNA.
It is interesting to notice that the CYP3A23-Luc and CYP3A23-198Luc reporters, although both activated synergistically by co-treatment, showed different responding patterns to esfenvalerate and dexamethasone (Figs. 6A and B). The CYP3A23-198Luc reporter, containing the basic promoter of the CYP3A23 gene (−193/+5), was activated to a higher extent by dexamethasone than esfenvalerate. In contrast, the CYP3A23-Luc reporter was activated higher by esfenvalerate (Figs. 6A and B). The CYP3A23-Luc reporter contains additional upstream and downstream sequences (−1445/+74). This difference suggests that other genomic elements, in addition to the basic promoter, plays a role in responding to dexamethasone and esfenvalerate. A previous study showed that liver nuclear extracts from dexamethasone treated rats had higher binding activity toward a C/EBP site in the upstream sequence of the CYP3A23 gene [15]. Importantly, a reporter containing this element was stimulated by over-expressing the corresponding protein C/EBPα. The CYP3A23-Luc but not CYP3A23-198Luc reporter contains the C/EBP element. Therefore, the activation of the CYP3A23-Luc reporter represented a composite effect of the C/EBP site with the proximal promoter. Namely, esfenvalerate might have modulated the expression or binding activity of C/EBPα, which in turn increased the activity of the CYP3A23-Luc reporter.
The CYP3A23-Luc reporter, on the other hand, contains several elements that are critical in the transactivation by dexamethasone: DexRE1 and DexRE2 sites [17]. Both sites were reportedly bound by PXR with the DexRE2 site (i.e., PXR element) being bound to a much higher extent [17]. Therefore, it is generally accepted that DexRE1 is a contributor to the full transactivation of the CYP3A23 gene in response to dexamethasone [17]. Interestingly, disruption of the DexRE1 site retained some responsiveness to dexamethasone but not esfenvalerate (Right of Fig. 6B). The precise mechanism on the differential effect remains to be determined. Given the fact that dexamethasone but not esfenvalerate is an inducer of PXR as well, it is likely that the DexRE1 site makes the DexRE2 site more accessible to PXR. This is of significance particularly when PXR is less abundant. Nevertheless, the responding pattern of the CYP3A23-Luc reporter was similar to the changes of CYP3A23 mRNA in primary hepatocytes (Figs. 5A and 6A), suggesting that the CYP3A23-Luc reporter, not the truncated or site-directed mutants, serves a better predicating tool for the induction of CYP3A23 in vivo.
The human counterpart of the CYP3A23-Luc reporter (CYP3A4-DP-Luc), on the other hand, was activated similarly as the CYP3A23-Luc reporter (Figs. 6A and 6C). Namely, it was activated the least by dexamethasone, the most by the co-treatment and in the middle by esfenvalerate. The similarity suggests that the synergistic activation by a glucocorticoid and a PXR ligand is conserved cross species. Indeed, dexamethasone has been shown to enhance the induction of several PXR target genes in multiple species [8, 17, 20, 21, 38, 39]. On the other hand, the mechanisms in the induction of PXR may vary depending on a species. In this study, we have provided several lines of evidence that support an involvement of transactivation in the induction of rat PXR. However, it appears that other mechanisms, probably in addition to transactivation, may account for the induction of human PXR. We have previously shown that dexamethasone significantly increased the expression of a human PXR transgene in 293T cells [18]. This transgene was driven by a cytomegalovirus promoter and contained the PXR coding sequence only. It has been reported that the cytomegalovirus promoter does not respond to glucocorticoids [40]. Therefore, induction of the PXR transgene was likely mediated by stabilizing PXR mRNA, protein or both, presumably by acting on part of the coding sequence and/or protein. However, it can not be excluded that mechanisms whereby dexamethasone induces human PXR vary from one cell type to another (hepatocytes versus 293T).
The synergistic induction of CYP enzymes may have profound toxicological consequences. Esfenvalerate is a widely used pyrethroid insecticide and the enhanced induction of CYP3A enzymes may increase its own clearance. Esfenvalerate is an ester thus a substrate of carboxylesterases [11, 41, 42]. On the other hand, esfenvalerate is also metabolized by cytochrome P450s [11, 42]. In rats, hydrolysis by carboxylesterases and oxidation by CYP enzymes contribute comparably to the overall clearance of this pyrethroid [42]. While it remains to be determined whether CYP3A23 oxidizes esfenvalerate, several rat CYPs are found to catalyze the oxidation of esfenvalerate including CYP3A2, a closely related CYP enzyme to CYP3A23 [42]. Nevertheless, it is conceivable that the synergistic induction of CYP3A23 plays a measurable role in the clearance of esfenvalerate in rats. In contrast to rats, synergistic induction of CYP3A4 certainly plays a greater role in the elimination of esfenvalerate in humans, as oxidation contributes as much as 95% to the clearance of esfenvalerate based on in vitro metabolism study [42].
In addition, several other human CYP enzymes, notably CYP2C class of proteins, have been shown to oxidize esfenvalerate with even higher activity than CYP3A enzymes [42]. PXR is a regulator on the expression of CYP2C genes and dexamethasone has been found to enhance the induction of these enzymes in human primary hepatocytes [38]. Therefore, it is expected that oxidative elimination of esfenvalerate would be markedly enhanced by conditions with increased glucocorticoids (stress condition). It is generally accepted that hydrolysis of pyre-throids is considered a detoxification process [43, 44]. However, oxidation of pyrethroids by CYPs, on the other hand, may have opposite toxicological consequences depending on a pyrethroid or even a test system. For example, CYP inducers phenobarbital and 3-methylcholanthrene potentiate deltamethrin (a pyrethroid) in the induction of neurobehavioral toxicity [45], whereas hepatoxicity of pyrethroid cypermethrin is markedly decreased in primary hepatocytes pretreated by phenobarbital [46].
In summary, our work points to several important conclusions. First, the induction of rat PXR by dexamethasone is abolished by the RNA synthesis inhibitor actinomycin D, and this glucocorticoid directly stimulates the rat PXR promoter, establishing that dexamethasone-induction of PXR is achieved by transactivation. Second, disruption of the element responding to the glucocorticoid receptor eliminates the stimulation of the promoter and the anti-glucocorticoid RU486 abolishes the induction, suggesting that the induction is a sequence-specific event and directed by the glucocorticoid receptor. Third, nanomolar dexamethasone synergistically enhances esfenvalerate in stimulating CYP3A23 promoter and inducing this enzyme, signifying the interaction between a PXR inducer and a PXR ligand in drug and insecticide elimination. PXR is recognized as a master regulator of genes involved in xenobiotic metabolism and transport. Secretion of glucocorticoids is increased under stress conditions. The enhanced PXR transactivation by glucocorticoids may have both pharmacological and toxicological significance.
Acknowledgments
This work was supported by NIH grants R01GM61988 and R01ES07965.
Abbreviation
- CYP
cytochrome P450
- DMEM
Dulbecco’s Modified Eagle Medium
- GAPDH
glyceradehyde-3-phosphate dehydrogenase
- HBSS
Hank’s Buffered Salt Solution
- ITS
Insulin-transferrin-sodium selenite
- PXR
pregnane X receptor
- RT-qPCR
quantitative reverse transcription-polymerase chain reaction
- Tat
tyrosine aminotransferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ma X, Idle JR, Gonzalez FJ. The pregnane X receptor: from bench to bedside. Expert Opin Drug Metab Toxicol. 2008;4:895–908. doi: 10.1517/17425255.4.7.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9:310–322. doi: 10.2174/138920008784220664. [DOI] [PubMed] [Google Scholar]
- 3.Goodwin B, Redinbo MR, Kliewer SA. Regulation of cyp3a gene transcription by the pregnane x receptor. Annu Rev Pharmacol Toxicol. 2002;42:1–23. doi: 10.1146/annurev.pharmtox.42.111901.111051. [DOI] [PubMed] [Google Scholar]
- 4.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 5.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. 2.1 A crystal structure of human PXR in complex with the St. John’s wort compound hyperforin. Biochemistry. 2003;42:1430–1438. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 6.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–828. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 7.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindley C, Hamilton G, McCune JS, Faucette S, Shord SS, Hawke RL, Wang H, Gilbert D, Jolley S, Yan B, LeCluyse EL. The effect of cyclophosphamide with and without dexamethasone on cytochrome P450 3A4 and 2B6 in human hepatocytes. Drug Metab Dispos. 2002;30:814–822. doi: 10.1124/dmd.30.7.814. [DOI] [PubMed] [Google Scholar]
- 9.Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, Stimmel JB, Goodwin B, Liddle C, Blanchard SG, Willson TM, Collins JL, Kliewer SA. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;275:15122–15127. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 10.Xie W, Barwick JL, Simon CM, Pierce AM, Safe S, Blumberg B, Guzelian PS, Evans RM. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 2000;14:3014–3023. doi: 10.1101/gad.846800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang D, Wang X, Wu Z, Deng R, Yan B. Pyrethroid insecticides: isoform-dependent hydrolysis, induction of cytochrome P450 3A4 and evidence on the involvement of the pregnane X receptor. Toxicol Appl Pharmacol. 2009;237:49–58. doi: 10.1016/j.taap.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kliewer SA, Willson TM. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J Lipid Res. 2002;43:359–464. [PubMed] [Google Scholar]
- 13.Moore LB, Goodwin B, Jones SA, Wisely GB, Serabjit-Singh CJ, Willson TM, Collins JL, Kliewer SA. St. John’s wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci U S A. 2000;97:7500–7502. doi: 10.1073/pnas.130155097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodrigues E, Vilarem MJ, Ribeiro V, Maurel P, Lechner MC. Two CCAAT/enhancer binding protein sites in the cytochrome P4503A1 locus. Potential role in the glucocorticoid response. Eur J Biochem. 2003;270:556–564. doi: 10.1046/j.1432-1033.2003.03413.x. [DOI] [PubMed] [Google Scholar]
- 16.El-Sankary W, Bombail V, Gibson GG, Plant N. Glucocorticoid-mediated induction of CYP3A4 is decreased by disruption of a protein: DNA interaction distinct from the pregnane X receptor response element. Drug Metab Dispos. 2002;30:1029–1034. doi: 10.1124/dmd.30.9.1029. [DOI] [PubMed] [Google Scholar]
- 17.Huss JM, Kasper CB. Two-stage glucocorticoid induction of CYP3A23 through both the glucocorticoid and pregnane X receptors. Mol Pharmacol. 2000;58:48–57. doi: 10.1124/mol.58.1.48. [DOI] [PubMed] [Google Scholar]
- 18.Song X, Xie M, Zhang H, Li Y, Sachdeva K, Yan B. The pregnane X receptor binds to response elements in a genomic context-dependent manner, and PXR activator rifampicin selectively alters the bindings among target genes. Drug Metab Dispos. 2004;32:35–42. doi: 10.1124/dmd.32.1.35. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, LeCluyse E, Liu L, Hu M, Matoney L, Yan B. Rat pregnane X receptor: molecular cloning, tissue distribution and xenobiotic regulation. Arch Biochem Biophys. 1999;368:14–22. doi: 10.1006/abbi.1999.1307. [DOI] [PubMed] [Google Scholar]
- 20.Cooper BW, Cho TM, Thompson PM, Wallace AD. Phthalate induction of CYP3A4 is dependent on glucocorticoid regulation of PXR expression. Toxicol Sci. 2008;103:268–77. doi: 10.1093/toxsci/kfn047. [DOI] [PubMed] [Google Scholar]
- 21.Pascussi JM, Drocourt L, Fabre JM, Maurel P, Vilarem MJ. Dexamethasone induces pregnane X receptor and retinoid X receptor-alpha expression in human hepatocytes: synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol Pharmacol. 2000;58:361–372. doi: 10.1124/mol.58.2.361. [DOI] [PubMed] [Google Scholar]
- 22.Sachdeva K, Yan B, Chichester CO. Lipopolysaccharide and cecal ligation/puncture differentially affect the subcellular distribution of the pregnane X receptor but consistently cause suppression of its target gene CYP3A. Shock. 2003;19:469–474. doi: 10.1097/01.shk.0000048903.46342.ec. [DOI] [PubMed] [Google Scholar]
- 23.Zhu W, Song L, Zhang H, Matoney L, LeCluyse E, Yan B. Dexamethasone differentially regulates expression of carboxylesterase genes in humans and rats. Drug Metab Dispos. 2000;28:186–191. [PubMed] [Google Scholar]
- 24.Xie M, Yang D, Wu M, Xue B, Yan B. Mouse liver and kidney carboxylesterase (M-LK) rapidly hydrolyzes antitumor prodrug irinotecan and the N-terminal three quarter sequence determines substrate selectivity. Drug Metab Dispos. 2003;31:21–7. doi: 10.1124/dmd.31.1.21. [DOI] [PubMed] [Google Scholar]
- 25.Radonić A, Thulke S, Mackay IM, Landt O, Siegert W, Nitsche A. Guideline to reference gene selection for quantitative real-time PCR. Biochem Biophys Res Commun. 2004;313:856–862. doi: 10.1016/j.bbrc.2003.11.177. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Yan B. Photochemotherapeutic agent 8-methoxypsoralen induces the expression of cytochrome P450 3A4 and carboxylesterase HCE2: evidence on a differential involvement of the pregnane X receptor. Toxicol Sci. 2007;95:13–22. doi: 10.1093/toxsci/kfl120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee CM, Pohl J, Morgan ET. Dual Mechanisms of CYP3A Protein Regulation by Proinflammatory Cytokine Stimulation in Primary Hepatocyte Cultures. Drug Metab Dispos. 2009;37:865–872. doi: 10.1124/dmd.108.026187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan WK, Klock G, Bernard HU. Progesterone and glucocorticoid response elements occur in the long control regions of several human papillomaviruses involved in anogenital neoplasia. J Virol. 1989;63:3261–3269. doi: 10.1128/jvi.63.8.3261-3269.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruland T, Lavik LAS, Dai HY, Dalen A. A glucocorticoid response element in the LTR U3 region of Friend murine leukaemia virus variant FIS-2 enhances virus production in vitro and is a major determinant for sex differences in susceptibility to FIS-2 infection in vivo. J Gen Virol. 2003;84:907–916. doi: 10.1099/vir.0.18625-0. [DOI] [PubMed] [Google Scholar]
- 30.Song X, Li Y, Liu J, Mukundan M, Yan B. Simultaneous substitution of phenylalaine-305 and aspartate-318 of rat PXR by the corresponding human residues abolishes the ability to transactivate the cytochrome P450 3A23 promoter. J Pharmacol Exp Ther. 2005;312:571–582. doi: 10.1124/jpet.104.074971. [DOI] [PubMed] [Google Scholar]
- 31.Liu F, Yang D, Song X, Deng R, Yan B. The far and distal enhancers in the CYP3A4 gene coordinates the proximal promoter in responding to the pregnane X receptor similarly but differentially to hepatocyte nuclear factor-4α. Biochem J. 2008;409:243–250. doi: 10.1042/BJ20070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Becker PB, Gloss B, Schmid W, Strahle U, Schutz G. In vivo protein-DNA interactions in a glucocorticoid response element require the presence of the hormone. Nature. 1996;324:686–688. doi: 10.1038/324686a0. [DOI] [PubMed] [Google Scholar]
- 33.Ottosson M, Mårin P, Karason K, Elander A, Björntorp P. Blockade of the glucocorticoid receptor with RU 486: effects in vitro and in vivo on human adipose tissue lipoprotein lipase activity. Obes Res. 1995;3:233–240. doi: 10.1002/j.1550-8528.1995.tb00143.x. [DOI] [PubMed] [Google Scholar]
- 34.Goecke A, Guerrero J. Glucocorticoid receptor beta in acute and chronic inflammatory conditions: clinical implications. Immunobiology. 2006;211:85–96. doi: 10.1016/j.imbio.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Shi D, Yang J, Yang D, You L, Yan B. Dexamethasone suppresses the expression of multiple rat carboxylesterases through transcriptional repression: evidence for an involvement of the glucocorticoid receptor. Toxicology. 2008;254:97–105. doi: 10.1016/j.tox.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adelsbach TL, Tjeerdema RS. Chemistry and fate of fenvalerate and esfenvalerate. Rev Environ Contam Toxicol. 2003;176:137–154. doi: 10.1007/978-1-4899-7283-5_3. [DOI] [PubMed] [Google Scholar]
- 37.Casida JE, Quistad GB. Golden age of insecticide research: past, present, or future? Annu Rev Entomol. 1998;43:1–16. doi: 10.1146/annurev.ento.43.1.1. [DOI] [PubMed] [Google Scholar]
- 38.Gerbal-Chaloin S, Pascussi JM, Pichard-Garcia L, Daujat M, Waechter F, Fabre JM, Carrère N, Maurel P. Induction of CYP2C genes in human hepatocytes in primary culture. Drug Metab Dispos. 2001;29:242–251. [PubMed] [Google Scholar]
- 39.Wang H, Faucette SR, Gilbert D, Jolley SL, Sueyoshi T, Negishi M, LeCluyse EL. Glucocorticoid receptor enhancement of pregnane X receptor-mediated CYP2B6 regulation in primary human hepatocytes. Drug Metab Dispos. 2003;31:620–630. doi: 10.1124/dmd.31.5.620. [DOI] [PubMed] [Google Scholar]
- 40.Burnstein KL, Maiorino CA, Dai JL, Cameron DJ. Androgen and glucocorticoid regulation of androgen receptor cDNA expression. Mol Cell Endocrinol. 1995;115:177–186. doi: 10.1016/0303-7207(95)03688-1. [DOI] [PubMed] [Google Scholar]
- 41.Godin SJ, Scollon EJ, Hughes MF, Potter PM, DeVito MJ, Ross MK. Species differences in the in vitro metabolism of deltamethrin and esfenvalerate: differential oxidative and hydrolytic metabolism by humans and rats. Drug Metab Dispos. 2006;34:1764–1771. doi: 10.1124/dmd.106.010058. [DOI] [PubMed] [Google Scholar]
- 42.Godin SJ, Crow JA, Scollon EJ, Hughes MF, DeVito MJ, Ross MK. Identification of rat and human cytochrome p450 isoforms and a rat serum esterase that metabolize the pyrethroid insecticides deltamethrin and esfenvalerate. Drug Metab Dispos. 2007;35:1664–1671. doi: 10.1124/dmd.107.015388. [DOI] [PubMed] [Google Scholar]
- 43.Casida JE, Gammon DW, Glickman AH, Lawrence LJ. Mechanisms of selective action of pyrethroid insecticides. Annu Rev Pharmacol Toxicol. 1983;23:413–438. doi: 10.1146/annurev.pa.23.040183.002213. [DOI] [PubMed] [Google Scholar]
- 44.Cantalamessa F. Acute toxicity of two pyrethroids, permethrin, and cypermethrin in neonatal and adult rats. Arch Toxicol. 1993;67:510–513. doi: 10.1007/BF01969923. [DOI] [PubMed] [Google Scholar]
- 45.Dayal M, Parmar D, Dhawan A, Ali M, Dwivedi UN, Seth PK. Effect of pretreatment of cytochrome P450 (P450) modifiers on neurobehavioral toxicity induced by deltamethrin. Food Chem Toxicol. 2003;41:431–437. doi: 10.1016/s0278-6915(02)00249-1. [DOI] [PubMed] [Google Scholar]
- 46.El-Tawil OS, Abdel-Rahman MS. The role of enzyme induction and inhibition on cypermethrin hepatotoxicity. Pharmacol Res. 2001;44:33–40. doi: 10.1006/phrs.2001.0826. [DOI] [PubMed] [Google Scholar]