

Abstract
A human p53 homologue, p63 (p40/p51/p73L/CUSP) that maps to the chromosomal region 3q27–29 was found to produce a variety of transcripts that encode DNA-binding proteins with and without a trans-activation domain (TA- or ΔN-, respectively). The p63 gene locus was found to be amplified in squamous cell carcinoma, and overexpression of ΔNp63 (p40) led to increased growth of transformed cells in vitro and in vivo. Moreover, p63-null mice displayed abnormal epithelial development and germ-line human mutations were found to cause ectodermal dysplasia. We now demonstrate that certain p63 isotypes form complexes with p53. p53 mutations R175H or R248W abolish the association of p53 with p63, whereas V143A or R273H has no effect. Deletion studies suggest that the DNA-binding domains of both p53 and p63 mediate the association. Overexpression of wild type but not mutant (R175H) p53 results in the caspase-dependent degradation of certain ΔNp63 proteins (p40 and ΔNp63α). The association between p53 and ΔNp63 supports a previously unrecognized role for p53 in regulation of ΔNp63 stability. The ability of p53 to mediate ΔNp63 degradation may balance the capacity of ΔNp63 to accelerate tumorigenesis or to induce epithelial proliferation.
The p53 gene is the most commonly inactivated gene in human cancers, and loss of critical p53 signaling pathways is central to tumorigenesis (1). In response to stress stimuli, the p53 transcription factor binds specific DNA sequences and activates responsive genes that induce cell-cycle arrest or programmed cell death (2). p53 mutations occurring in human cancers produce abnormal p53 proteins that are unable to bind DNA and regulate the transcription of its target genes (3). p53 mutants also heterodimerize with wild-type p53 (p53WT), which results in a conformational change of the protein complex such that it no longer binds to p53-regulated cis-acting elements (4, 5). As a result of this heteromeric association, p53 mutants inhibit the function of p53WT and its ability to regulate cell proliferation, oncogenesis, and apoptosis (6).
Recently cloned genes p73 (located on chromosomal locus 1p36) and p63 (locus 3q27–9) encode proteins with sequence and functional homology to p53 and share a similar exon/intron organization (7). p73 (1p36) was shown to inhibit cell growth and induce apoptosis in cell lines, but it has variable trans-activation activities on known p53 promoters (8).
p63 plays an essential role in epidermal–mesenchymal interactions during embryonic development, where it is required for regenerative proliferation of limb, craniofacial, and epithelial development and for differentiation of stratified squamous epithelia (9, 10). Mutations of this gene in humans cause an autosomal dominant disorder characterized by ectrodactyly, ectodermal dysplasia, and a limb mammary syndrome (11). Autoantibodies recognizing p63 isotypes in nuclei of stratified squamous epithelium were found in patients with chronic ulcerative stomatitis (12). We demonstrated that p63 is amplified in squamous cell carcinoma cells, and certain ΔNp63 isotypes (which lack a trans-activation domain—i.e., p40) encoded by this gene display oncogenic properties (13).
Proteins encoded by p53 homologues exhibit a modular structure similar to p53, and some isotypes contain an N-terminal trans-activation domain (TA isotypes), whereas others do not (ΔN isotypes). Although they all contain DNA-binding and oligomerization domains, some contain additional C-terminal sequences (8, 14, 15). TAp63 isotypes are designated as TAp63α (p51B), TAp63β, and TAp63γ (p51A). The ΔNp63 isotypes include ΔNp63α (p73L, CUSP), ΔNp63β, ΔNp63γ, and p40 (14, 16–18).
Although, The TAp63 isotypes display strong to intermediate trans-activation activities, ΔNp63 isotypes display dominant-negative activities (14). TAp63γ can activate a pro-apoptotic signaling pathway similar to p53, although the degree of apoptotic changes was much less in cells expressing ΔNp63γ, consistent with its reduced trans-activating ability. The opposing trans-activating properties of ΔNp63 (or ΔNp73) isotypes vs. TAp63 (TAp73) isotypes raise the possibility that isotypes lacking the trans-activation domain might display some oncogenic/anti-apoptotic potentials in contrast to other isotypes with tumor suppressor/pro-apoptotic potentials (14, 19).
p40 represents the p63 core domain and consists of a DNA-binding domain and a short oligomerization region (18). The similarities between the DNA-binding and oligomerization domains of p63 isotypes and p53 raise the possibility that these proteins may also bind p53-specific DNA-binding sites in the human genome or physically interact with p53.
Materials and Methods
Cell Lines, Antibodies, and Reagents.
FaDu cells (expressing endogenous p53 and p63), SaOs2 cells (p53-null, no p63 expression), human embryonic kidney (HEK)-293 cells, human small cell lung cancer (SCLC) cell lines, H1628, H2106, H1881 (expressing mutant p53, R175H, R248W, R273H, respectively, and p63), and non-small cell lung carcinoma (NSCLC, L953, 1012, expressing mutant p53, R175H, R248W, respectively, and lacking the second p53 allele) cells were purchased from the American Type Culture Collection. SCLC cells and HNSCC primary cell lines (i.e., 022) were maintained in RPMI medium 1640/10% fetal bovine serum (FBS). FaDu cells were grown in minimal Eagle's medium/10% FBS. SaOs2 cells were cultured in McCoy's 5A medium/10% FBS. HEK-293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM)/10% FBS. Normal and tumor prostate tissue specimens were obtained from the Pathology Tissue Bank at the Johns Hopkins Hospital.
A polyclonal antibody against an N-terminal p40 specific peptide (residues 5–18) present in all ΔNp63 isotypes was used (13). The monoclonal antibodies to p53 (BP53–12, against the N-terminal residues 20–27, and PAb122, against the C-terminal residues 370–378) were purchased from Labvision NeoMarkers (Fremont, CA). A 26S-proteasome inhibitor, MG-132 (American Peptide, Sunnyvale, CA); calpain inhibitor Ac-Leu-Leu-norleucinal (Sigma); and caspase inhibitors, Z-VAD-fmk, Ac-YVAD-CHO, Ac-DEVD-CHO, Ac-VEID-CHO, and Ac-IETD-CHO (Enzyme Systems Products, Livermore, CA; Z = benzyloxycarbonyl, fmk = fluoromethyl ketone, CHO = aldehyde analogue) were used. Caspases 1, 3, 6, and 8 were obtained from Biomol (Plymouth Meeting, PA).
Yeast Two-Hybrid Screening.
We screened a mouse embryonic cDNA library (B6: C57BL/6, embryonic day 14.5; Stratagene). The bait plasmid, pGal4-BD-p40, encoded a fusion protein composed of the Gal4 DNA-binding domain and a p40 protein. A two-hybrid screen was performed as described previously (20). Plasmids representing the human p53WT or its mutants (p53143, p53175, p53248, and p53273) were generated as fusions with the Gal4 activation domain (AD). The pGal4-BD-p40 (or pGal4-BD-ΔNp63α) construct was cotransformed into yeast with the pGal4-AD constructs encoding either p53WT or p53 mutants. Final colonies were analyzed by a β-galactosidase quantitative liquid assay using o-nitrophenyl β-d-galactopyranoside (ONPG) as a substrate (20).
Coprecipitation Assays.
SaOs2 cells were infected with an empty adenovirus, Ad5 (multiplicity of infection, MOI, = 1), or with Ad-p40 (MOI = 1) for 1 h. Cells were lysed 18 h after infection with buffer A (100 mM Tris⋅HCl, pH 7.5/150 mM NaCl/2 mM EDTA/0.5% Triton X-100/0.5% Brij-50 containing 1 mM PMSF, 4 μg/ml aprotinin, 1 μg/ml pepstatin A, 1 μg/ml leupeptin, 1 mM Na3VO4, and 50 mM NaF). Complexes were precipitated from precleared cell lysates by using antibody to ΔNp63 or antibody to p53 (BP53–12) as indicated.
Tissue samples were homogenized with buffer B (10 mM Hepes–KOH, pH 7.9/10 mM KCl/1.5 mM MgCl2/0.5 mM DTT/0.5 mM PMSF) for 15 min on ice, then Nonidet P-40 was added to a final concentration of 0.2% for 10 min. Nuclear and cytoplasmic lysates were prepared and used for coprecipitation experiments as described (ref. 21, pp. 4–22).
Transfections.
The mammalian expression cassettes pCEP4-p53WT, pCEP4-p53143, pCEP4-p53175, pCEP4-p53248, pCEP4-p53273, pCEP4-ΔNp63α, and pCEP4 (empty vector) were used for transient transfection of SaOs2 cells with 5 μg of DNA and 10 μl of FuGene-6 (Boehringer Mannheim). Cells were collected after 24 h and subsequently analyzed.
Construction of p40 and p53 Adenoviruses.
A full-length p40 cDNA and p53WT were subcloned into pAdTrack-CMV, and recombinant adenovirus was prepared accordingly (13). Final yields of adenoviruses were generally 1011 to 1012 plaque-forming units.
Northern Blot Analysis.
For cancer cell lines, total RNA was isolated with the Trizol reagent (GIBCO/BRL). Northern blot hybridization was performed as described (ref. 21, pp. 9–13).
Immunofluorescence Analysis.
FaDu or 022 cells were plated on culture slides (Becton Dickinson) for 48 h. 022 cells were further infected with Ad-p53 (MOI = 1) for an additional 18 h before fixation (22). Cells were then incubated with an affinity-purified antibody to ΔNp63 (1:500) and antibody to p53 (PAb122, 1:100) overnight at 4°C. Slides were incubated with FITC-conjugated goat anti-mouse Ig (1:1000) and rhodamine-conjugated goat anti-rabbit Ig (1:1000) for 2 h in the dark. DNA in nuclei was stained with 4′,6-diamidino-2-phenylindole (DAPI; 1:10,000) for 5 min. Immunofluorescence was examined under a Zeiss Axiophot epifluorescence microscope.
Metabolic Cell Labeling.
Cells were infected with recombinant adenoviruses for 18 h, washed with PBS, preincubated with methionine-free medium for 30 min, and labeled for 1 h with [35S]methionine at 200 μCi/ml (1 μCi = 37 kBq). Labeled cells were washed twice in PBS and then maintained in nonradioactive medium for 0, 1, 1.5, 2, 3, or 6 h. Total cell lysates were precipitated with antibody to ΔNp63 and resolved by SDS/10% PAGE, and dried gels were visualized by autoradiography.
Protease Cleavage Assay.
Pure caspases 1, 3, 6 and 8 expressed in bacteria were used (PharMingen or Biomol). To test for protease cleavage, [35S]methionine-labeled p40 protein was generated by transcription-translation kit (Promega). Five microliters of radioactive mix was incubated with purified caspases for 2 h at 37°C. Cleavage reactions were performed as described (23).
Results
ΔNp63 Isotypes Interact with p53 in Yeast.
To explore the function of p63, we examined the ability of the ΔNp63 core domain (p40) to associate with other proteins by using a two-hybrid yeast genetic strategy. We screened a mouse embryonic cDNA library with p40 as the bait. A total of 1.6 × 106 yeast transformants were placed under triple selection (for tryptophan, leucine, and histidine independence). Twenty-one clones that activated the yeast chromosomal HIS3 and lacZ reporter genes in the presence of p40 were sequenced and analyzed by using the National Center for Biotechnology Information database. The most common target identified was p53, yielding three independent clones. The interaction between the p53 clones and p40 was quite strong compared with the p53/simian virus 40 (SV40) positive control pair or the lamin C/SV40 negative control pair (data not shown).
Because p53 mutations identified in various human cancers may affect the binding of p53 to ΔNp63, we examined the ability of the most common p53 mutants (as prey plasmids) to interact with ΔNp63α (as bait plasmid), the most abundant p63 isotype in human cancer cells. Interestingly, a strong interaction persisted between ΔNp63α and p53WT, p53143, or p53273 mutants, whereas mutations R175H or R248W of p53 abolished the interaction (see Table 1, which is published as supplemental data on the PNAS web site, www.pnas.org).
To confirm the data obtained from the two-hybrid screens, 35S-labeled p40 was mixed with glutathione S-transferase (GST)-p53WT followed by precipitation with glutathione-agarose. As negative controls, radioactive mixes were precipitated with glutathione-agarose alone or in the presence of GST-midkine (GST-MK). We showed that His-tagged p40 strongly associates with p53WT, p53143, or p53273, whereas a dramatic decrease of binding was observed between p40 and p53175 or p53248 under these conditions (data not shown).
In Vivo Complexes Between p53 and ΔNp63 Colocalize to Nuclei.
Because the ΔNp63α isotype is predominantly expressed in human tumor cells (13), we investigated whether ΔNp63α associates with p53 in transfected cells. SaOs2 cells (p53−/−, no p63 expression) were cotransfected with a pCMV-GFP-ΔNp63α expression cassette alone and together with either p53WT or p53 mutants. Expression of p53WT, p53 mutants, or ΔNp63α was analyzed by immunoblotting (Fig. 1A Top and Middle, respectively). ΔNp63α/p53 complexes were precipitated from total cell lysates with antibody to ΔNp63 followed by immunoblotting with antibody to p53 (Fig. 1A Bottom). In these experiments we observed that ΔNp63α binds to p53WT, p53143, or p53273, whereas it does not bind to p53175 or p53248 (Fig. 1A).
Figure 1.

Interaction of ΔNp63α and p53 in vivo. (A) Association of ΔNp63α and p53 in transfected cells. SaOs2 cells were transiently transfected with the indicated plasmids. Coprecipitation with antibody to ΔNp63 followed by immunoblotting with antibody to p53 reveals a specific band in lanes 4, 5, and 8. ΔNp63α did not interact with p53 mutants R175H or R248W (lanes 6 and 7). (B) FaDu cells. Total lysates were assayed for expression of ΔNp63α (Upper) and p53 (Lower), and p63/p53 complexes were immunoprecipitated (IP) with antibody to either ΔNp63α or p53. Negative control is anti-hemagglutinin (HA) antibody. (C) Human normal prostate (lanes 1 and 2) or primary prostate tumor (lanes 3 and 4) were lysed, and nuclear (lanes 1 and 3) and cytoplasmic (lanes 2 and 4) fractions were analyzed. Left and Center show the p53 and ΔNp63α protein levels in cell lysates, respectively. Antibodies used are indicated. (Right) Immunoprecipitation (IP) of ΔNp63α/p53 complexes with antibody to ΔNp63 followed by blotting with antibody to p53 (BP53–12), with higher levels in the tumor.
Because we previously demonstrated a high level of p63 mRNA expression in normal human prostate (18), cell lysates from normal prostate and prostate tumor representing nuclear and cytoplasmic fractions were used for coprecipitation assays to localize the complexes. As a positive control, we used a total lysate obtained from FaDu cells (Fig. 1B). As negative controls, we used total protein lysates obtained from human NSCLC cells, which express ΔNp63α but harbor p53 mutations R248W or R175H (samples 1012 or L953, respectively; data not shown). Although, the p53 level is much higher in the normal prostate than in the tumor (Fig. 1C Left), the ΔNp63α protein levels demonstrate a reverse pattern when an antibody that recognizes all ΔNp63 isotypes is used (Fig. 1C Center). Both proteins (p53 and ΔNp63α) are localized mainly in the nuclear fraction (Fig. 1C Left and Center), and as shown, physical complexes between endogenous p53 and ΔNp63α are formed in vivo (Fig. 1C Right).
To further examine whether p53 mutations (R175H or R248W) affect the ability of endogenous p53 and p63 proteins to form physical complexes, we prepared nuclear lysates from human SCLC cell lines H1628, H2106, and H1881 (harboring p53 mutations R175H, R248W, R273H, respectively). Complexes between p53 and ΔNp63 were precipitated with antibody to ΔNp63 and immunoblotted with antibody to p53 (PAb122). Our data showed that either R175H or R248W mutation abolished the association of mutant p53 with ΔNp63 proteins in vivo, whereas the R273H mutation had no effect on the interaction between p53 and ΔNp63 (data not shown).
Finally, by immunofluorescence microscopy, we found that p53 and ΔNp63α were localized in the interphase cell nuclei of 022 cells infected with Ad-p53WT (exogenous p53 and endogenous ΔNp63α) and FaDu cells (where p53 and ΔNp63α proteins are expressed endogenously). Both proteins (p53 and ΔNp63α) showed a diffuse pattern of granular foci. Merging of the ΔNp63α and p53 images demonstrates that their localization patterns clearly overlap (Fig. 2).
Figure 2.
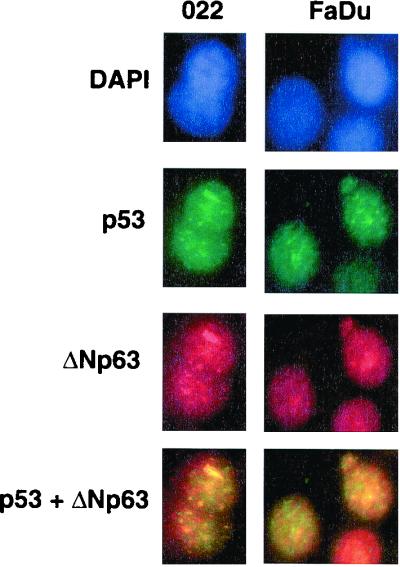
Nuclear colocalization of ΔNp63 and p53. (Left) 022 cells expressing endogenous ΔNp63 were infected with Ad-p53WT (MOI = 1) for 18 h. (Right) FaDu cells expressing endogenous p53 and ΔNp63. DAPI shows nuclear staining with 4′,6-diamidino-2-phenylindole. Slides were incubated with antibody to ΔNp63 or with antibody to p53 (PAb122). Expression of ΔNp63 and p53 was visualized with rhodamine-conjugated anti-rabbit Ig (red) or with FITC-conjugated anti-mouse Ig (green), respectively. The merged images shown indicate the nuclear colocalization of ΔNp63 and p53 (yellow).
Deletion Analysis of p53/p63 Interaction.
To further examine the regions required for the p53 and p63 interaction, we generated truncated variants of p53WT and the p63 core domain (see Table 2, which is published as supplemental data on the PNAS web site, www.pnas.org). We selected three modular blocks from the p53 tumor suppressor (trans-activation and proline-rich domains, residues 1–101; DNA-binding domain, residues 102–293; oligomerization and C-terminal domains, residues 294–393). The regions for the p63 core domain were selected based on homology to p53 (residues 1–74, 75–266, 267–356). Truncated fragments of the p63 core domain or p53WT were fused to the Gal4-binding domain or Gal4-activation domain of the yeast plasmids. Using the two-hybrid assay, we found that one protein domain of p53 (residues 102–293) is sufficient to mediate the interaction with p63. Likewise, the putative DNA-binding domain of p63 (residues 75–266) is essential for the interaction with p53.
p53 Regulates p40 or ΔNp63α Stability and Targets It into a Protein Degradation Pathway.
To examine whether the physical association with p53 can affect p40/ΔNp63α levels or stability, SaOs2 cells infected with a constant amount of Ad-p40 (MOI = 1) alone or with various concentrations (MOI = 0 to 2) of Ad-p53WT, Ad-p53175, or empty adenovirus Ad5. Coexpression with p53WT, p53175 (Fig. 3A), or Ad5 (data not shown) did not affect the p40 mRNA level. However, the p40 protein levels were markedly reduced (by ≈10-fold) in parallel with an increase of Ad-p53WT (Fig. 3A), whereas no change of p40 was observed with the mutant Ad-p53175 (Fig. 3A) or Ad5 (data not shown) at similar MOI. To further examine this phenomenon, 022 cells (which express endogenous ΔNp63α) were infected with Ad-p53WT, Ad-p53175, or Ad5 (all at MOI = 1), and changes in ΔNp63α expression at 0, 4, 8, and 24 h after infection were measured by immunoblotting with corresponding antibodies. As expected, endogenous ΔNp63α protein levels were moderately diminished at 8 h (by ≈50%) and more dramatically at 24 h (by ≈90%) after coexpression with the Ad-p53WT. In contrast, expression of mutant Ad-p53175 (Fig. 3B) or Ad5 (data not shown) demonstrated no changes in ΔNp63α levels. Interestingly, other p53 mutants, including p53143 and p53273, that retain ΔNp63α-binding capacity also mediated degradation of endogenous ΔNp63α (Fig. 4A).
Figure 3.

p53 targets ΔNp63 (p40 or ΔNp63α) into a protein degradation pathway. (A) Down-regulation of the exogenous p40 protein is mediated by p53WT expression. SaOs2 cells were coinfected with Ad-p40 (MOI = 1) and either Ad-p53WT or Ad-p53175 (MOI = 0–2) for 14 h, and p40 expression was examined at the RNA (5 μg) and protein (20 μg) level (from total cell lysates). Decreasing amounts of p40 were seen with p53WT (lanes 2 and 3), but not mutant p53175 (lanes 5 and 6). (B) p53 coexpression mediates degradation of endogenous ΔNp63α protein. 022 cells were infected with Ad-p53WT (MOI = 1) or Ad-p53175 (MOI = 1). ΔNp63α protein levels were determined at 0, 4, 8, and 24 h after infection with antibody to ΔNp63. Decreasing amounts of p40 were seen with p53WT at 8 and 24 h but not with the p53175.
Figure 4.

p53 mutants that bind to ΔNp63α mediate its degradation. (A) p53143 or p53273 mediates degradation of ΔNp63α in transfected cells. 022 cells were transfected with either pCEP4-p53143 (1 μg, 5 μg) or pCEP4-p53273 (1 μg, 5 μg). Total lysates were resolved by SDS/PAGE and probed with antibodies to p53, ΔNp63, or β-actin. (B) The half-life of p40 in the presence of overexpressed p53WT or p53175. Cells infected with adenoviruses for 18 h were labeled with 200 μCi/ml [35S]methionine for 1 h. Labeled cells were then maintained in nonradioactive medium for 0 min, 60 min, 90 min, 120 min, 3 h, or 6 h before collecting (data for 3 and 6 h not shown). Total lysates were precipitated with antibody to ΔNp63 and resolved by SDS/PAGE followed by autoradiography. The immunoprecipitates were normalized by trichloroacetic acid-precipitable radioactive material, and equal amounts of total radioactive lysates (dpm/mg of total protein) were used for experiments. p53WT overexpression decreases the p40 half-life (by ≈80%).
To examine the timing of p63 protein degradation in more detail, we analyzed the half-life of ΔNp63 proteins in the absence and presence of p53 overexpression. SaOs2 cells infected with Ad-p40 alone or together with either Ad-p53WT or Ad-p53175 were grown in medium containing [35S]methionine for 1 h, and then maintained in a fresh methionine-free medium for 0 min, 60 min, 90 min, 120 min, 3 h, and 6 h. Total cell lysates were precipitated with antibody to ΔNp63 and resolved by SDS/PAGE. Our results led us to estimate a half-life for p40 expressed alone in SaOs2 cells (p53−/−) at greater than 2 h (data not shown). However, in the presence of overexpressed p53, the half-life of p40 was less than 45 min, whereas coexpression with Ad-p53175 had no effect on p40 half-life (Fig. 4B).
To elucidate the mechanism underlying p53-dependent p40/ΔNp63α degradation we studied the effect of the 26S-proteasome inhibitor MG-132 (Fig. 5). SaOs2 cells infected with Ad-p53WT and Ad-p40 were treated with MG-132 (20 μM) for 6 h before preparation of cell lysates. We found that the proteasome inhibitor MG-132 increased the p53 protein level (by ≈2-fold), as expected because it is degraded through a proteasome pathway (ref. 24; Fig. 5A). Interestingly, the proteasome inhibitor also stabilized the overall protein level of p40 (Fig. 5A, compare lanes in the absence or presence of MG-132). However, MG-132 had very little inhibitory effect on the p53-mediated degradation of the exogenous p40 protein (Fig. 5A). Similarly, the calpain inhibitor I (Ac-Leu-Leu-norleucinal) had no affect on the degradation of p40 (data not shown). 022 cells were infected with Ad-p53WT (MOI = 1) and then incubated with or without MG-132 under similar conditions. We again found that ectopic expression of p53 mediated degradation of endogenous ΔNp63α via a proteasome-independent pathway (Fig. 5B). Thus, it appears unlikely that a proteasome- or calpain-dependent pathway is involved in the ΔNp63 degradation process mediated by p53 overexpression.
Figure 5.

p53 mediates ΔNp63 (p40 or ΔNp63α) degradation through a caspase pathway. (A) The addition of MG-132 does not affect the p53WT-mediated p40 degradation, although overall levels of p53 and p40 are stabilized. SaOs2 cells were coinfected with a constant amount of Ad-p40 and various concentrations of Ad-p53WT for 14 h, followed by incubation without or with 20 μM MG-132 for 6–7 h. (B) MG-132 does not affect p53-mediated degradation of endogenous ΔNp63α protein in 022 cells. 022 cells were infected with Ad-p53WT (lane 1, MOI = 0; lane 2, MOI = 0.5; lane 3, MOI = 1) for 14 h, followed by incubation without or with 20 μM MG-132 for 6–7 h. (C) Z-VAD-fmk inhibits p53-mediated degradation of p40. SaOs2 cells were coinfected with Ad-p40 and Ad-p53WT (MOI = 0–1) for 14 h, followed by incubation without or with 10 μM Z-VAD-fmk for 6–7 h. Probes and antibodies used for detection are shown on the left. Under control conditions, p40 degradation continues, whereas it is inhibited by Z-VAD-fmk (p40 row). Levels of β-actin and poly(ADP-ribose) polymerase (PARP) were measured as internal controls.
Recently, p53 was shown to induce a caspase-like activity that degrades the Mdm2 protein, although cellular apoptotic changes were not detected (25). Similarly, our studies showed the presence of stable poly(ADP-ribose) polymerase, which indicates an absence of initiation of cellular apoptosis upon p53 overexpression (Fig. 5C). Of note, the infection with all adenoviruses had no effect on cell morphology within 24 h, whereas infected cells usually demonstrated apoptotic changes after 48–72 h (data not shown).
A p53-induced caspase was also shown to cleave the retinoblastoma protein (Rb) (26). Hence, to explore whether caspases are involved in the p40 degradation mediated by p53, we examined the effect of the caspase inhibitor Z-VAD-fmk on p40 degradation. SaOs2 cells infected with Ad-p53WT and Ad-p40 were treated with Z-VAD-fmk (10 μM) for 7 h before preparation of the cell lysates. We found that Z-VAD-fmk inhibited the degradation of p40 in the presence of p53WT expression (Fig. 5C). These data suggest that a caspase-like activity is apparently present in cell lysates expressing p53WT, and it might be involved in the ΔNp63 degradation. These studies further suggest that p53WT associates with ΔNp63 isotypes and also promotes their degradation through a caspase-mediated pathway.
Computer analysis of the p40 and ΔNp63α protein sequence revealed a potential cleavage site for caspase 1 (i.e., YVED, residues 182–185) predicted to yield fragments identical to those observed (Fig. 6A). To directly address this possibility, we incubated the 35S-labeled p40 protein with total lysates from SaOs2 cells infected with Ad-p53WT in the absence and presence of various substrate–inhibitors for caspases 1, 3, 6, or 8. We found that Ac-YVAD-CHO (an inhibitor of caspase 1) prevented the degradation of p40, whereas the other inhibitors had no effect (Fig. 6B).
Figure 6.

Caspase 1 degrades p40. (A) Schematic representation of caspase 1 cleavage site in p40. (B) p53-expressing lysates contain caspase 1, which cleaves p40 in vitro. Total lysates from SaOs2 cells infected with Ad-p53 were incubated with [35S]p40 or 35S-labeled mutated p40 (YVEA185) in the absence or presence of 5 μM each caspase substrate–inhibitors (for caspase 1, Ac-YVAD-CHO; caspase 3, Ac-DEVD-CHO; caspase 6, Ac-VEID-CHO; caspase 8, Ac-IETD-CHO). (C) Purified caspase 1 cleaves p40 in vitro. For protease cleavage, [35S]p40 or 35S-labeled mutated p40 (YVEA) polypeptides were incubated with purified caspases for 2 h at 37°C and analyzed by SDS/PAGE followed by autoradiography. p40 (residues 1–356, 44 kDa) is cleaved in half, generating two fragments (residues 1–184 and 185–356) with an apparent molecular mass of 20 kDa.
To further examine the ability of caspases to cleave p63, 35S-labeled p40 was incubated with purified preparations of various caspases (1, 3, 6, or 8) expressed in bacteria (Fig. 6C). We found that caspase 1 cleaved p40 into two fragments of similar mobility (Fig. 6C). Similarly, caspase 1 was shown to cleave ΔNp63α in vitro (data not shown). When we genetically mutated this putative cleavage site in p40 from YVED to YVEA, the total lysate from p53WT-expressing SaOs2 cells (Fig. 6B) or purified caspase 1 (Fig. 6C) lost the ability to cleave p40 in vitro.
Discussion
We have presented several lines of evidence indicating a physical and functional relationship between p53WT and ΔNp63. To our knowledge, this is the first unbiased two-hybrid screen that unequivocally identified p53 as a true protein-binding partner. The physical interaction of p40 (or ΔNp63α) with p53 in vivo supports an intimate association between these members of the p53 family. Although p40 (or ΔNp63α) binds strongly to p53WT, some mutants (p53175 and p53248) abolish this interaction in various experimental conditions.
By deletion analysis, we have further shown that this interaction is mediated by the p53 DNA-binding domain (residues 102–293) and the presumed DNA-binding domain of p40 (residues 75–266). This is an interesting observation, since p53 has been shown to form homotetramers or heterooligomers with p53 mutants only with the oligomerization domain (4, 5). However, recent reports pointed to a weak or absent association between p53 and its homologues by means of this domain in vitro (27). The ability to form heterodimers with p53WT was also shown for p73β, whereas p73α preferentially associated with p53175 (8, 28).
Our data support a novel type of physical association at the DNA-binding domains between p53 homologues. We demonstrated that at least two residues (R175 and R248) are involved in the interaction between p53 and ΔNp63 in the absence of DNA.
These results further support the concept that other members of the p53 family may form heterodimers in different combinations, which might affect p53 physiological activity and/or stability of p53 homologues. Depending on the individual components of the heterodimers, the protein complexes might activate certain downstream genes or lead to a decrease in p53 trans-activation activity. For example, ΔNp63 can actively repress p53-mediated trans-activation of a reporter gene when tethered to p53 during binding to target sequence (14). Moreover, we have observed that the p63 core domain negatively affects p53-induced expression of specific endogenous genes (data not shown). This effect can be explained by the interaction of ΔNp63 with p53 and/or by competitive binding of both transcription factors to the same promoter region of target genes.
We further report here that p53 overexpression promotes a p53-dependent degradation of ΔNp63 isotypes in a dose- and time-dependent manner. This striking effect was shown for p53WT but not for the p53175 mutant or empty adenovirus. The proteasome inhibitor MG-132 and the calpain inhibitor Ac-Leu-Leu-norleucinal had very little effect on the ΔNp63 degradation mediated by p53WT overexpression. However, the proteasome pathway might still be involved in degradation of endogenous p63 proteins, because MG-132 stabilizes overall levels of p63. In contrast, the general caspase inhibitor Z-VAD-fmk abolished the ΔNp63 degradation in the presence of p53WT, suggesting that ΔNp63 isotypes may be substrates for a p53WT-mediated caspase-like activity.
Although we failed to detect the p63 cleavage products in vivo, these products might be rapidly degrading through other proteolytic pathways. Moreover, our study with individual caspase substrate–inhibitors and purified caspases identifies caspase 1 as a candidate for cleavage of ΔNp63 in vitro. Recent reports suggest that caspase 1 (interleukin-1β-converting enzyme, ICE) plays an important role in various pathological cellular processes beyond its primary function in generating mature IL-1β or IL-18 (29–31). Similarly, a caspase-3-like activity was involved in the p53-mediated degradation of Mdm2 and Rb proteins (25, 26). Apparently, various caspase-like proteases are involved in the p53-mediated degradation of Mdm2, Rb, or p63, because these proteins contain different recognition sites and their cleavage is inhibited by various substrate–inhibitors.
The mechanism by which p53 activates caspase activity is not yet understood. The association of p53 with p63 may affect the three-dimensional configuration of p63 proteins, leading to exposure of the p63 caspase-sensitive cleavage site on the surface and its targeting for caspase-mediated degradation. A few reports suggest that p53 may activate caspase signaling through a Bax/cytochrome c-independent pathway, yet through a Fas/APO-dependent pathway (32, 33).
We have previously shown that ΔNp63 plays an oncogenic role in human cancers (13). p63 is frequently amplified and ΔNp63 is overexpressed in primary lung cancers and in HNSCC cell lines and squamous cell cancers. Moreover, p40 overexpression in Rat-1a cells leads to more rapid growth of transformed cells and tumor growth in athymic mice. In addition, proliferating human keratinocytes and various epithelial neoplastic cells and lesions predominantly express the ΔNp63 isotypes (34–36). Overall, these data suggest a critical role for p63 in control of cell proliferation during oncogenesis.
Others have shown that some p73 and p63 isotypes containing a putative trans-activation domain are capable of inducing apoptosis and, perhaps, tumor suppression (8, 14). However, these isotypes were shown to have quite distinct effects on various p53 cis-elements (37), and inactivating mutations were not identified in human tumors (38). Moreover, ΔN isotypes can act as dominant-negative mutants to diminish transactivation by p53, suggesting that ΔN isotypes may behave as oncoproteins or anti-apoptotic factors (14, 19). Recently, ΔNp73 was reported to play an anti-apoptotic role during developmental neuronal death or p53-mediated apoptosis in vitro (19), whereas ΔNp63 was shown to induce a tumorigenic phenotype (13). The specific association between the p53 tumor suppressor and the putative ΔNp63 oncoprotein, and subsequent degradation of p63 isotypes, implies that regulation of cell growth may depend on a balance between tumor suppressor and oncogenic members of the p53 family.
The physiologic response of the p63 isotypes or p53/p63 binding to outside stimuli remains to be fully explored. p53, p73, and TAp63γ levels and stability are elevated in response to DNA damage induced by various stimuli (2, 22). However, ΔNp63 levels were suppressed by UV irradiation of cultured keratinocytes, and transgenic overexpression of ΔNp63α inhibited UV-induced apoptosis (39). Furthermore, a negative correlation between overexpression of p53 and suppression of ΔNp63 levels has been shown, but no mechanism was postulated for this phenomenon (39). Our work demonstrating a role for p53 in ΔNp63 degradation now sheds light on the molecular mechanisms underlying these observations.
Recent studies with p63-null mice suggested that this gene plays a critical role in regulating epidermal stem cell renewal (9, 10). Moreover, irreversible growth arrest and differentiation of keratinocytes is associated with the disappearance of ΔNp63 isotypes (35, 36). While p63 is critical for epithelial proliferation (10), p53 is implicated in the control of differentiation of several cell types (40). Although overexpression of p53 results in a differentiated phenotype, dominant-negative variants of p53 inhibit differentiation (40). p53 levels are also down-regulated during embryogenesis, while proliferative processes are taking place (40). These findings suggest a cell- and stage-specific role for p53 in differentiation, implicating p53 as a regulator (perhaps through p63) in the early differentiation process, controlling a balance between differentiated cells and cells undergoing proliferation.
This emerging model supports the presence of transcriptional cross-talk between p53 and p63, suggesting that p63 is functioning as a negative regulator of some p53-responsive genes, while p53 plays an important role in control of p63 stability. This critical interaction may balance the oncogenic and growth-stimulating activity of p63 in tumorigenesis with its ability to induce epithelial proliferation during development.
Supplementary Material
Acknowledgments
We thank Dr. Bert Vogelstein for various plasmids and critical discussions. We are also indebted to Dr. David Hill for generating the antibody to ΔNp63 (Oncogene Research, Boston, MA). We thank Dr. L. Lee for CUSP (ΔNp63α) cDNA. This work was supported in part by National Cancer Institute Lung Cancer Specialized Program of Research Excellence Grant CA-58184–01 (D.S.), Grant R01-DE-012588-01 from the National Institute of Dental and Craniofacial Research (D.S.), and Grant R01-AI-47224-01 from the National Institute of Allergy and Infectious Diseases (E.A.R.)
Abbreviations
- TA
trans-activation
- MOI
multiplicity of infection
- Z
benzyloxycarbonyl
- fmk
fluoromethyl ketone
- CHO
aldehyde analogue
References
- 1.Hollstein M, Sidransky D, Vogelstein B, Harris C. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 2.Polyak K, Xia Y, Zweier J, Kinzler K, Vogelstein B. Nature (London) 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 3.Kern S, Pietenpol J, Thiagalingam S, Seymour A, Kinzler K, Vogelstein B. Science. 1992;256:827–830. doi: 10.1126/science.1589764. [DOI] [PubMed] [Google Scholar]
- 4.Milner J, Medcalf E. Cell. 1991;65:765–774. doi: 10.1016/0092-8674(91)90384-b. [DOI] [PubMed] [Google Scholar]
- 5.McLure K, Lee P. EMBO J. 1999;18:763–770. doi: 10.1093/emboj/18.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gualberto A, Aldape K, Kozakiewicz K, Tisty T. Proc Natl Acad Sci USA. 1998;95:5166–5171. doi: 10.1073/pnas.95.9.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaelin W G., Jr Oncogene. 1999;18:7701–7705. doi: 10.1038/sj.onc.1202955. [DOI] [PubMed] [Google Scholar]
- 8.Kaghad M, Bonnet H, Yang A, Creancier L, Biscan J, Valent A, Minty A, Chalon P, Lelias J, Dumont X, et al. Cell. 1997;90:809–881. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 9.Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson R, Tabin C, Sharpe A, Caput D, Crum C, McKeon F. Nature (London) 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 10.Mills A, Zheng B, Wang Z, Vogel H, Roop D, Bradley A. Nature (London) 1999;398:708–714. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 11.Celli J, Dujif P, Hamel B, Bamshad M, Kramer B, Smits A, Newbury-Ecob R, Hennekam R, Van Buggenhout G, Van Haeringen A, et al. Cell. 1999;99:143–153. doi: 10.1016/s0092-8674(00)81646-3. [DOI] [PubMed] [Google Scholar]
- 12.Lee L, Walsh P, Prater C, Su L, Marchbank A, Egbert T, Dellavalle R, Targoff I, Kaufman K, Chorzelski T, Jablonska S. J Invest Dermatol. 1999;113:146–151. doi: 10.1046/j.1523-1747.1999.00651.x. [DOI] [PubMed] [Google Scholar]
- 13.Hibi K, Trink B, Patturajan M, Westra W H, Caballero O L, Hill D E, Ratovitski E A, Jen J, Sidransky D. Proc Natl Acad Sci USA. 2000;97:5462–5467. doi: 10.1073/pnas.97.10.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang A, Kaghad M, Wang Y, Gillett E, Fleming M, Dotsch V, Andrews N, Caput D, McKeon F. Mol Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 15.Chi S, Ayed A, Arrowsmith C. EMBO J. 1999;18:4438–4445. doi: 10.1093/emboj/18.16.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osada M, Ohba M, Kawahara C, Ishioka C, Kamamaru R, Katoh I, Ikawa Y, Nimura Y, Nakagawara A, Obinata M, Ikawa S. Nat Med. 1998;4:839–843. doi: 10.1038/nm0798-839. [DOI] [PubMed] [Google Scholar]
- 17.Senoo M, Seki N, Ohira M, Sugano S, Watanabe M, Inuzuka S, Okamoto T, Tachibana M, Tanaka T, Shinkai Y, Kato H. Biochem Biophys Res Commun. 1998;250:536–541. doi: 10.1006/bbrc.1998.9013. [DOI] [PubMed] [Google Scholar]
- 18.Trink B, Okami K, Wu L, Sriuranpong V, Jen J, Sidransky D. Nat Med. 1998;4:747–748. doi: 10.1038/nm0798-747. [DOI] [PubMed] [Google Scholar]
- 19.Pozniak C, Radinovic S, Yang A, McKeon F, Kaplan D, Miller F. Science. 2000;289:304–306. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- 20.Ratovitski E, Alam M, Quick R, McMillan A, Bao C, Kozlovsky C, Hand T, Johnson R, Mains R, Eipper B, Lowenstein C. J Biol Chem. 1999;274:993–999. doi: 10.1074/jbc.274.2.993. [DOI] [PubMed] [Google Scholar]
- 21.Ausubel F, Brent R, Kingston R, Moore D, Sidman J, Smith J, Struhl K. Short Protocols in Molecular Biology. 4th Ed. New York: Wiley; 1997. [Google Scholar]
- 22.Gong J G, Costanzo A, Yang H Q, Melino G, Kaelin W G, Jr, Levrero M, Wang J Y. Nature (London) 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 23.Casciola-Rosen L, Andrade F, Ulanet D, Wong W, Rosen A. J Exp Med. 1999;190:815–826. doi: 10.1084/jem.190.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haupt Y, Maya R, Kazaz A, Oren M. Nature (London) 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 25.Pochampally R, Fodera B, Chen L, Lu W, Chen J. J Biol Chem. 1999;274:15271–15277. doi: 10.1074/jbc.274.21.15271. [DOI] [PubMed] [Google Scholar]
- 26.Gottlieb E, Oren M. EMBO J. 1998;17:3587–3596. doi: 10.1093/emboj/17.13.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davison T, Vagner C, Kaghad M, Ayed A, Caput D, Arrowsmith C. J Biol Chem. 1999;274:18709–18714. doi: 10.1074/jbc.274.26.18709. [DOI] [PubMed] [Google Scholar]
- 28.Di Como C, Gaiddon C, Prives C. Mol Cell Biol. 1999;19:1438–1449. doi: 10.1128/mcb.19.2.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dinarello C. Ann NY Acad Sci. 1998;856:1–11. doi: 10.1111/j.1749-6632.1998.tb08307.x. [DOI] [PubMed] [Google Scholar]
- 30.Li M, Ona V, Guegan C, Chen M, Jackson-Lewis V, Andrews L, Olszewski A, Stieg P, Lee J, Przedborski S, Friedlander R. Science. 2000;288:335–339. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- 31.Mao P, Jiang Y, Wee B, Porter A. J Biol Chem. 1998;273:23621–23624. doi: 10.1074/jbc.273.37.23621. [DOI] [PubMed] [Google Scholar]
- 32.Ding H, McGill G, Rowan S, Schmaltz C, Shimamura A, Fischer D. J Biol Chem. 1998;273:28378–28383. doi: 10.1074/jbc.273.43.28378. [DOI] [PubMed] [Google Scholar]
- 33.Kim A, Raffo A, Brandt-Rauf P, Pincus M, Monaco B, Abarzua P, Fine R. J Biol Chem. 1999;274:34924–34931. doi: 10.1074/jbc.274.49.34924. [DOI] [PubMed] [Google Scholar]
- 34.Park B, Lee S, Kim J, Lee S, Lee C, Chang S, Park J, Chi S. Cancer Res. 2000;60:3370–3374. [PubMed] [Google Scholar]
- 35.Parsa R, Yang A, McKeon F, Green H. J Invest Dermatol. 1999;113:1099–1105. doi: 10.1046/j.1523-1747.1999.00780.x. [DOI] [PubMed] [Google Scholar]
- 36.Nylander K, Coates P, Hall P. Int J Cancer. 2000;87:368–372. [PubMed] [Google Scholar]
- 37.Shimada A, Kato S, Enjo K, Osada M, Ikawa Y, Kohno K, Obionata M, Kanamaru K, Ikawa S, Ishioka C. Cancer Res. 1999;59:2781–2786. [PubMed] [Google Scholar]
- 38.Hagiwara K, McMenamin M, Miura K, Harris C C. Cancer Res. 1999;59:4165–4169. [PubMed] [Google Scholar]
- 39.Liefer K, Koster M, Wang X, Yang A, McKeon F, Roop D. Cancer Res. 2000;60:4016–4020. [PubMed] [Google Scholar]
- 40.Louis J, McFarland V, May P, Mora P. Biochim Biophys Acta. 1988;950:395–402. doi: 10.1016/0167-4781(88)90136-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.