

Abstract
Mammalian colon harbors trillions of bacteria, yet there is no undue inflammatory response by the host against these bacteria under normal conditions. The bacterial fermentation products acetate, propionate, and butyrate are believed, at least in part, to be responsible for these immunosuppressive effects. Dendritic cells play an essential role in presentation of antigens to T lymphocytes and initiation of adaptive immune responses. Here we report that butyrate and propionate block the generation of dendritic cells from bone marrow stem cells, without affecting the generation of granulocytes. This effect is dependent on the Na+-coupled monocarboxylate transporter Slc5a8, which transports butyrate and propionate into cells, and on the ability of these two bacterial metabolites to inhibit histone deacetylases. Acetate, which is also a substrate for Slc5a8 but not an inhibitor of histone deacetylases, does not affect dendritic cell development, indicating the essential role of histone deacetylase inhibition in the process. The blockade of dendritic cell development by butyrate and propionate is associated with decreased expression of the transcription factors PU.1 and RelB. Butyrate also elicits its biologic effects through its ability to activate the G-protein-coupled receptor Gpr109a, but this mechanism is not involved in butyrate-induced blockade of dendritic cell development. The participation of Slc5a8 and the non-involvement of Gpr109a in butyrate effects have been substantiated using bone marrow cells obtained from Slc5a8−/− and Gpr109a−/− mice. These findings uncover an important mechanism underlying the anti-inflammatory functions of the bacterial fermentation products butyrate and propionate.
Keywords: Bacteria, Bone Marrow, Colon Cancer, Histone Deacetylase, Immunology, Immunosupressor, Inflammation, Slc5a8, Butyrate, Propionate
Introduction
Dendritic cells (DCs)2 function as sentinels in peripheral tissues and lymphoid organs guarding against pathogens (1–3). When encountered with pathogens, immature DCs differentiate into antigen-presenting cells with the up-regulation of major histocompatibility complex class II and costimulatory signals such as B7 and IL-12. Mature DCs potently stimulate naive T cells to initiate T cell responses (2, 3). Targeted depletion of DCs results in abrogation of T cell responses against pathogens (4). Because of the essential role of DCs in initiating adaptive immune responses, microorganisms have evolved various mechanisms to suppress or block DC maturation and function (5–7).
Normally, microorganism-induced immunosuppression is associated with harmful effects in host. In certain cases however, the same process may have beneficial effects on host. Mammalian colon harbors trillions of bacteria without eliciting significant immune activation. There is a mutual beneficial relationship between the gut microbiota and the host (8, 9). The bacterial fermentation products acetate, propionate, and butyrate (collectively called short-chain fatty acids) are believed to be the mediators of the beneficial effects of gut bacteria on the host (10, 11). Among these short-chain fatty acids, butyrate has received the most attention for its biologic effects. The beneficial effects of butyrate on colon range from being a nutrient for colonocytes to being an anti-inflammatory and antitumor agent (12, 13). Although the anti-inflammatory function of butyrate is well known at the phenomenological level, the molecular mechanisms underlying the process are not fully understood.
Butyrate is an inhibitor of histone deacetylases (HDACs) (12, 13). The Na+-coupled monocarboxylate transporter SLC5A8 (human ortholog is in uppercase letters, and rodent ortholog is in lowercase letters) is necessary for the entry of butyrate into colon cells for subsequent inhibition of HDACs (14, 15). Recently we have shown that propionate, another short-chain fatty acid produced in the colon by bacterial fermentation, is also an inhibitor of HDACs (16). SLC5A8 has been shown to function as a tumor suppressor in the colon (17) and other tissues (13, 18, 19); the ability of this transporter to mediate the Na+-coupled entry of HDAC inhibitors (butyrate, propionate, and pyruvate) into cells underlies the tumor-suppressive function of this transporter (15, 16). While SLC5A8 is necessary for the intracellular actions of butyrate, butyrate also elicits biologic effects on colon cells extracellularly by serving as an agonist for the cell surface G-protein-coupled receptor GPR109A, but without involving HDAC inhibition (20). GPR109A was originally described as the receptor for nicotinate, and the activation of the receptor by nicotinate provided the molecular basis for the anti-lipolytic actions of this vitamin (21, 22). Subsequent studies have shown that the ketone body β-hydroxybutyrate is the physiologic ligand for the receptor (23). Butyrate is also able to activate the receptor but with a low affinity; millimolar concentrations of butyrate are needed to activate the receptor (20, 23). This indicates that butyrate might serve as a ligand for GPR109A in the colon under physiologic conditions because of the presence of high levels of this fatty acid in the colonic lumen (∼20 mm) due to bacterial fermentation of dietary fiber. We have shown that GPR109A is indeed expressed in the colon and that the expression is restricted to the lumen-facing apical membrane of the colonic epithelial cells where the receptor has direct access to luminal butyrate (20). The expression of the receptor is silenced in colon cancer and in colon cancer cell lines; re-expression of the receptor in colon cancer cell lines leads to cell death in the presence of the receptor ligands nicotinate and butyrate (20). Thus, butyrate functions as a tumor-suppressive agent by two independent mechanisms, first via SLC5A8-mediated entry into colon cells with subsequent inhibition of HDACs and second via activation of GPR109A. This provides a molecular basis for the tumor-suppressive functions of SLC5A8 and GPR109A in the colon.
Chronic inflammation in the colon increases the risk for colon cancer. The incidence of colon cancer is much higher in patients with ulcerative colitis than in control subjects (24, 25). Butyrate has been shown to suppress inflammation in colonic epithelial cells (20, 26), indicating that the immunosuppressive functions of this bacterial fermentation product may also play a role in the ability of this short-chain fatty acid to protect against colon cancer. DCs constitute an important component of mucosal immunity. In the present study, we explored the influence of butyrate and propionate, both being products of bacterial metabolism in colon, on DC development from mouse bone marrow precursor cells and the involvement of Slc5a8/HDAC inhibition and Gpr109a in the process. These studies have uncovered a novel mechanism underlying the anti-inflammatory functions of these bacterial fermentation products.
EXPERIMENTAL PROCEDURES
Animals
C57BL/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME). Slc5a8−/− mice and Gpr109a−/− mice have been described previously (22, 27). Use of animals in these studies adhered to the “Principles of Laboratory Animal Care” (NIH publication no. 85-23, revised in 1985) and was approved by the Institutional Committee for Animal Use in Research and Education.
Bone Marrow Cultures
Bone marrow cells were flushed out from femur and tibia with phosphate-buffered saline, and single cell suspensions were prepared. Red blood cells were lysed with ACK lysis buffer and Lineage negative (Lin−) cells were prepared by immunopanning to deplete CD4-positive cells (anti-CD4 antibody, clone GK1.5), CD8-positive cells (anti-CD8 antibody, clone TIB105), B220-positive cells (anti-B220 antibody, clone RA3–6B2), CD11c-positive cells (anti-CD11c antibody, clone HL3), and CD11b-positive cells (anti-CD11b antibody, clone M1/70). Lin− bone marrow cells were cultured (106 cells/35-mm dish) in the presence of 25 ng/ml of GM-CSF in RPMI 1640 medium, containing 10% fetal calf serum, 10 mm Hepes (pH 7.4), 2 mm glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 50 μm 2-mercaptoethanol. Acetate, propionate, butyrate, and nicotinate were added at a concentration of 0.5 mm, unless indicated. The pan-HDAC inhibitor N1-hydroxy-N8-phenyloctanediamide (SAHA) was used at a concentration of 1 μm. On days 4 and 6 following the initial seeding, non-adherent cells were harvested and analyzed for granulocytes and DCs. On day 4, following the removal of non-adherent cells, cultures were replenished with fresh medium containing GM-CSF and short-chain fatty acids.
Antibodies and Flow Cytometry
Cells were stained with antibodies (BD Biosciences, San Diego, CA) against Gr-1-biotin (clone RB6–8C5), CD11c-phycoerythrine (PE) (clone HL-3), and streptavidin-allophyocyanin. Stained cells were acquired and analyzed by FACS Caliber and FACS Canto (Becton Dickinson).
Western Blotting
Cells were lysed in Laemmli buffer, boiled, and frozen at −70 °C until analysis. Lysates were loaded in each well of SDS-PAGE (5–16%), followed by electrophoresis and blotting onto PVDF membrane. Membranes were probed with antibodies specific for acetyl-Lys12-histone H4, and histone H4 (Sigma), followed by appropriate horseradish peroxidase-conjugated secondary antibodies (Cell Signal, Danvers, MA). The signals were detected by enhanced chemiluminescence (GE Health Care).
Measurement of HDAC Activity
This was done using a commercially available kit (BioVision, Inc., Mountain View, CA) (28, 29). Lysates from SW480 cells (a human colon cancer cell line) were used as the source of HDACs. The activity was monitored in the presence of increasing concentrations of short-chain fatty acids.
Mouse Genotyping
Mice with different genotypes were identified by PCR. In the case of Slc5a8, the primers for wild-type genotype were 5′-AAGTATACTCTGAACACATTTCAGG-3′ and 5′-TGTCACTATATGCAGAGGATACACG-3′, and the primers for knock-out genotype were 5′-AAGTATACTCTGAACACATTTCAGG-3′ and 5′-ATCTTTCCACTTCTTAGAAAGCTGG-3′. In the case of Gpr109a, the primers for wild type genotype were 5′-TCAGATCTGACTCGTCCACC-3′ and 5′-CCATTGCCCAGGAGTCCGAAC-3′, and the primers for knock-out genotype were 5′-TCAGATCTGACTCGTCCACC-3′ and 5′-CCTCTTCGCTATTACGCCAGC-3′.
RT-PCR
The expression of Slc5a8 and Gpr109a in bone marrow cells from wild type, Slc5a8−/−, and Gpr109a−/− mice was analyzed by RT-PCR. The PCR primers for Slc5a8 were 5′-TTATGGGCGGTCGCAGTA-3′ and 5′-CAGAGGCCCACAAGGTTGACAT-3′. The primers for Gpr109a were 5′-GCCTTGAGCCTTCGCTAGGT-3′ and 5′-AGTTCCAAGAAAGCGTAAGCCA-3′.
Bioluminescence Resonance Energy Transfer (BRET) Assay
This assay monitors the dissociation of G protein heterotrimers upon activation of GPR109A with a ligand (30). HEK293 cells were transfected overnight with human GPR109A cDNA along with constructs coding for Gαi1, venus-Gβ1, venus-Gγ2, and GRK3-luciferase. Cells were then harvested in phosphate-buffered saline containing 5 mm EDTA. Cell suspension was transferred to a 96-well plate (100 μl/well), and exposed to short-chain fatty acids at different concentrations. The luciferase substrate benzyl colenterazine (5 μm) was then added to the wells in dark. Steady-state BRET measurements were made within 15 min of exposure of the cell suspensions to short-chain fatty acids using a photon-counting multimode plate reader. The BRET signal was calculated as the ratio of the emission intensity at 520–545 nm to the emission intensity at 475–495 nm.
Chromatin Immunoprecipitation (ChIP)
This was done using a kit from Millipore Corp. (Billerica) and an antibody specific for acetylated histone H4. Lin− bone marrow cells were cultured (106 cells/35-mm dish) in the presence of 25 ng/ml of GM-CSF in RPMI 1640 medium, containing 10% fetal calf serum, 10 mm Hepes (pH 7.4), 2 mm glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 50 μm 2-mercaptoethanol. Acetate, propionate, and butyrate were added at a concentration of 0.5 mm. Following 4 days in culture, the cells were harvested and subjected to ChIP assay. The genomic DNA associated with the immunoprecipitates was used for PCR with primers specific for the promoters of two transcription factors, namely PU.1 and RelB, which are known to regulate DC differentiation. Three different primer sets specific for three distinct regions of the PU.1 promoter (P1, P2, and P3) and two different primer sets specific for two distinct regions of the RelB promoter (R1 and R2) were used in the assay. The sequences of the primers are as follows: 5′-CAGCTGTGGTACTTAGCTGG-3′ and 5′-CTTGCTGAAGTAGTCCACTG-3′ (P1), 5′-GCAGGGCCTACAGGAAGAGC-3′ and 5′-AGGAAGTCTCTGGCCGGCTG-3′ (P2), 5′-CCAGTTTCCTCTGGGCAG-3′ and 5′-GGTGGGGATCAAGGCGGATC-3′ (P3), 5′-GCCTGGAATTGCATAATTGTCCAGGAA-3′ and 5′-CCCTTGTCTCAACGCAAACCAAC-3′ (R1), and 5′-CCTGCCTCTGTCTGTCCAGAATGG-3′ and 5′-AGGACCAGAAACAGGGATGTGGCT-3′ (R2).
Statistics
Experiments were repeated 2–4 times, and the data are presented as means ± S.D. Statistical significance was determined by Student's t test.
RESULTS AND DISCUSSION
Blockade of GM-CSF-induced Development of DCs from Bone Marrow Precursor Cells by Butyrate
GM-CSF induces the development of granulocytes at early stage (on days 2–4) and relatively higher proportion of functionally immature DCs at a later stage (on day 6) from bone marrow stem cells (31). To test the effect of butyrate on GM-CSF-induced development of DCs, we cultured Lin− bone marrow stem cells with GM-CSF in the presence or absence of butyrate. 4 and 6 days later, cultures were analyzed for DCs and granulocytes using specific markers CD11c (DCs) and Gr-1 (granulocytes). Presence of butyrate (0.5 mm) drastically reduced the GM-CSF-induced development of DCs (CD11c+Gr-1−) from 20% to 1% on day 4, and from 65% to 4% on day 6 (Fig. 1, A and B). Butyrate did not affect the development of granulocytes (Gr-1+CD11c−). The blockade of DCs differentiation by butyrate was dose-dependent, with 50% of the maximum inhibition (EC50) observed at 30 ± 8 μm (Fig. 1C). Contrary to its effect on GM-CSF-induced development of DCs, butyrate did not affect the LPS-induced up-regulation of CD86 and I-Ad in functionally immature DCs (Fig. 2), suggesting that the effects of butyrate are restricted to the development of DCs.
FIGURE 1.

Blockade of GM-CSF induced DC development from bone marrow precursors by butyrate. Lin− bone marrow cells were cultured with GM-CSF (25 ng/ml) in the presence or absence of butyrate (0.5 mm). On days 4 and 6, cultures were harvested and analyzed for CD11c and Gr-1 expression by flow cytometry. A, expression of CD11c and Gr-1 on cells with and without butyrate. B, quantification of butyrate effect on the development of granulocytes and DCs. C, dose-dependent effect of butyrate on the development of granulocytes and DCs.
FIGURE 2.

Lack of effect of butyrate on functional maturation of DCs. DCs (CD11c+Gr-1− cells), obtained on day 6 from bone marrow cell cultures in the presence of GM-CSF were cultured with or without LPS (lipopolysaccharide) (100 ng/ml) and in the presence or absence of butyrate (0.5 mm). Twenty-four hours later, cells were harvested and analyzed for CD86 (B7.2) and CD11c (A) or for I-Ad (MHC-class II) and CD11c (B). The numbers represent the percentage of positive cells in the corresponding quadrants.
Differential Effects of Short-chain Fatty Acids on DC Development
We examined the effects of acetate and propionate, the other two bacterial fermentation products, on GM-CSF-induced development of DCs to determine if these short-chain fatty acids also inhibit DC development. At a concentration of 0.5 mm, propionate blocked DC development whereas acetate did not (Fig. 3, A and B). In addition to the bacterial fermentation products, SLC5A8 also transports nicotinate, another monocarboxylate (32, 33). But, this compound did not block the development of the DCs (Fig. 3, A and B).
FIGURE 3.

Differential effects of monocarboxylates on DC development. A, Lin− bone marrow cells were cultured in the presence of GM-CSF (25 ng/ml) with and without different monocarboxylates (0.5 mm). After 6 days of culture, cells were analyzed for expression of CD11c and Gr-1 by flow cytometry. B, quantification of the effects of various monocarboxylates on the development of granulocytes and DCs.
The differential effects of these monocarboxylates on the development of DCs from mouse bone marrow precursor cells cannot be explained at the level of their interaction with Slc5a8 because all four monocarboxylates examined are substrates for the transporter. There are two potential mechanisms by which butyrate might block DC development, one by its ability to inhibit HDACs following its entry into cells via Slc5a8 and the other by its ability to activate the cell surface G-protein-coupled receptor Gpr109a. To differentiate between these two possible mechanisms, we examined the effects of acetate, propionate, butyrate, and nicotinate on HDAC activity using cell lysates from SW480 cells as the source of HDAC activity. These studies showed that acetate and nicotinate did not inhibit HDACs whereas propionate and butyrate did (Fig. 4A). These differential effects on HDAC activity mirrored the differential effects on DC development, suggesting that HDAC inhibition might be involved in the blockade of DC development by butyrate and propionate. We also examined the abilities of these monocarboxylates to serve as agonists for GPR109A. These studies showed that only butyrate and nicotinate served as agonists for human GPR109A whereas acetate and propionate did not (Fig. 4B). The results of HDAC inhibition and GPR109A activation along with the substrate specificity of SLC5A8 for the four monocarboxylates examined in the study are summarized in Fig. 4C. By comparing these data with the differential effects of these monocarboxylates on DC development, we conclude that inhibition of HDACs is the most likely mechanism underlying the blockade of DC development by butyrate and propionate.
FIGURE 4.

Interaction of monocarboxylates with SLC5A8 and GPR109A. A, selectivity of monocarboxylates as inhibitors of HDAC. SW480 cell lysates were used as the source of HDAC activity. The dose-dependent effects of acetate, propionate, butyrate, and nicotinate on HDAC activity were monitored in a cell-free system using a commercially available kit (BioVision). B, selectivity of monocarboxylates as agonists for human GPR109A. The agonist selectivity of acetate, propionate, butyrate, and nicotinate on GPR109A was monitored using the BRET assay. The concentration of monocarboxylates in the assay was 5 mm. C, differential features of monocarboxylates in terms of selectivity as substrates of human SLC5A8 and agonists for human GPR109A and ability to inhibit HDACs.
Involvement of HDAC Inhibition and Subsequent Suppression of PU.1 and RelB in the Blockade of DC Development
To confirm the involvement of HDAC inhibition in the blockade of DC development by butyrate, Lin− bone marrow cells were treated with GM-CSF in the presence or absence of butyrate and propionate, which are HDAC inhibitors, and acetate, which has no HDAC inhibition activity, for 4 h; cell lysates were then analyzed for acetylation status of histone H4. These studies were done with bone marrow cells cultured for 4 days in the presence of GM-CSF. Treatment with butyrate and propionate increased the acetylation status of histone H4, indicating inhibition of HDACs (Fig. 5A). On the other hand, treatment with acetate did not affect the acetylation status of H4. We then examined the effects of a structurally unrelated pan-HDAC inhibitor (SAHA) on the histone acetylation status as well as on DC development. Treatment with SAHA (1 μm) increased the acetylation of histone H4 (Fig. 5A). This was accompanied with a marked reduction (∼90%) in DC development without any significant effect on the development of granulocytes (Fig. 5B). These results provide convincing evidence for the involvement of HDAC inhibition in butyrate/propionate-induced blockade of DC development.
FIGURE 5.

Role of HDACs in DC development. A, Lin− bone marrow cells were cultured for 4 days in the presence or absence of GM-CSF (25 ng/ml), and then exposed for 4 h to acetate (0.5 mm), propionate (0.5 mm), butyrate (0.5 mm), or SAHA (1 μm). Cell lysates were analyzed for total and acetylated form of Lys12-histone H4. B, Lin− bone marrow cells were cultured for 6 days with GM-CSF (25 ng/ml) in the presence or absence of SAHA (1 μm) and then analyzed by flow cytometry for dendritic cells and granulocytes.
PU.1 and RelB are the two important transcription factors that regulate DC development, and HDAC activity is required for their expression (34–37). Therefore, we examined the effects of various monocarboxylates on PU.1 and RelB expression in developing DCs (Fig. 6A). Real-time RT-PCR for the expression levels of PU.1 mRNA and RelB mRNA showed that the HDAC inhibitors propionate (0.5 mm) and butyrate (0.5 mm), which are also substrates for SLC5A8, suppressed PU.1 and RelB expression whereas acetate (0.5 mm), which does not inhibit HDACs but a substrate for SLC5A8, did not have any effect. We then used ChIP assay to determine if histone H4 that is bound to PU.1 and RelB promoters is differentially acetylated following treatment with butyrate and propionate. The locations of the promoter regions examined by PCR are shown in Fig. 5B. The ChIP assay showed that acetylated H4 binds to PU.1 promoter, confirmed by three different primer sets selected in the promoter (Fig. 5C). The differential effects of propionate/butyrate versus acetate in the ChIP assay were most evident for the primer set specific for the PU.1 promoter region P1. The effects were relatively less robust for the regions P2 and P3. In the case of the RelB promoter, treatment of bone marrow cells with the HDAC inhibitors propionate and butyrate resulted in the loss of the promoter in the acetylated histone H4 immunoprecipitates (Fig. 5C). The effect was more robust for the promoter region R1 than for the promoter region R2. These data indicate that butyrate and propionate, through their ability to inhibit HDACs, suppress PU.1 and RelB expression and thus block DC development.
FIGURE 6.

Suppression of PU. 1 and RelB expression in bone marrow cells by butyrate and propionate. A, bone marrow cells were cultured with GM-CSF (25 ng/ml) for 4 days and then treated with or without monocarboxylates (0.5 mm) for 6 h. Expression levels of PU.1 mRNA and RelB mRNA were assessed by real-time RT-PCR. Glyceraldehyde-3-phosphate dehydrogenase expression was used as the internal control. B, specific regions of the PU.1 and RelB promoters that are monitored by ChIP assay with primer sets P1, P2, and P3 (PU.1) and R1 and R2 (RelB). C, day 4 GM-CSF bone marrow cultures were treated with or without acetate (0.5 mm), propionate (0.5 mm), or butyrate (0.5 mm) for 6 h and then used for ChIP analysis with an antibody against acetylated histone H4 and different primer sets specific for PU.1 and RelB promoters.
Butyrate and propionate inhibit two specific isoforms of HDAC, namely HDAC1 and HDAC3 (28). HDACs associate with the transcription complexes of specific promoter targets and influence the acetylation status of histones. HDACs also modulate the recruitment of repressors/co-repressors to the transcription complexes (38, 39). The acetylation status of histones determines the accessibility of the promoters to transcription complexes. Normally, acetylation of histones is associated with induction of gene expression. However, HDAC inhibitors have been shown to either induce or suppress the expression of specific genes (40). Traditionally, HDACs have been considered as the transcriptional co-repressors, meaning that inhibition of HDACs results in increased expression of genes. However, recent studies showing that the expression of several genes is suppressed as a consequence of HDAC inhibition have brought a paradigm shift in the control of transcription by HDACs (41). HDACs can also function as co-activators. Present studies show that inhibition of HDACs with butyrate and propionate in developing DCs leads to suppression of PU.1 and RelB, two of the transcription factors necessary for DC development. Interestingly, the mechanisms linking HDAC inhibitors to suppression of PU.1 and RelB expression in developing DCs seem distinct. Previous studies with the nonspecific HDAC inhibitor trichostatin A have shown that HDAC activity is necessary for the expression of PU.1 (35). Inhibition of HDACs leads to increased acetylation of histone H4 on PU.1 promoter and consequently loss of RNA polymerase II from the gene (35). Our present studies with butyrate and propionate, specific inhibitors of HDAC1 and HDAC3, also show increased acetylation of histone H4 on PU.1 promoter with a consequent decrease in gene expression, suggesting that a similar mechanism might be involved in the suppression of PU.1 expression by these bacterial fermentation products. In contrast, inhibition of HDACs with butyrate and propionate causes the loss of the RelB promoter in the acetylated histone H4 immunoprecipitate, indicating that treatment with these HDAC inhibitors leads to decreased acetylation of histone H4 on RelB promoter. Previous studies have shown that several genes are silenced by butyrate via decreased histone acetylation around the transcription start sites of these genes (42). In these cases, HDAC inhibitors seem to target non-histone proteins rather than the histones associated with the genes. An example in this group of genes is SRC (42, 43). The activity of SRC promoter is dependent on TAF1, a subunit of the basal transcription factor TFIID, and TAF1 plays a critical role in the suppression of SRC expression by HDAC inhibitors (44). This factor possesses histone acetylase activity, which is negatively regulated by acetylation. TAF1 may be the non-histone protein that is a direct target for HDACs, explaining why HDAC inhibitors decrease the acetylation of histones associated with the SRC promoter. A similar mechanism might be involved in the decreased acetylation of histone H4 associated with the RelB promoter and consequent suppression of its expression.
Relevance of Slc5a8 to Butyrate-induced Blockade of DC Development
Slc5a8, a Na+-coupled high-affinity transporter, is essential for the entry of butyrate into cells, particularly at low concentrations, to inhibit HDACs (14, 15). To establish the role of Slc5a8 in butyrate-induced blockade of DC development, we examined the effects of butyrate on DC development from Lin− bone marrow cells from wild-type and Slc5a8−/− mice (Fig. 7A). Butyrate (50 μm) caused 75% inhibition of DC development in bone marrow cells from wild-type mice. The effect was significantly blunted in Slc5a8−/− bone marrow cells. The inhibition was decreased from 75% to 40% (p < 0.02). In contrast, the inhibitory potency of butyrate remained essentially the same as that of wild type bone marrow cells when Gpr109a−/− bone marrow cells were used (p > 0.05) (Fig. 7A), indicating that there is no role for this receptor in the butyrate-induced blockade of DC development. The blockade of DC development by butyrate is not complete in Slc5a8−/− bone marrow cells most likely because this fatty acid diffuses into cells to an appreciable extent even in the absence of Slc5a8, causing a significant inhibition of HDACs. RT-PCR showed that wild-type bone marrow cells express Slc5a8 as well as Gpr109a (Fig. 7B). However, the blocking effects of butyrate and propionate on DC development from these cells involve only the transporter but not the receptor.
FIGURE 7.
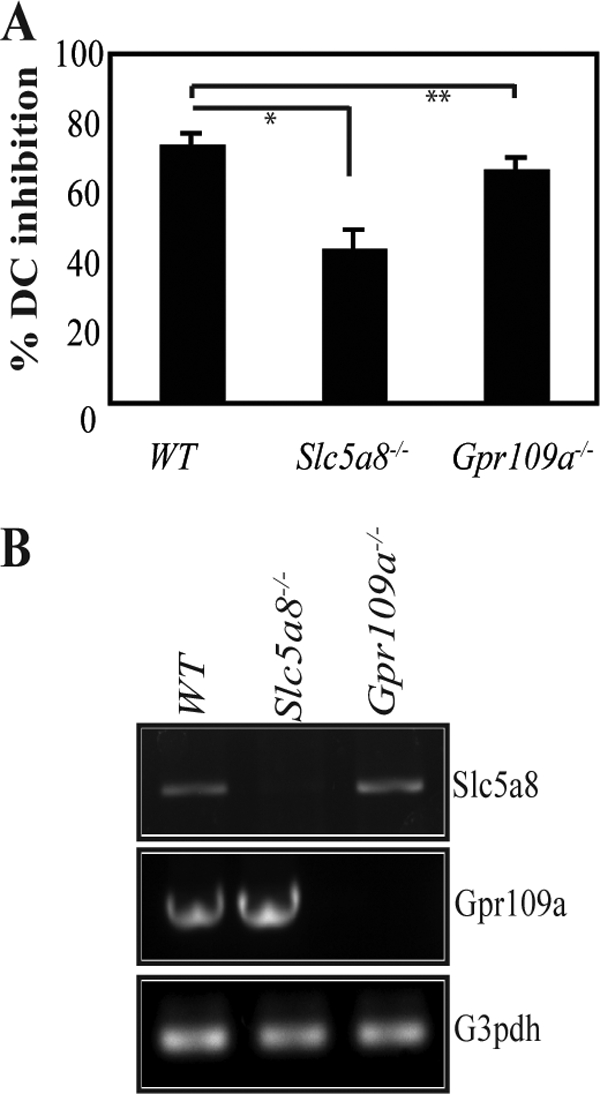
Obligatory role of Slc5a8 in butyrate-induced blockade of DC development. Lin− bone marrow cells from wild type, Slc5a8−/−, and Gpr109a−/− mice were cultured with GM-CSF (25 ng/ml) in the presence or absence of 50 μm butyrate. On day 6, cultures were analyzed for DCs. A, percent inhibition of DC development was calculated based on the number of DCs recovered on day 6 without treatment with butyrate. *, p < 0.02; **, p > 0.05. B, expression of Slc5a8 and Gpr109a in bone marrow cells from wild type, Slc5a8−/−, and Gpr109a−/− mice analyzed by RT-PCR. G3pdh, glyceraldehyde-3-phosphate dehydrogenase.
Our results demonstrate that while HDACs play a critical role in generation of DCs, they are not required for development of granulocytes. A recent study showed that trichostatin A, a pan-HDAC inhibitor, blocked the effects of GM-CSF on macrophage proliferation as well as on generation of DCs (45). Our studies focused on the bacterial fermentation products acetate, propionate, and butyrate. Propionate and butyrate function as HDAC inhibitors but, unlike trichostatin A, propionate and butyrate inhibit specifically HDAC1 and HDAC3 (28), suggesting that these two HDAC isoforms may play a critical role in DC development. The findings that the bacterial metabolites propionate and butyrate effectively suppress the development of DCs have immense clinical relevance. Even though mammalian colon harbors trillions of bacteria, there is no evidence of immune activation in the intestinal tract against these bacteria, suggesting that the host immune system is kept under control to facilitate the bacterial colonization of the colon. The present study provides the first glimpse of the mechanisms involved in the process at the molecular level. Colonic bacteria suppress the development of DCs through the ability of their fermentation products propionate and butyrate to inhibit HDACs in the host. This mechanism requires Slc5a8; without this transporter, these inhibitors cannot gain access to their intracellular targets HDAC1 and HDAC3 in DC progenitor cells. There are ∼800 different strains of bacteria in human colon, and not all of them generate propionate and butyrate. The findings that acetate does not have any effect on DC development suggest that the ability of colonic bacteria to suppress DC development is likely to be influenced by the presence of specific strains of bacteria in the colon depending on the fermentation products generated by these bacteria. It is known that the bacterial strain composition varies from person to person, determined by diet and environmental factors specific to a given individual and that the end products of fermentation depend on the specific strains of bacteria in the colon and also on the chemical nature of the fiber that is fermented. Consequently, the relative concentrations of acetate, propionate, and butyrate in the colonic lumen also vary from person to person. Therefore, the ability of colonic bacteria to suppress immune function through the blockade of DC development may not be uniform across the population. This has significant clinical implications in the pathogenesis of inflammatory bowel diseases, particularly ulcerative colitis. Based on our present findings, we speculate that the colonic colonization of bacterial strains that generate propionate and butyrate may provide protection against inflammatory bowel diseases such as ulcerative colitis.
HDACs have been implicated in the control of expression of several transcription factors involved in development of DCs (46–50). It is possible that the bacterial fermentation products propionate and butyrate block DC development by affecting the expression of specific transcription factors in DC precursor cells. Our studies indicate that suppression of the transcription factors PU.1 and RelB is at least partly responsible for this process. We have shown recently that the colonic expression of Slc5a8, which is necessary for butyrate and propionate to elicit their biologic effects through HDAC inhibition, is regulated by gut bacteria (51). Conventional mice, which harbor bacteria, express Slc5a8 in the colon robustly. In contrast, the colonic expression of the transporter is markedly suppressed in germ-free mice, which do not have bacteria in the colon. However, when the colon of the germ-free mice is colonized with bacteria, the expression of the transporter returns to normal levels comparable to that seen in conventional mice. Based on these findings, we conclude that conventional bacteria suppress immune function in the intestinal tract through their fermentation products propionate and butyrate at least partly via inhibition of DC development. This process involves Slc5a8-dependent entry of propionate and butyrate into DC precursor cells with subsequent inhibition of HDACs.
Footnotes
- DC
- dendritic cell
- HDAC
- histone deacetylase
- SAHA
- N1-hydroxy-N8-phenyloctanediamide
- BRET
- bioluminescence resonance energy transfer.
REFERENCES
- 1.Banchereau J., Steinman R. M. (1998) Nature 392, 245–252 [DOI] [PubMed] [Google Scholar]
- 2.Kaisho T., Akira S. (2003) Curr. Mol. Med. 3, 759–771 [DOI] [PubMed] [Google Scholar]
- 3.Reis e Sousa C. (2004) Curr. Opin. Immunol. 16, 21–25 [DOI] [PubMed] [Google Scholar]
- 4.Jung S., Unutmaz D., Wong P., Sano G., De los Santos K., Sparwasser T., Wu S., Vuthoori S., Ko K., Zavala F., Pamer E. G., Littman D. R., Lang R. A. (2002) Immunity 17, 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Kooyk Y., Appelmelk B., Geijtenbeek T. B. (2003) Trends Mol. Med. 9, 153–159 [DOI] [PubMed] [Google Scholar]
- 6.Larsson M., Beignon A. S., Bhardwaj N. (2004) Semin. Immunol. 16, 147–161 [DOI] [PubMed] [Google Scholar]
- 7.Trifilo M. J., Hahm B., Zuniga E. I., Edelmann K. H., Oldstone M. B. (2006) J. Infect. Dis. 194, Suppl. 1, S3–S10 [DOI] [PubMed] [Google Scholar]
- 8.Hooper L. V., Midtvedt T., Gordon J. I. (2002) Annu. Rev. Nutr. 22, 283–307 [DOI] [PubMed] [Google Scholar]
- 9.Bäckhed F., Ley R. E., Sonnenburg J. L., Peterson D. A., Gordon J. I. (2005) Science 307, 1915–1920 [DOI] [PubMed] [Google Scholar]
- 10.Topping D. L., Clifton P. M. (2001) Physiol. Rev. 81, 1031–1064 [DOI] [PubMed] [Google Scholar]
- 11.Wong J. M., de Souza R., Kendall C. W., Emam A., Jenkins D. J. (2006) J. Clin. Gastroenterol. 40, 235–243 [DOI] [PubMed] [Google Scholar]
- 12.Bordonaro M., Lazarova D. L., Sartorelli A. C. (2008) Cell Cycle 7, 1178–1183 [DOI] [PubMed] [Google Scholar]
- 13.Gupta N., Martin P. M., Prasad P. D., Ganapathy V. (2006) Life Sci. 78, 2419–2425 [DOI] [PubMed] [Google Scholar]
- 14.Miyauchi S., Gopal E., Fei Y. J., Ganapathy V. (2004) J. Biol. Chem. 279, 13293–13296 [DOI] [PubMed] [Google Scholar]
- 15.Thangaraju M., Cresci G., Itagaki S., Mellinger J., Browning D. D., Berger F. G., Prasad P. D., Ganapathy V. (2008) J. Gastrointest. Surg. 12, 1773–1782 [DOI] [PubMed] [Google Scholar]
- 16.Thangaraju M., Gopal E., Martin P. M., Ananth S., Smith S. B., Prasad P. D., Sterneck E., Ganapathy V. (2006) Cancer Res. 66, 11560–11564 [DOI] [PubMed] [Google Scholar]
- 17.Li H., Myeroff L., Smiraglia D., Romero M. F., Pretlow T. P., Kasturi L., Lutterbaugh J., Rerko R. M., Casey G., Issa J. P., Willis J., Willson J. K., Plass C., Markowitz S. D. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 8412–8417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ganapathy V., Thangaraju M., Gopal E., Martin P. M., Itagaki S., Miyauchi S., Prasad P. D. (2008) AAPS J. 10, 193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ganapathy V., Thangaraju M., Prasad P. D. (2009) Pharmacol. Ther. 121, 29–40 [DOI] [PubMed] [Google Scholar]
- 20.Thangaraju M., Cresci G. A., Liu K., Ananth S., Gnanaprakasam J. P., Browning D. D., Mellinger J. D., Smith S. B., Digby G. J., Lambert N. A., Prasad P. D., Ganapathy V. (2009) Cancer Res. 69, 2826–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wise A., Foord S. M., Fraser N. J., Barnes A. A., Elshourbagy N., Eilert M., Ignar D. M., Murdock P. R., Steplewski K., Green A., Brown A. J., Dowell S. J., Szekeres P. G., Hassall D. G., Marshall F. H., Wilson S., Pike N. B. (2003) J. Biol. Chem. 278, 9869–9874 [DOI] [PubMed] [Google Scholar]
- 22.Tunaru S., Kero J., Schaub A., Wufka C., Blaukat A., Pfeffer K., Offermanns S. (2003) Nat. Med. 9, 352–355 [DOI] [PubMed] [Google Scholar]
- 23.Taggart A. K., Kero J., Gan X., Cai T. Q., Cheng K., Ippolito M., Ren N., Kaplan R., Wu K., Wu T. J., Jin L., Liaw C., Chen R., Richman J., Connolly D., Offermanns S., Wright S. D., Waters M. G. (2005) J. Biol. Chem. 280, 26649–26652 [DOI] [PubMed] [Google Scholar]
- 24.Feagins L. A., Souza R. F., Spechler S. J. (2009) Nat. Rev. Gastroenterol. Hepatol. 6, 297–305 [DOI] [PubMed] [Google Scholar]
- 25.Terzić J., Grivennikov S., Karin E., Karin M. (2010) Gastroenterology 138, 2101–2114.e5 [DOI] [PubMed] [Google Scholar]
- 26.Hamer H. M., Jonkers D., Venema K., Vanhoutvin S., Troost F. J., Brummer R. J. (2008) Aliment. Pharmacol. Ther. 27, 104–119 [DOI] [PubMed] [Google Scholar]
- 27.Frank H., Gröger N., Diener M., Becker C., Braun T., Boettger T. (2008) J. Biol. Chem. 283, 24729–24737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thangaraju M., Carswell K. N., Prasad P. D., Ganapathy V. (2009) Biochem. J. 417, 379–389 [DOI] [PubMed] [Google Scholar]
- 29.Thangaraju M., Karunakaran S. K., Itagaki S., Gopal E., Elangovan S., Prasad P. D., Ganapathy V. (2009) Cancer 115, 4655–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollins B., Kuravi S., Digby G. J., Lambert N. A. (2009) Cell Signal. 21, 1015–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R. M. (1992) J. Exp. Med. 176, 1693–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gopal E., Fei Y. J., Miyauchi S., Zhuang L., Prasad P. D., Ganapathy V. (2005) Biochem. J. 388, 309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gopal E., Miyauchi S., Martin P. M., Ananth S., Roon P., Smith S. B., Ganapathy V. (2007) Pharm. Res. 24, 575–584 [DOI] [PubMed] [Google Scholar]
- 34.Anderson K. L., Perkin H., Surh C. D., Venturini S., Maki R. A., Torbett B. E. (2000) J. Immunol. 164, 185501861. [DOI] [PubMed] [Google Scholar]
- 35.Laribee R. N., Klemsz M. J. (2005) Biochim. Biophys. Acta 1730, 226–234 [DOI] [PubMed] [Google Scholar]
- 36.Koski G. K., Lyakh L. A., Cohen P. A., Rice N. R. (2001) Crit. Rev. Immunol. 21, 179–189 [PubMed] [Google Scholar]
- 37.Cejas P. J., Carlson L. M., Kolonias D., Zhang J., Lindner I., Billadeau D. D., Boise L. H., Lee K. P. (2005) Mol. Cell Biol. 25, 7900–7916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones P. L., Shi Y. B. (2003) Curr. Top. Microbiol. Immunol. 274, 237–268 [DOI] [PubMed] [Google Scholar]
- 39.Cunliffe V. T. (2008) Curr. Opin. Genet. Dev. 18, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu W. S., Parmigiani R. B., Marks P. A. (2007) Oncogene 26, 5541–5552 [DOI] [PubMed] [Google Scholar]
- 41.Smith C. L. (2008) BioEssays 30, 15–24 [DOI] [PubMed] [Google Scholar]
- 42.Rada-Iglesias A., Enroth S., Ameur A., Koch C. M., Clelland G. K., Respuela-Alonso P., Wilcox S., Dovey O. M., Ellis P. D., Langford C. F., Dunham I., Komorowski J., Wadelius C. (2007) Genome Res. 17, 708–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kostyniuk C. L., Dehm S. M., Batten D., Bonham K. (2002) Oncogene 21, 6340–6347 [DOI] [PubMed] [Google Scholar]
- 44.Dehm S. M., Hilton T. L., Wang E. H., Bonham K. (2004) Mol. Cell Biol. 24, 2296–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sebastián C., Serra M., Yeramian A., Serrat N., Lloberas J., Celada A. (2008) J. Immunol. 180, 5898–5906 [DOI] [PubMed] [Google Scholar]
- 46.Dong X., Lutz W., Schroeder T. M., Bachman L. A., Westendorf J. J., Kumar R., Griffin M. D. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 16007–16012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koipally J., Renold A., Kim J., Georgopoulos K. (1999) EMBO J. 18, 3090–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nencioni A., Beck J., Werth D., Grünebach F., Patrone F., Ballestrero A., Brossart P. (2007) Clin. Cancer Res. 13, 3933–3941 [DOI] [PubMed] [Google Scholar]
- 49.Wu L., Liu Y. J. (2007) Immunity 26, 741–750 [DOI] [PubMed] [Google Scholar]
- 50.Onai N., Manz M. G. (2008) Immunity 28, 490–492 [DOI] [PubMed] [Google Scholar]
- 51.Cresci G. A., Thangaraju M., Mellinger J. D., Liu K., Ganapathy V. (2010) J. Gastrointest. Surg. 14, 449–461 [DOI] [PubMed] [Google Scholar]