

Abstract
In atrial and nodal cardiac myocytes, M2 muscarinic receptors activate inhibitory G-proteins (Gi/o), which in turn stimulate G-protein-gated inwardly rectifying K+ channels through direct binding of the Gβγ subunit. Despite also releasing Gβγ, Gs-coupled receptors such as the β-adrenergic receptor are not able to prominently activate this current. An appealing hypothesis would be if components were sequestered in membrane domains such as caveolae/rafts. Using biochemical fractionation followed by Western blotting and/or radioligand binding experiments, we examined the distribution of the components in stable HEK293 and HL-1 cells, which natively express the transduction cascade. The channel, M2 muscarinic, and A1 adenosine receptors were located in noncaveolar/nonraft fractions. Giα1/2 was enriched in both caveolar/raft and noncaveolar/nonraft fractions. In contrast, Gsα was only enriched in caveolar/raft fractions. We constructed YFP-tagged caveolin-2 (YFP-Cav2) and chimeras with the M2 (M2-YFP-Cav2) and A1 (A1-YFP-Cav2) receptors. Analysis of gradient fractions showed that these receptor chimeras were now localized to caveolae-enriched fractions. Microscopy showed that M2-YFP and A1-YFP had a diffuse homogenous membrane signal. YFP-Cav2, M2-YFP-Cav2, and A1-YFP-Cav2 revealed a more punctuate pattern. Finally, we looked at the consequences for signaling. Activation via M2-YFP-Cav2 or A1-YFP-Cav2 revealed substantially slower kinetics compared with M2-YFP or A1-YFP and was reversed by the addition of methyl-β-cyclodextrin. Thus the localization of the channel signal transduction cascade in non-cholesterol rich domains substantially enhances the speed of signaling. The presence of Gsα solely in caveolae may account for signaling selectivity between Gi/o and Gs-coupled receptors.
Keywords: Caveolae, G-protein-coupled Receptors (GPCR), G Proteins, Membrane Function, Potassium Channels, Signal Transduction
Introduction
Agonist stimulation of G-protein-coupled receptors (GPCRs)2 leads to numerous cellular responses via activation of heterotrimeric G-proteins and thereby modulation of downstream signaling components. In atrial and nodal cardiac myocytes binding of acetylcholine to M2 muscarinic receptors activates inhibitory G-proteins, which in turn stimulate G-protein-gated inward rectifying K+ channels (GIRK) through direct binding of the Gβγ subunit (1). In the heart GIRK channels are thought to be a heterotetramer of the inwardly rectifying K+ channel subunits Kir3.1 and Kir3.4 (2–5). This cellular cascade underlies heart rate slowing through the parasympathetic system (6, 7). These channels and analogous signaling systems are also present in central neurons where they are responsible for late inhibitory postsynaptic potentials and in neuroendocrine cells where activation inhibits hormone release (1, 8, 9). Despite also releasing Gβγ, GPCRs coupled to the stimulatory G-protein (Gs) such as the β-adrenergic receptors are not able to prominently activate this current (10). An appealing hypothesis to explain these observations would be if components were sequestered in membrane domains such as caveolae/rafts.
Caveolae are invaginations, of about 50–100 nm in size, of the plasma membrane and play a role in a range of biological processes such as transmembrane signaling, lipid and protein sorting, and intracellular trafficking (11). Caveolae are characterized by being rich in cholesterol and sphingolipids. Rafts are less physically defined but are thought to be smaller membrane microdomains defined also by high cholesterol content (12). Whether caveolae are localized solely in the plasma membrane and as well as in other intracellular membranes is still an open question. Because of their high lipid-to-protein ratio they are relatively insoluble at 4 °C in detergents like Triton X-100. This Triton insolubility is a physical property of their molecular organization and produces a liquid-ordered membrane domain unlike a fluid one. Membrane fractionation by sucrose density gradient centrifugation localizes the detergent insoluble caveolae/lipid rafts in the buoyant, upper fraction. Caveolae characteristically are defined by the presence of the protein caveolin, which consists of a family of caveolin-1, caveolin-2, and caveolin-3 (Cav-1, Cav-2, Cav-3) (13–15) (16). Cav-1 and Cav-2 are widely expressed in many cell types, whereas Cav-3 is a muscle-specific isoform. Caveolin defines the structure of caveolae but can also interact with signaling molecules such as eNOS, HaRas, GPCRs, and G-proteins and can potentially concentrate these proteins in these structures (16).
One paradox in the activation of GIRK channels is that all GPCRs, whichever G-protein family they couple to, might be expected to activate the current as they all release free Gβγ upon activation. However Gs (and Gq)-coupled GPCRs such as the β-adrenergic receptors are not able to prominently activate this current even in heterologous expression systems (10, 17) and activation seems to be mediated by Gi/o-coupled receptors in the heart and central neurons. This selectivity is physiologically important in the heart as normal increases in heart rate in response to increased sympathetic tone would be counteracted by simultaneous GIRK channel activation. One appealing hypothesis to explain these observations would be if components were selectively sequestered in membrane domains such as caveolae/rafts. Here we investigate whether the relevant GPCRs, G-proteins, and channel subunits are present or not in cholesterol rich membrane domains. We show that only G-proteins were enriched in caveolar/raft fractions, though for Giα1/2 a significant fraction was also present in noncaveolar/nonraft domains. In contrast Gsα was only enriched in caveolar/raft fractions. Furthermore, we looked at the physiological consequences for signaling if we drove one of the other components into caveolae. Fusing M2-YFP or A1-YFP with Cav-2 led to localization in caveolae and significantly slower signaling.
EXPERIMENTAL PROCEDURES
Molecular Biology
YFP-Cav2 was constructed by direct fusion of Cav-2 to the C terminus of YFP without a stop codon and without a linker by a PCR-based overlap extension method using AgeI/NotI to clone YFP-Cav2 in pEYFP-N1 (Clontech). RFP-Cav2 was constructed using the same method as mentioned above using NheI/HindIII. M2 was cloned into pEYFP-N1 on BglII/HindIII ends and the same way into pEYFP-Cav2 (in-frame and without a stop codon). A1-YFP-Cav2 was created by cloning A1 from A1-YFP into pEYFP-Cav2 on HindIII/KpnI (in-frame and without a stop codon). All constructs were verified by automated sequencing.
Cell Culture
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (Gibco-Invitrogen), supplemented with 100 units/ml penicillin G sodium, 100 mg/ml streptomycin (Invitrogen), and 10% fetal bovine serum (FBS). Cells were maintained in a humidified incubator at 37 °C with 95% O2 and 5% CO2. Cells were passaged up to twice a week. Unless otherwise stated, 800 ng of cDNA per vector was transfected using Lipofectamine (Invitrogen) or we used 5 μl of Fugene HD in 97 μl of cell culture medium containing no serum or antibiotics and 800 or 1600 ng of DNA. We always added 40 ng of EGFP DNA to the cells we used for electrophysiology, to visualize transfected cells under epifluorescence. The HL-1 cell line (22) was a gift from Prof. William C. Claycomb (Louisiana State University, Baton Rouge, LA). HL-1 cells were cultured under 5% C02 atmosphere in Claycomb medium supplemented with 10% fetal bovine serum, 0.2 mm l-glutamine, 0.1 mm norepinephrine, 100 units/ml penicillin, and 100 μg/ml streptomycin (all from Sigma). The medium was replaced every day. Cells were grown onto T75 culture flasks precoated overnight with 0.03% fibronectine/0.02% gelatin solution. For electrophysiology and imaging experiments, cells were grown on uncoated 13-mm glass coverslips (BDH).
Microscopy
Confocal images were taken using a Bio-Rad Radiance 2100 laser scanning confocal Nikon TE300 microscope (Bio-Rad), EYFP was excited using an argon 488 nm laser and emission recorded using a HQ515/15 filter. Wide field images were acquired using a 20× objective, and individual cells were visualized using a 40 or 60× oil-immersion objective. Images were median-filtered for display.
Confocal images for co-localization experiments were taken using an Olympus IX81 microscope and images scanned sequentially. EYFP was excited using an argon 488-nm laser and emission was recorded between 510 and 555 nm. RFP was excited using a 559-nm laser and emission was recorded between 575 and 675 nm. Individual cells were visualized using a 60× oil-immersion objective (NA 1.35). Images were pseudo-colored and median-filtered for display.
Total internal reflection fluorescence (TIRF) microscopy was performed using a Nikon Eclipse TE200E inverted microscope using a 100× objective (Apo TIRF, NA 1.49). Cells were excited using an argon 514-nm laser and a long pass 520-nm filter was used to collect the emitted light. A micrometer allowed adjustment of the angle of illumination, and it was possible to obtain images under epifluorescence and TIRF illumination. A digital EMCCD camera (iXon897BV, Andor) was used to acquire images, and they were stored and analyzed on a computer using the NIS elements software. Images were median-filtered for presentation but no other form of image manipulation was performed.
Membrane Fractionation
Cells were fractionated using the detergent-containing (1% Triton X-100) method by modifying published protocols (18, 19). All steps were carried out at 4 °C. The plasma membrane fractions were prepared from a T-75 flask of confluent HEK293 or HL-1 cells. Each flask was washed twice with ice-cold PBS, and cells were then collected by scraping in 10 ml of ice-cold PBS. After a 2 min 300 × g centrifugation step the pellets were homogenized in TNE buffer (25 mm Tris-HCl, 150 mm NaCl, and 5 mm EDTA) containing 1% Triton X-100 and protease inhibitor mix (Roche). The homogenates were mixed with OptiPrep (Sigma) to a final concentration of 40% OptiPrep. A discontinuous OptiPrep gradient was generated by layering 2-ml layers of 35% OptiPrep in TNE buffer, followed by 30, 25, and 20% OptiPrep in TNE buffer and finally topped up with TNE buffer and centrifuged at 190,000 × g for 4 h at 4 °C using the TI 41.14 rotor. Samples were fractionated in 1-ml aliquots from the top to bottom to form 12 fractions (fraction 1 equates to the top fraction and fraction 12 to the bottom one). When the fractions were prepared for SDS-PAGE they were mixed with 3× reducing SDS-PAGE loading buffer (R-STB) and incubated for 10 min at 95 °C.
Subcellular Fractionation
All steps were carried out at 4 °C. The fractions were prepared from a T-75 flask of confluent HEK293 cells. Cells have been washed twice with ice-cold PBS and collected by scraping in 1 ml of homogenization buffer (3 mm imidazole pH 7.4; 250 mm sucrose; 1 mm EDTA; 10 μg/ml cycloheximide; protease inhibitor (Roche) (20). After 2 min of sonication, the postnuclear supernatant (PNS) was prepared by a 10-min centrifugation step at 1000 × g. Centrifugation of the PNS for 1 h at 200,000 × g lead to membrane fraction (pellet) and cytosolic fraction (supernatant). For SDS-PAGE fractions were mixed with R-STB and incubated for 10 min at 95 °C. The ratio of analyzed membrane to cytosol fraction was 1:10.
Radioligand Binding Experiments
Cells were incubated with [3H]quinuclidinyl benzylate (QNB), N-[3H]methyl scopolamine (NMS) or 8-[3H]cyclopentyl-1,3-dipropylxanthine (DPCPX). Experiments to detect M2 muscarinic receptor binding were performed with [3H]QNB (can cross the membrane) or [3H]NMS (cannot cross the membrane). To measure nonspecific binding, experiments were done in parallel with 10 μm atropine. [3H]DPCPX binds to the A1 receptor and to determine the nonspecific binding, cells were incubated with 10 μm DPCPX. Every experiment was done in triplicate.
SDS-PAGE and Western Blotting
15 μl of each OptiPrep density fraction was separated on Laemmli gels (21). Gels were transferred to PVDF membrane and blocked in Tris-buffered saline (TBS) (pH 7.4) containing 5% dried milk powder. After blocking, membranes were incubated with anti-caveolin (rabbit polyclonal, 1:10000, (BD Transduction Laboratories)), anti-flotillin-1 (rabbit polyclonal, 1:5000, (Sigma)), anti-Giα-1 and Giα-2 subunits (rabbit polyclonal, 1:5000, (Calbiochem)), anti-Kir3.1 (GIRK1) (rabbit polyclonal, 1:2000, (Alomone Labs)), anti-Kir3.4 (GIRK4) (rabbit polyclonal, 1:2000, (Alomone Labs)), anti-mAChR M2 (rabbit polyclonal, 1:200, (Santa Cruz Biotechnology)) or anti-Gαs (rabbit polyclonal, 1:100, (Santa Cruz Biotechnology) antibodies for 2 h. Membranes were then washed with TBS three times for 5 min. After washing, the primary antibody was detected by adding an HRP-conjugated goat anti-rabbit specific secondary antibody (1:5000, Santa Cruz Biotechnology (sc-2054)) for 2 h. The membranes were then washed three times with TBS for 5 min and developed using the ECLTM Western blotting chemiluminescent reagent kit (Amersham Biosciences) as per manufacturer's instructions. Western blot band intensity was determined and quantified using ImageJ software (NIH, Bethesda, MD). The region of interest was marked and measured in every lane, and the background was subtracted to give the final band intensity. Statistical comparisons were made using a two-tailed Student's t test. All data are represented as mean ± S.E. where n indicates the number of experiments.
Electrophysiology
Cells were seeded on 13-mm glass coverslips for electrophysiological recordings. For the evaluation of current kinetics a fast perfusion system was used to apply drugs (Rapid Solution Changer, RSC-160, Bio-Logic France). Cells were clamped at −60 mV, the extracellular solution was (mm): NaCl 80, KCl 60, CaCl2 2, MgCl2 1, HEPES 10, NaH2PO4 0.33, glucose 10, pH 7.4; while the intracellular solution was (mm): K gluconate 110, KCl 20, NaCl 10, MgCl2 1, MgATP 2, EGTA 2, GTP 0.3, pH 7.4. After agonist application, current activated with a delay “lag” followed by a rapid rise to peak amplitude “time to peak.” After removal of the agonist, the current decay back to baseline. Current activation and deactivation were fitted by a single exponential function A × exp(−t/τ) + C (where A is the current amplitude at the start of the fit, t is time, τ is the activation or the deactivation time constant, and C is the steady-state asymptote). For each cell we assessed whether there were any flow artifacts resulting from the pressure of drug application. We did this by applying bath solution from one of the sewer pipes at the beginning of the recordings. Furthermore, to control for variations in positioning of the sewer-pipe system relative to the cell, we calibrate the system by using the kinetics of channel block by 1 mm barium. Block of the current occurred with an initial delay “lag” before reaching equilibrium; it was assumed that this lag reflected the intrinsic delivery time to the cell and was 61 ± 6 ms in these experiments (n = 57). All data are represented as mean ± S.E. where n indicates the number of cells recorded. Statistical analyses were performed using nonparametric tests specifically with a Kruskal-Wallis test with a Dunn post-hoc test or a Mann-Whitney test as appropriate. ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
RESULTS
Distribution of Signaling Components in Triton-soluble and -insoluble Fractions
Caveolin-enriched membranes are characteristically insoluble in buffer containing Triton X-100 when prepared in the cold because of their high content of cholesterol and sphingolipids. We used discontinuous OptiPrep gradient ultracentrifugation after solubilization of membranes in Triton X-100-containing buffer. We analyzed membranes from HEK293 cells stably expressing Kir3.1/Kir3.4 and the M2 receptor. We also used HL-1 cells, which are one of the better models of native cardiac myocytes (22). We have recently analyzed the GPCR-G-protein-Kir3.0 signaling pathway in these cells. We found that it exists and approximates to that present in mouse native atrial myocytes (23). Aliquots of fractions per condition were analyzed by SDS-PAGE, transferred onto PVDF membranes, and immunoblotted with anti-caveolin, anti-flotillin-1, anti-M2-receptor, anti-Kir3.1, anti-Kir3.4, anti-Giα1/2, and anti-Gsα antibodies. The distribution of M2 receptors and K+ channel (Kir3.1/Kir3.4) were analyzed in stable HEK293 cells heterologously expressing the relevant components following OptiPrep density fractionation. We detected both, the M2 receptor and the channel proteins in the heavy fractions. In contrast, caveolin and flotillin-1, usually used as caveolar markers, are localized in the buoyant membrane fractions in HEK293 cells. The analysis of the distribution of different G-proteins showed that Giα1/2 is enriched in caveolae but is also present in the heavy fractions. In contrast Gsα is only detectable in the buoyant, caveolin-containing fractions (Fig. 1A). We also examined the cascade in HL-1 cells and as in HEK293 cells M2 receptors and Kir3.4 were detected in the heavy fractions. In contrast to HEK293 cells, in HL-1 cells both caveolin and flotillin were detected throughout every fraction. Other investigators have made similar observations on the localization of caveolin and flotillin in adult cardiomyocytes (19), and it is indicative of a non-plasma membrane localization of these proteins in these cells. As in HEK293 cells Giα1/2 is enriched in buoyant as well as in heavy fractions (Fig. 1B). We repeated these experiments on both cell lines at least three times with similar results.
FIGURE 1.

The M2 muscarinic receptor and the K+ channel subunits Kir3.1 and Kir3.4 are localized in noncaveolar/nonraft fractions. A, expression and localization of caveolin, flotillin-1, Giα1/2, Gsα, the muscarinic receptor M2, Kir3.4, and Kir3.1 after OptiPrep density fractionation of HEK293 cells stably expressing M2 Kir3.1/Kir3.4 as described under “Experimental Procedures.” The fractions were collected from the gradient and were subjected to SDS-PAGE and immunoblot analysis. B, expression and localization of caveolin, flotillin-1, Giα1/2, the muscarinic receptor M2, and Kir3.4 upon OptiPrep density fractionation done with HL-1 cells as described under “Experimental Procedures.” The fractions were collected from the gradient and were subjected to SDS-PAGE and immunoblot analysis. C, expression and localization of caveolin, Giα1/2, Gsα, and the muscarinic receptor M2 upon OptiPrep density fractionation of HEK293 cells stably expressing M2 Kir3.1/Kir3.4 after the incubation with 2 mm MβCD for 90 min. The fractions were collected from the gradient and were subjected to SDS-PAGE and immunoblot analysis. Additionally, the quantification of the Western blots for Giα1/2 (n = 3) and Cav (n = 4) are shown. The error bars represent the standard error.
Cells were incubated with 2 mm methyl-β-cyclodextrin (MβCD) for 90 min at 37 °C to remove cholesterol from the cells and therefore disrupt caveolae and lipid rafts. Western blot analyses of OptiPrep density fractions, prepared after incubation with MβCD compared with controls (without MβCD incubation) showed reduced caveolin in the upper fractions (Fig. 1C). The analysis of G-proteins showed that there was some residual expression of Gαi1/2 (Fig. 1C, right) but complete loss of Gαs in the buoyant fractions after MβCD incubation. Combining the signal of fraction 2 to 4 the reduction of caveolin and Gαi1/2 after MβCD treatment was significant (caveolin: before MβCD: 71.8 ± 6.4 after MβCD: 36.7 ± 7.9, p = 0.007 (n = 4); Gαi1/2: before MβCD: 49.6 ± 9.4 after MβCD: 9.4 ± 5.4, p = 0.011 (n = 3)). The translocation of M2 receptors was not influenced by MβCD (Fig. 1C).
Further analysis of the localization of the M2 muscarinic receptor was performed using radioligand binding. The distribution of M2 receptors in percent along a OptiPrep density gradient was detected by using [3H]QNB as well as [3H]NMS. Both are specific muscarinic receptor antagonists but [3H]QNB is membrane permeable while [3H]NMS is not. As a control for the specificity of the ligands, experiments were performed by co-incubating with 1 μm atropine, a muscarinic receptor antagonist. In HEK293 cells stably expressing Kir3.1/Kir3.4 and M2, the M2 receptor is mainly present in non-buoyant fractions (Fig. 2, A and B). HL-1 cells showed a similar pattern (Fig. 2C). Finally, in an analogous manner using [3H]DPCPX to detect A1 adenosine receptors, we found these receptors were also located endogenously in non-buoyant fractions in HL-1 cells (Fig. 2D).
FIGURE 2.

Radioligand binding experiments show that M2 muscarinic receptors are in noncaveolae/nonraft fractions. Distribution of M2 muscarinic (A, B, C) and A1 adenosine (D) receptors in percent along an OptiPrep density gradient after cold Triton extraction of the membranes. Fraction 1 refers to the top of the gradient. For each condition nonspecific binding was measured in the presence of 10 μm atropine (A, B, C) or 10 μm DPCPX (D). The error bars represent the standard error. A, fractions of HEK293 cells stably expressing M2 Kir3.1/Kir3.4. The percent of M2 receptors in each fraction is determined by the amount of specific [3H]QNB binding. The data are expressed as specific [3H]QNB binding and represent three experiments each measured in triplicate. B, fractions of HEK293 cells stably expressing M2 Kir3.1/Kir3.4. The percent of M2 receptors in each fraction is determined by the amount of specific [3H]NMS binding. The data are expressed as the percent of total specific [3H]NMS binding and represent six experiments each measured in triplicate. C, fractions of HL-1 cells. The percent of M2 receptors in each fraction is determined by the amount of specific [3H]NMS binding. The data are expressed as the percent of total specific [3H]NMS binding and represent five experiments each measured in triplicate. D, fractions of HL-1 cells. The percent of A1 receptors in each fraction is determined by the amount of specific [3H]DPCPX binding. The data are expressed as specific [3H]DPCPX binding and represent four experiments each measured in triplicate.
The results suggest that the M2 muscarinic receptor as well as the A1 adenosine receptor is not localized in caveolae-corresponding fractions in both HEK293 cells and HL-1 cells. Does this change on agonist application? We prepared OptiPrep density fractions before and after incubation with 10 μm of the muscarinic receptor agonist carbachol and analyzed them using Western blotting. There was no change in the distribution of most of the proteins of interest (Fig. 3). The absolute expression of Gsα in caveolar fractions was weaker but the relative distribution through the gradient was not altered (Fig. 3B). Further analysis via cytosol membrane fractions showed that after carbachol incubation Gsα is detached to the membrane (Fig. 3C).
FIGURE 3.

A, carbachol has no influence on the localization of M2 muscarinic receptors and the K+ channel subunits Kir3.1 and Kir3.4. Expression and localization of caveolin, Giα1/2, Gsα, Kir3.4, Kir3.1, and the muscarinic receptor M2 upon OptiPrep density fractionation done with HEK293 cells stably expressing M2 Kir3.1/Kir3.4 before and after the addition of 10 μm carbachol (CCh). The fractions were collected from the gradient and were subjected to SDS-PAGE and immunoblot analyses. B, quantification of Gsα, Western blots (n = 5), the error bars represent the standard error. C, analysis of cytosol and membrane fractions shows that carbachol does not lead to detachment of Gsα from the membrane. Caveolin as well as Gsα are localized in the membrane fraction. c, cytosol; m, membrane.
Receptor-Caveolin 2 fusion constructs
To explore if the non-caveolar distribution might have functional consequences we adopted a previously published strategy and fused M2-YFP and A1-YFP to caveolin-2 (24). We first analyzed whether these receptors were now located in buoyant membrane fractions. We found indeed this was the case for M2-YFP-Cav2 (Fig. 4A) and A1-YFP-Cav2 (Fig. 4B) and that this was reversed by methyl-β-cyclodextrin in the case of M2-YFP-Cav2 (Fig. 4C).
FIGURE 4.

Receptors fused to Cav-2 are localized in caveolin corresponding fractions. A, fractions of HEK293 cells stably expressing Kir3.1/Kir3.4 transfected with M2-YFP-Cav2. The diagram shows the distribution of M2-YFP-Cav2 in percent along an OptiPrep density gradient. For each condition nonspecific binding was measured in the presence of 10 μm atropine. The percent of M2 receptors in each fraction is determined by the amount of specific [3H]NMS binding. The data are expressed as the percent of total specific [3H]NMS binding and represent four experiments each measured in triplicates. The error bars represent the standard error. Fraction 1 refers to the top of the gradient. B, fractions of HEK293 cells stably expressing Kir3.1/Kir3.4 transfected with A1-YFP-Cav2. The diagram shows the distribution of A1-YFP-Cav2 in percent along an OptiPrep density gradient. For each condition nonspecific binding was measured in the presence of 10 μm DPCPX. The percent of A1 receptors in each fraction is determined by the amount of specific [3H]DPCPX binding. The data are expressed as the percent of total specific [3H]DPCPX binding and represent three experiments each measured in triplicates. The error bars represent the standard error. Fraction 1 refers to the top of the gradient. C, comparison of the sum of fraction 2, 3, and 4 of HEK293 cells stably expressing Kir3.1/Kir3.4 transfected with M2/YFP/Cav2 without methyl-β-cyclodextrin and after the addition of 2 mm MβCD. D, analysis of HEK293 cells stably expressing Kir3.1/Kir3.4 transfected either with M2-YFP or M2-YFP-Cav2 (M2/Cav2). The data are expressed as the percent of total specific [3H]QNB binding of M2-YFP compared with M2-YFP-Cav2 and represent three experiments. The error bars represent the standard error. E, analysis of HEK293 cells stably expressing Kir3.1/Kir3.4 transfected either with M2-YFP or M2-YFP-Cav2. The data are expressed as the percent of total specific [3H]NMS binding of 800 ng of transfected M2-YFP (M2 800 ng) compared with 160 ng of transfected M2-YFP (M2 160 ng) and 800 ng of transfected M2-YFP-Cav2 (M2/Cav2 800 ng) and represent three experiments. The error bars represent the standard error.
Using radioligand binding we quantified the relative expression of M2-YFP and M2-YFP-Cav2. Using [3H]QNB it was found that the total levels of expression do not vary much between the two constructs with equivalent transfection efficiencies (Fig. 4D). However estimating membrane expression using [3H]NMS radioligand binding we found with M2-YFP-Cav2 that this was clearly reduced compared with M2-YFP (Fig. 4E). The receptor surface expression level is likely to be the most important factor governing signaling. We therefore titrated plasmid concentration for M2-YFP to give comparable membrane expression and found that reducing the amount of DNA to ∼160 ng under our standard transfection conditions maintained transfection efficiency while reducing total expression (Fig. 4E).
We analyzed the subcellular distribution of M2-YFP, A1-YFP, YFP-Cav2, M2-YFP-Cav2, and A1-YFP-Cav2 using confocal and total internal reflection fluorescence microscopy in HEK293 cells. M2-YFP and to a lesser extent A1-YFP revealed a peripheral ring consistent with diffuse membrane localization when studied with confocal or standard wide field epifluorescence (Fig. 5, A and B). When the footprint of the cell was studied using TIRFM a diffuse homogenous signal was present (Fig. 5A). In contrast, YFP-Cav2-, M2-YFP-Cav2-, and A1-YFP-Cav2-expressing HEK293 cells revealed a much more punctuate pattern (Fig. 5, A and B). This pattern was also present in cells imaged using TIRFM suggesting a proportion of these punctuate structures were present within or just beneath the membrane (Fig. 5A). TIRF microscopy of HEK293 cells expressing either YFP-Cav2, M2-YFP-Cav2, or A1-YFP-Cav2 after MβCD incubation showed a reduced punctuate pattern and more uniform expression pattern (Fig. 5C). HL-1 cells showed a similar pattern of distribution after heterologous expression of the constructs (Fig. 5B). Co-localization studies were performed in HEK293 cells expressing both, RFP-Cav2 and A1-YFP-Cav2, M2-YFP-Cav2 or as a control, YFP-Cav2. The merged pictures clearly show a co-localization of the receptors fusions with RFP-Cav2 (Fig. 6).
FIGURE 5.

Receptors targeted to caveolae show a punctuate fluorescence pattern. A, epifluorescence, TIRF, and confocal microscopy images of HEK293 cells expressing A1-YFP, M2-YFP, YFP-Cav2, A1-YFP-Cav2, or M2-YFP-Cav2. Bar, 10 μm. B, confocal microscopy images of M2-YFP- and M2-YFP-Cav2-expressing HL-1 cells. Bar, 10 μm. C, epifluorescence and TIRF images of HEK293 cells expressing YFP-Cav2, A1-YFP-Cav2, or M2-YFP-Cav2 after incubation with 2 mm MβCD. Bar, 10 μm. Images were acquired as detailed under “Experimental Procedures.”
FIGURE 6.
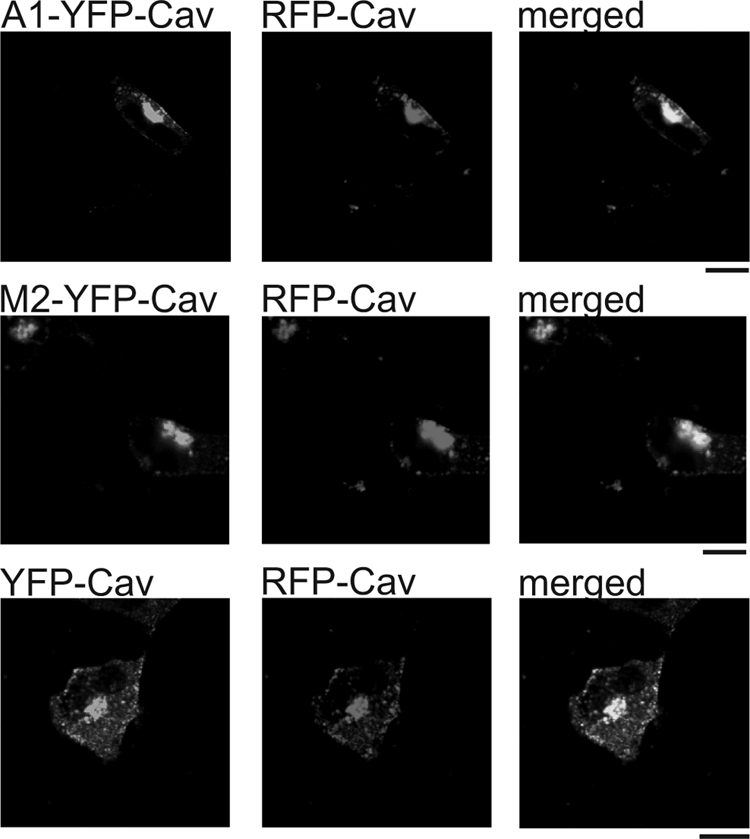
Confocal microscopy images of HEK293 cells expressing A1-YFP-Cav2, M2-YFP-Cav2, or YFP-Cav2 together with RFP-Cav2. Merged images showing co-localization of A1-YFP-Cav2 with RFP-Cav2, M2-YFP-Cav2 with RFP-Cav2 and as a positive control YFP-Cav2 with RFP-Cav2. Bar, 10 μm.
Functional Comparison of Channel Regulation by the Receptor Fusions
We asked whether there were any differences in signaling via M2-YFP and A1-YFP and their corresponding caveolin-2 fusions. The most revealing assay is one where we measure the kinetics of current activation of Kir3.1/Kir3.4 channels on agonist application and withdrawal (25–28). In addition, we measure the magnitude of the agonist induced current. To apply agonists we used a rapid and localized drug-perfusion system that enabled us to apply and remove agonists in under 250 ms. We observed an initial lag (lag) followed by a rapid rise to a peak amplitude of current (time to peak). With prolonged agonist application current amplitude wanes as the response desensitizes, and upon removal of agonist it deactivates back to baseline levels. Fig. 7 shows representative current recordings from the stable Kir3.1/Kir3.4 cell line transfected with M2, M2-YFP-Cav2, A1, and A1-YFP-Cav2. We measured the magnitude of the peak current in the presence of agonist, channel activation (lag+time to peak and τac) and deactivation rates (τdeac) as currents return to baseline after the removal of agonist. We also measured desensitization of the current in the presence of the agonist as a percentage of current decrease after 20 s of drug application. The mean data are shown in Table 1. Cells expressing the YFP-Cav2 receptor fusions show much slower activation kinetics compared with the cells expressing the YFP tagged receptors. Both parameters lag+time to peak as well as τac were significantly slower (Fig. 7 and Table 1) for both tested receptors. Inactivation kinetics of the current tended to be faster in the cells expressing the caveolar-targeted receptor, although data did not reach statistical significance. The GPCRs targeted to caveolae showed less desensitization compared with the receptors fused to YFP, although the M2-YFP-Cav2 data did not reach statistical significance (Table 1 and Fig. 7). These differences persisted even on transfection of lower M2-YFP cDNA (160 ng) concentrations that gives comparable membrane expression to the one achieved with the caveolar-targeted receptor (Table 1). However, when cells expressing the M2-YFP-Cav2, were incubated with 2 mm MβCD for 90 min at 37 °C, recovery of the lag+time to peak and the τac were obtained to levels equivalent to that measured after transfection of 160 ng of M2-YFP as well as to data measured after transfection of 160 ng of M2-YFP and MβCD incubation (Table 1).
FIGURE 7.

Receptors targeted to caveolae show slower GIRK channel activation kinetics. Representative current traces recorded from a HEK293 stable line expressing Kir3.1/Kir3.4 (GIRK1/4) transiently transfected with either M2 (upper left panel), M2-YFP-Cav2 (upper right panel), A1 (lower left panel), or A1-YFP-Cav2 (lower right panel). Agonists (carbachol and NECA, respectively) were applied for 20 s as indicated.
TABLE 1.
Characteristics of GIRK channel activation (HEK293 cells stably expressing Kir3.1/Kir3.4) with the various receptor constructs
Currents density and kinetics of activation and inactivation of the currents are summarized. Statistical analyses were performed using nonparametric tests. For the comparison of M2-YFP (low concentration) with M2-YFP (low concentration) treated with MβCD, M2-YFP-Cav2, and also with M2-YFP-Cav2 treated with MβCD a Kruskal-Wallis test was used with a Dunn post-hoc test. For the comparison of A1 and A1-YFP-Cav2 a Mann-Whitney test was used. ns, not significant; *, p < 0.05; **, p < 0.01.
| Receptor | I basal pA/pF | I agonist pA/pF | Lag+TTP | τac | % desensitization | τdeac |
|---|---|---|---|---|---|---|
| s | ms | s | ||||
| M2 (n = 6) | 27.6 ± 6.1 | 99.6 ± 19.9 | 1.26 ± 0.2 | 536 ± 114 | 24 ± 5.4 | 12.3 ± 2.4 |
| M2-YFP (n = 8) | 23.1 ± 3.6 | 93.8 ± 14.2 | 1.73 ± 0.2 | 638.7 ± 93.2 | 19.1 ± 4.5 | 31.2 ± 8.5 |
| M2-YFP low (n = 8) | 51.7 ± 10.3 | 47.6 ± 13.2 | 3.71 ± 0.6 | 1105 ± 182.8 | 19.5 ± 4.5 | 5 ± 0.7 |
| M2-YFP low MCD (n = 9) | 42.7 ± 7.6 | 54.9 ± 12.2 | 2.2 ± 0.2 | 736 ± 88.3 | 27.8 ± 3.2 | 8.4 ± 1.3 |
| M2-YFP-Cav2 (n = 11) | 40 ± 7.6 | 28.7 ± 6.5 | 13.7 ± 2.1* | 4504 ± 748* | 3.9 ± 1.9 | 6.2 ± 1.3 |
| M2-YFP-Cav2 MCD (n = 10) | 55.4 ± 9.5 | 22.2 ± 2.4 | 3.86 ± 0.54 | 1106 ± 142 | 7.05 ± 2.4 | 5.5 ± 0.7 |
| A1 (n = 6) | 19.4 ± 3.6 | 70.8 ± 14.5 | 0.79 ± 0.2 | 441 ± 94.2 | 43 ± 4.3 | 41.4 ± 9.4 |
| A1-YFP-Cav2 (n = 6) | 37.4 ± 5.6* | 45.9 ± 10.1 | 4.59 ± 1** | 3479 ± 746.5** | 21.6 ± 5.8* | 24.6 ± 4.7 |
DISCUSSION
The activation of G-protein-gated K+ channels is well studied in native and heterologous expression systems. There are numerous strands of experimental data that support a model in which agonist activation of a Gi/o-coupled receptor, such as the M2 muscarinic or the GABA-B receptor, leads to the release of free Gβγ from G-protein heterotrimers, which in turn binds to cytosolic domains on the Kir3.0 tetramer leading to channel activation (29–32). There are two issues with this model. Kir3.0 activation is relatively rapid given the number of steps that are required after agonist binding at the GPCR. Furthermore, activation is largely sensitive to pertussis toxin implicating inhibitory G-proteins (33) and does not occur through Gs-coupled GPCRs under normal circumstances (10, 34). One appealing model to explain both these observations would be if the components were scaffolded together in a macromolecular complex and there is emerging evidence that this may be the case (27, 35–39). However is it possible that there are other factors at work? In particular, cholesterol rich domains such as caveolae and rafts have been proposed to exist, and they are an appealing mechanism for potentially partitioning signaling generating selectivity and rapid kinetics. One condition that would have to be operative for such schemes to work is that all components of the transduction cascade would have to be resident in the same compartment. Using biochemical techniques our results show that the relevant GPCRs (M2 and A1) and the channel complex (Kir3.1/Kir3.4) are essentially excluded from caveolin-containing domains. Furthermore, the application of the receptor agonist carbachol does not alter the M2 distribution. It is worth noting that our biochemical approach using cold Triton insolubility will isolate both caveolae and rafts (11, 40). In contrast the inhibitory G-protein, specifically Giα1 and Giα2 as detected by the antibody, shows significant distribution both to noncaveolar/nonraft and caveolar/raft domains. This distribution also does not change dramatically with agonist application. Thus a plausible inference from these studies is that active signaling to the Kir3.0 channel involves sequential interactions outside of caveolae and rafts including that fraction of Giα1 and Giα2 resident there. The study of the distribution of Gsα revealed a further interesting difference in that this G-protein subunit seemed to reside predominantly if not solely in a caveolar/raft fraction. This is interesting in the context of the model above in that Gsα (and associated Gβγ in the resting condition) would be excluded from interacting with Kir3.0 transduction cascade and thus might contribute to Gi/Gs selectivity. It is also intriguing that some investigators have seen adenylate cyclase together with Gsα predominantly located in caveolae/rafts and suggested that this results in more efficacious signaling for the β1 adrenergic receptor (41, 42). Others groups have also shown a co-localization of the β2 adrenergic receptor with Cav-3 (43).
To investigate the consequences for signaling of caveolar localization we adopted a strategy to target G-protein-coupled receptors to these domains by fusing to caveolin-2 (24). This fusion dramatically changed the distribution of the receptor in the membrane from a diffuse and homogenous signal to a patchier one. In the case of the M2 receptor overall expression was not affected however there was less at the plasma membrane therefore a lower DNA concentration was transfected for M2 control experiments. It is known that caveolae internalize and form a unique intracellular vesicle population (44). Both the M2 and A1 receptor showed much slower activation kinetics after such fusion. In the case of the M2 receptor it was possible to control for both total and membrane expression and even in these circumstances this pattern persisted. One interpretation of these results is that sequestration of the receptor away from the channel and perhaps the G-protein accrues a kinetic penalty as now either the receptor or G-protein subunits must leave caveolae to engage with and activate the channel. The slower activation time possibly reflects transition of signaling components across this boundary. This idea was supported by the use of methyl-β-cyclodextrin: treatment of M2-YFP-Cav2 expressing cells led to an acceleration of the activation kinetics comparable to that of equivalent noncaveolar membrane expression of M2-YFP. It is worth noting that the biochemical assays reflect the steady-state distribution and do not address the kinetics of entry or exit from cholesterol rich domains for a noncaveolar/nonraft protein and visa versa. Our experimental results are in agreement with modeling studies showing that segregation of components in caveolae significantly slows signaling (45). We also noted that the degree of rapid desensitization was attenuated through these caveolin-2 receptor fusions. A number of alternative explanations have been advanced to explain the phenomenon of rapid desensitization (46, 47) however we favor the idea that it is related to the dynamics of the G-protein cycle (28). Fast robust activation leads to prominent desensitization whereas slower and less complete activation does not. The observation of slower activation with the caveolin-2 receptor fusions and thus as a result less prominent fast desensitization are consistent with this hypothesis.
There is a substantial body of work examining the distribution of GPCRs and G-proteins in caveolae and rafts and a hypothesis has emerged of caveolae as a universal regulator of signaling (44, 48). The detection of the G-protein Giα1/2 in both buoyant and heavy fractions is consistent with previous studies (49). Gsα seems to have a cell-specific distribution (48). One particularly intriguing idea is that there is a direct interaction between caveolin and the inactive G-protein and that caveolae act as cellular reserve for these proteins (50). Furthermore, it has also been suggested that the binding of activated Gα to caveolin may lead to desensitization of the cellular response of other receptors which are coupled to the same G-protein (51) and such mechanism may be a molecular mechanism to attenuate the signal strength (52). We were not able to see pronounced translocation of Giα1/2 to a heavy fraction on agonist stimulation. The distribution of GPCRs is more controversial and inconsistent between different laboratories though conditions, isolation methods, and specific tissues studied vary. For example, some investigators have identified M2 muscarinic receptors in caveolar fractions isolated from cardiac myocytes after carbachol incubation (53) while others suggest that the M2 receptor is internalized into clathrin-coated vesicles rather than in caveolae (54). Again further investigators have shown that the M2 muscarinic receptor undergoes an agonist-induced internalization through an unidentified pathway, neither clathrin-coated pits nor caveolae seem to be involved (55). It is worth stating that we used both antibody detection and radioligand binding to assay the distribution particularly for the M2 receptor. In addition, we saw clear receptor localization in buoyant fractions when fused to caveolin-2. As regards the distribution of K+ channels, in particular Kir3.0 in caveolae less is known. Kir3.1/Kir3.2 channels were detected in rafts corresponding fractions when expressed in CHO cells as well as in fractions prepared from mouse forebrain homogenates (56).
In summary, we provide evidence that the Kir3.0 transduction cascade is located outside caveolae and rafts and that this optimizes the kinetics of the response. The localization of Gsα, and perhaps other components such as adenylate cyclase, in caveolae and rafts may act as a mechanism for selectivity in that free Gβγ released from caveolae is much less efficacious in activating GIRK channels (located outside caveolae) compared with other effectors located within caveolae.
Acknowledgment
We thank Prof. William Claycomb for providing the HL-1 cell line.
This work was supported by the Wellcome Trust.
- GPCR
- G-protein-coupled receptor
- GIRK
- G-protein-gated inward rectifying K+ channel
- HEK
- human embryonic kidney
- MβCD
- methyl-β-cyclodextrin
- TIRF
- total internal reflection fluorescence
- DPCPX
- 8-[3H]cyclopentyl-1,3-dipropylxanthine
- QNB
- [3H]quinuclidinyl benzylate.
REFERENCES
- 1.Yamada M., Inanobe A., Kurachi Y. (1998) Pharmacol. Rev. 50, 723–760 [PubMed] [Google Scholar]
- 2.Kubo Y., Reuveny E., Slesinger P. A., Jan Y. N., Jan L. Y. (1993) Nature 364, 802–806 [DOI] [PubMed] [Google Scholar]
- 3.Dascal N., Lim N. F., Schreibmayer W., Wang W., Davidson N., Lester H. A. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 6596–6600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krapivinsky G., Gordon E. A., Wickman K., Velimirović B., Krapivinsky L., Clapham D. E. (1995) Nature 374, 135–141 [DOI] [PubMed] [Google Scholar]
- 5.Fleischmann B. K., Duan Y., Fan Y., Schoneberg T., Ehlich A., Lenka N., Viatchenko-Karpinski S., Pott L., Hescheler J., Fakler B. (2004) J. Clin. Invest. 114, 994–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drici M. D., Diochot S., Terrenoire C., Romey G., Lazdunski M. (2000) Br. J. Pharmacol. 131, 569–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wickman K., Nemec J., Gendler S. J., Clapham D. E. (1998) Neuron 20, 103–114 [DOI] [PubMed] [Google Scholar]
- 8.Kuzhikandathil E. V., Oxford G. S. (2000) J. Gen. Physiol. 115, 697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sodickson D. L., Bean B. P. (1996) J. Neurosci. 16, 6374–6385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leaney J. L., Milligan G., Tinker A. (2000) J. Biol. Chem. 275, 921–929 [DOI] [PubMed] [Google Scholar]
- 11.Thomas C. M., Smart E. J. (2008) J. Cell. Mol. Med. 12, 796–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown D. A., London E. (1998) Annu. Rev. Cell. Dev. Biol. 14, 111–136 [DOI] [PubMed] [Google Scholar]
- 13.Okamoto T., Schlegel A., Scherer P. E., Lisanti M. P. (1998) J. Biol. Chem. 273, 5419–5422 [DOI] [PubMed] [Google Scholar]
- 14.Scherer P. E., Okamoto T., Chun M., Nishimoto I., Lodish H. F., Lisanti M. P. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 131–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parton R. G. (1996) Curr. Opin. Cell. Biol. 8, 542–548 [DOI] [PubMed] [Google Scholar]
- 16.Couet J., Li S., Okamoto T., Ikezu T., Lisanti M. P. (1997) J. Biol. Chem. 272, 6525–6533 [DOI] [PubMed] [Google Scholar]
- 17.Rusinova R., Mirshahi T., Logothetis D. E. (2007) J. Biol. Chem. 282, 34019–34030 [DOI] [PubMed] [Google Scholar]
- 18.Smart E. J., Ying Y. S., Mineo C., Anderson R. G. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 10104–10108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Head B. P., Patel H. H., Roth D. M., Lai N. C., Niesman I. R., Farquhar M. G., Insel P. A. (2005) J. Biol. Chem. 280, 31036–31044 [DOI] [PubMed] [Google Scholar]
- 20.Fialka I., Pasquali C., Lottspeich F., Ahorn H., Huber L. A. (1997) Electrophoresis. 18, 2582–2590 [DOI] [PubMed] [Google Scholar]
- 21.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 22.Claycomb W. C., Lanson N. A., Jr., Stallworth B. S., Egeland D. B., Delcarpio J. B., Bahinski A., Izzo N. J., Jr. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 2979–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nobles M., Sebastian S., Tinker A. (2010) Pflugers Arch. 460, 99–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rimoldi V., Reversi A., Taverna E., Rosa P., Francolini M., Cassoni P., Parenti M., Chini B. (2003) Oncogene 22, 6054–6060 [DOI] [PubMed] [Google Scholar]
- 25.Benians A., Leaney J. L., Milligan G., Tinker A. (2003) J. Biol. Chem. 278, 10851–10858 [DOI] [PubMed] [Google Scholar]
- 26.Benians A., Leaney J. L., Tinker A. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 6239–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benians A., Nobles M., Hosny S., Tinker A. (2005) J. Biol. Chem. 280, 13383–13394 [DOI] [PubMed] [Google Scholar]
- 28.Leaney J. L., Benians A., Brown S., Nobles M., Kelly D., Tinker A. (2004) Am. J. Physiol. Cell. Physiol 287, 182–191 [DOI] [PubMed] [Google Scholar]
- 29.Soejima M., Noma A. (1984) Pflugers Arch. 400, 424–431 [DOI] [PubMed] [Google Scholar]
- 30.Logothetis D. E., Kurachi Y., Galper J., Neer E. J., Clapham D. E. (1987) Nature 325, 321–326 [DOI] [PubMed] [Google Scholar]
- 31.Reuveny E., Slesinger P. A., Inglese J., Morales J. M., Iñiguez Lluhi J. A., Lefkowitz R. J., Bourne H. R., Jan Y. N., Jan L. Y. (1994) Nature 370, 143–146 [DOI] [PubMed] [Google Scholar]
- 32.Sadja R., Alagem N., Reuveny E. (2003) Neuron 39, 9–12 [DOI] [PubMed] [Google Scholar]
- 33.Pfaffinger P. J., Martin J. M., Hunter D. D., Nathanson N. M., Hille B. (1985) Nature 317, 536–538 [DOI] [PubMed] [Google Scholar]
- 34.Wellner-Kienitz M. C., Bender K., Pott L. (2001) J. Biol. Chem. 276, 37347–37354 [DOI] [PubMed] [Google Scholar]
- 35.Clancy S. M., Fowler C. E., Finley M., Suen K. F., Arrabit C., Berton F., Kosaza T., Casey P. J., Slesinger P. A. (2005) Mol. Cell. Neurosci. 28, 375–389 [DOI] [PubMed] [Google Scholar]
- 36.Riven I., Iwanir S., Reuveny E. (2006) Neuron 51, 561–573 [DOI] [PubMed] [Google Scholar]
- 37.Nobles M., Benians A., Tinker A. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 18706–18711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peleg S., Varon D., Ivanina T., Dessauer C. W., Dascal N. (2002) Neuron 33, 87–99 [DOI] [PubMed] [Google Scholar]
- 39.Zhang Q., Pacheco M. A., Doupnik C. A. (2002) J. Physiol. 545, 355–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mercier I., Jasmin J. F., Pavlides S., Minetti C., Flomenberg N., Pestell R. G., Frank P. G., Sotgia F., Lisanti M. P. (2009) Lab. Invest. 89, 614–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ostrom R. S., Violin J. D., Coleman S., Insel P. A. (2000) Mol. Pharmacol. 57, 1075–1079 [PubMed] [Google Scholar]
- 42.Ostrom R. S., Gregorian C., Drenan R. M., Xiang Y., Regan J. W., Insel P. A. (2001) J. Biol. Chem. 276, 42063–42069 [DOI] [PubMed] [Google Scholar]
- 43.Gratton J. P., Bernatchez P., Sessa W. C. (2004) Circ. Res. 94, 1408–1417 [DOI] [PubMed] [Google Scholar]
- 44.Parton R. G., Simons K. (2007) Nat. Rev. Mol. Cell. Biol. 8, 185–194 [DOI] [PubMed] [Google Scholar]
- 45.Chen C. Y., King J. R. (2006) Bull. Math. Biol. 68, 863–888 [DOI] [PubMed] [Google Scholar]
- 46.Kobrinsky E., Mirshahi T., Zhang H., Jin T., Logothetis D. E. (2000) Nat. Cell. Biol. 2, 507–514 [DOI] [PubMed] [Google Scholar]
- 47.Bender K., Wellner-Kienitz M. C., Bösche L. I., Rinne A., Beckmann C., Pott L. (2004) J. Physiol. 561, 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel H. H., Murray F., Insel P. A. (2008) Annu. Rev. Pharmacol. Toxicol. 48, 359–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sargiacomo M., Sudol M., Tang Z., Lisanti M. P. (1993) J. Cell. Biol. 122, 789–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li S., Okamoto T., Chun M., Sargiacomo M., Casanova J. E., Hansen S. H., Nishimoto I., Lisanti M. P. (1995) J. Biol. Chem. 270, 15693–15701 [DOI] [PubMed] [Google Scholar]
- 51.Murthy K. S., Makhlouf G. M. (2000) J. Biol. Chem. 275, 30211–30219 [DOI] [PubMed] [Google Scholar]
- 52.Allen J. A., Yu J. Z., Dave R. H., Bhatnagar A., Roth B. L., Rasenick M. M. (2009) Mol. Pharmacol. 76, 1082–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feron O., Smith T. W., Michel T., Kelly R. A. (1997) J. Biol. Chem. 272, 17744–17748 [DOI] [PubMed] [Google Scholar]
- 54.Yamanushi T. T., Shui Z., Leach R. N., Dobrzynski H., Claydon T. W., Boyett M. R. (2007) Am. J. Physiol. Heart Circ. Physiol. 292, H1737–H1746 [DOI] [PubMed] [Google Scholar]
- 55.Roseberry A. G., Hosey M. M. (1999) J. Biol. Chem. 274, 33671–33676 [DOI] [PubMed] [Google Scholar]
- 56.Delling M., Wischmeyer E., Dityatev A., Sytnyk V., Veh R. W., Karschin A., Schachner M. (2002) J. Neurosci. 22, 7154–7164 [DOI] [PMC free article] [PubMed] [Google Scholar]