

Abstract
Mitochondrial reactive oxygen species (ROS) play an important role in both physiological cell signaling processes and numerous pathological states, including neurodegenerative disorders such as Parkinson disease. While mitochondria are considered the major cellular source of ROS, their role in ROS removal remains largely unknown. Using polarographic methods for real-time detection of steady-state H2O2 levels, we were able to quantitatively measure the contributions of potential systems toward H2O2 removal by brain mitochondria. Isolated rat brain mitochondria showed significant rates of exogenous H2O2 removal (9–12 nmol/min/mg of protein) in the presence of substrates, indicating a respiration-dependent process. Glutathione systems showed only minimal contributions: 25% decrease with glutathione reductase inhibition and no effect by glutathione peroxidase inhibition. In contrast, inhibitors of thioredoxin reductase, including auranofin and 1-chloro-2,4-dinitrobenzene, attenuated H2O2 removal rates in mitochondria by 80%. Furthermore, a 50% decrease in H2O2 removal was observed following oxidation of peroxiredoxin. Differential oxidation of glutathione or thioredoxin proteins by copper (II) or arsenite, respectively, provided further support for the thioredoxin/peroxiredoxin system as the major contributor to mitochondrial H2O2 removal. Inhibition of the thioredoxin system exacerbated mitochondrial H2O2 production by the redox cycling agent, paraquat. Additionally, decreases in H2O2 removal were observed in intact dopaminergic neurons with thioredoxin reductase inhibition, implicating this mechanism in whole cell systems. Therefore, in addition to their recognized role in ROS production, mitochondria also remove ROS. These findings implicate respiration- and thioredoxin-dependent ROS removal as a potentially important mitochondrial function that may contribute to physiological and pathological processes in the brain.
Keywords: Antioxidant, Brain, Mitochondria, Neurodegeneration, Parkinson Disease, Reactive Oxygen Species (ROS), Peroxiredoxin, Thioredoxin
Introduction
Since the discovery that electron leak and incomplete reduction of oxygen occurs in the respiration chain (1, 2), mitochondria have been considered a major contributor to cellular oxidative damage through the generation of reactive oxygen species (ROS),2 including superoxide (O2˙̄), H2O2, and hydroxyl radical (HO·). Mitochondrial ROS production has been implicated in numerous pathological processes, including the etiology of various acute and chronic neuronal disorders (3), as well as aging (4). More recent studies suggest that ROS, such as H2O2, also serve an important role in cell signaling pathways and thereby regulate a diverse set of physiological processes (5). Therefore, maintaining the delicate balance between pathological and physiological levels of H2O2 is critical for proper cell function and survival.
The biological significance of mitochondrial ROS are highlighted by targeted deletion or overexpression of antioxidant enzymes: 1) thioredoxin 2 (Trx2) knock-out mice present an embryonic lethal phenotype (6), 2) manganese superoxide dismutase knock-out mice typically die within 3 weeks after birth with severe neurodegeneration and mitochondrial oxidative damage (7, 8), and 3) lifespan is increased in transgenic mice overexpressing catalase targeted to mitochondria (9). The vast majority of studies related to ROS metabolism have focused on mitochondria as a source of ROS. Detoxification of mitochondrial ROS has largely focused on manganese superoxide dismutase, a critically important antioxidant enzyme that scavenges O2˙̄. Mitochondria also possess a multilevel network of both enzymatic and non-enzymatic antioxidant systems for the detoxification of H2O2. However, the one or more mechanisms and enzymatic systems involved in mitochondrial H2O2 detoxification are poorly understood. Rare attempts to address this issue have produced intriguing results that demonstrate the removal of exogenous ROS by actively respiring mitochondria (10, 11). The expression and contributions of enzymatic systems for H2O2 detoxification vary widely between tissues. Catalase, for example, is highly expressed in mitochondria from liver and heart (12, 13). Meanwhile, catalase in the brain is confined to peroxisomes, and there is little, if any, expression in mitochondria (14). The major enzymes believed to be responsible for H2O2 detoxification in the brain are peroxidase systems: GSH/glutathione peroxidase (GPx) and Trx2/peroxiredoxin 3 and 5 (Prx3 and Prx5). Zoccarato et al. (11) first demonstrated that brain mitochondria removed exogenously added H2O2 in a respiration-dependent manner, implicating GPx as the major enzymatic pathway in the process. However, a quantitative analysis to determine the involvement of potential enzymatic pathways and particularly the role of the Trx/Prx system in mitochondrial H2O2 detoxification remains to be examined. Identifying the enzymatic pathways by which mitochondrial H2O2 detoxification occurs is critical given the important physiological and pathological roles of H2O2.
In this study, we used a novel, polarographic method to quantitatively measure the ability of mitochondria to remove exogenously added H2O2. Because H2O2 is freely permeable to cell membranes, this method of addition was hypothesized to reflect mitochondrial metabolism of H2O2 arising from various cellular sources, both intra- and extramitochondrial. Here, we demonstrate that rat brain mitochondria remove H2O2 in a unique respiration-dependent manner primarily via the Trx/Prx system.
EXPERIMENTAL PROCEDURES
Chemical Reagents
Auranofin (S-triethylphosphinegold (I)-2,3,4,6-tetra-O-acetyl-1-thio-β-d-glucopyranoside) was obtained from Alexis Biochemicals (San Diego, CA). All other chemicals unless otherwise noted were obtained from Sigma. Cell culture reagents were obtained from Invitrogen (Carlsbad, CA). N27 cells were obtained from Drs. Kedar Prasad and Wenbo Zhou at the University of Colorado Denver.
Isolation of Purified Rat Brain and Liver Mitochondria
Brain mitochondria were isolated from male Sprague-Dawley rats (2–3 months old) using Percoll density gradient centrifugation (15) with slight modification (16). For liver-derived mitochondria, crude mitochondrial fractions were prepared (13), then applied to Percoll density gradient centrifugation as described for brain mitochondria for further purification. Purity and viability of mitochondrial fractions were assessed via Western blot analysis and oxygen consumption rates, respectively (17). Coupled mitochondria with respiratory control ratios greater than 5 were used in all experiments. For assays of enzymatic activity, isolated mitochondria were subjected to three freeze-thaw cycles and centrifuged at 8000 × g for 15 min at 4 °C to obtain supernatant. At least three independent mitochondrial preparations were used in all experiments.
Cell Culture
The T-antigen-immortalized N27 cell line described previously (18) was maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (v/v), penicillin (100 units/ml), streptomycin (100 μg/ml), and 2 mm l-glutamine at 37 °C in a 5% CO2 humidified atmosphere.
Polarographic Measurement of Exogenous H2O2 Removal
Mitochondrial H2O2 removal was measured using an Apollo 4000 Free Radical Analyzer equipped with a 100-μm Clark-type H2O2 electrode (World Precision Instruments, Inc., Sarasota, FL). Mitochondria (0.1 mg/ml) were incubated in an open, thermostatted chamber at 30 °C in incubation buffer (100 mm KCl, 75 mm mannitol, 25 mm sucrose, 10 mm Tris-HCl, 10 mm KH2PO4, 50 μm EDTA, and 600 μm MgCl2, pH 7.4). After obtaining a stable signal baseline, 2–3 μm (except where indicated) H2O2 was added exogenously, followed by the reagent or inhibitor under study (titrated to achieve maximal response), isolated mitochondria, and lastly respiration substrates (2.5 mm malate plus 10 mm glutamate, or 10 mm succinate) at 1-min intervals (see Fig. 1). This 1-min interval was necessary to allow the polarographic signal to stabilize between additions and achieve accurate measurements. H2O2 removal rates were calculated based on the linear signal decay for 1–2 min following the addition of substrates. Values were converted to nanomoles of H2O2/min/mg of protein using a predetermined H2O2 standard curve. The addition of some reagents/inhibitors to the incubation buffer caused spiking or baseline shifts in signal current that were typically attributed to minute differences in pH or temperature. Such changes were taken into consideration when calculating removal rates. The addition of exogenous catalase (40 units/ml) caused a rapid and complete decrease in signal to initial baseline levels, whereas superoxide dismutase (500 units/ml) had no effect (data not shown). This demonstrated that the electrode was specific for H2O2 and not other species, namely O2˙̄.
FIGURE 1.
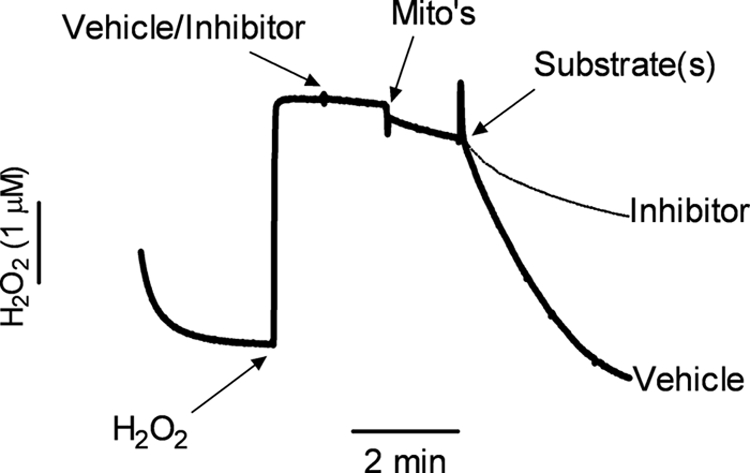
Representative polarographic traces of mitochondrial H2O2 removal. Exogenous H2O2 (3 μm) was added following baseline stabilization of the H2O2 electrode in incubation buffer. Subsequent additions were as follows: vehicle/inhibitor under study, mitochondria (Mito's, 0.1 mg/ml), and respiration substrate (malate (2.5 mm)/glutamate (10 mm), or succinate (10 mm)). Typical traces in the presence of vehicle (DMSO) or inhibitor (auranofin (1 μm)) are shown. H2O2 decay/removal rates were determined following the addition of substrate and adjusted to baseline rates prior to addition.
To decrease oxygen (O2) tension in the system for select experiments, nitrogen (N2) gas was bubbled through a side-port into the open, thermostatted incubation chamber. O2 levels were measured using a 2-mm Clark-type O2 electrode (WPI, Inc.). Using this method, experiments involving low O2 tension were conducted in ∼2% O2. During these experiments, O2 and H2O2 levels were measured simultaneously to ensure that decreased O2 levels were maintained throughout.
N27 cells were collected via trypsin and resuspended in Dulbecco's phosphate buffered saline (D-PBS) containing 1000 mg/liter d-glucose and 36 mg/liter sodium pyruvate. Following stabilization of H2O2 electrode in D-PBS, 1 × 106 N27 cells were added, followed by vehicle (DMSO) or any inhibitor under study. 3 μm exogenous H2O2 was added last, and removal rates were measured as described above.
Measurement of Thioredoxin Reductase Activity
Thioredoxin reductase (TrxR) activity was measured using an insulin-reduction assay in the presence of Escherichia coli recombinant thioredoxin (19). Reduced thiols were measured using 5,5′-dithio-bis(2-nitrobenzoic acid) (Ellman's reagent) at an absorbance of 412 nm on a Versamax microplate reader (Molecular Devices, Sunnyvale, CA).
Measurement of Coupled Reductase/Peroxidase Activity
The coupled activities of glutathione reductase (GR)/GPx or TrxR/Prx were assessed by following the decrease of NADPH absorbance at 340 nm (20, 21). The reaction was supplemented with glutathione (GSH, 2 mm) or E. coli recombinant Trx (5 μm), respectively, and initiated by the addition of H2O2 (500 μm).
Fluorometric Measurement of Mitochondrial H2O2 Production
Mitochondrial ROS production was assessed using a fluorometric method. Extramitochondrial release of H2O2 from isolated mitochondria (0.1 mg/ml) incubated with PQ and/or auranofin was measured using horseradish peroxidase-linked Amplex Red fluorescence (Invitrogen, Carlsbad, CA) (22).
Statistics
Statistical analysis was performed using Prism 5.0 (GraphPad Inc., San Diego, CA).
RESULTS
Measurement/Characterization of Brain Mitochondrial H2O2 Removal via Polarography
A polarographic method that quantitatively measures steady-state H2O2 levels in real-time was utilized to determine whether exogenous H2O2 removal occurs in isolated brain mitochondria under various respiration states and conditions. The results are summarized in Table 1. Fig. 1 shows a representative polarographic trace obtained during the measurement of exogenous H2O2 removal in brain mitochondria in the presence of vehicle (thick line) or inhibitor (thin line). Under resting conditions with no added substrates (state 1), brain mitochondria consumed H2O2 at a slow rate, which increased 10-fold with the addition of respiration substrates (malate plus glutamate; state 2 conditions). Heat inactivation (90 °C for 2 min) completely abolished H2O2 removal by respiring mitochondria, whereas the state 1 rate was unaffected. This indicates that a small proportion of H2O2 removal in brain mitochondria occurs via non-enzymatic scavenging. Highest rates of removal occurred upon addition of malate/glutamate, which feeds electrons to Complex I (NADH dehydrogenase) of the respiratory chain. The presence of ADP (state 3 conditions) caused a slight increase in malate/glutamate-dependent removal rates. H2O2 removal under succinate-supported respiration was slightly decreased compared with malate/glutamate. Succinate, which feeds electrons to the respiratory chain via Complex II (succinate dehydrogenase), also produces H2O2 via reverse electron transport through Complex I (23). Using rotenone as a Complex I inhibitor to block this effect caused an increase in H2O2 removal rate (9.7 ± 0.5 versus 8.3 ± 0.7; p < 0.05). These data suggest that intrinsic succinate-supported H2O2 production may compete with exogenous H2O2 in these experiments and lead to lower observed rates of net H2O2 removal. Addition of an uncoupler, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, 1 μm), significantly decreased malate/glutamate-stimulated removal rates by 20%. Further exploration into the role of mitochondrial bioenergetics revealed small, but insignificant decreases (∼5–15%) in H2O2 removal rates following inhibition of the respiratory chain or TCA cycle enzymes, such as aconitase, isocitrate dehydrogenase, and malate dehydrogenase (data not shown). With some minor exceptions, these results agree with previous studies examining the active, enzymatic removal of H2O2 in respiring mitochondria using a fluorometric method (11).
TABLE 1.
H2O2 removal by brain mitochondria under varying respiration states and conditions
Rates of H2O2 removal by isolated brain mitochondria (0.1 mg/ml) in incubation buffer with H2O2 (3 μm) under the indicated conditions. Low O2 conditions were performed under ∼2% O2 levels using N2 gas bubbled into the incubation chamber. Rates are expressed as mean ± S.E. (n = 3–6).
| Substrate/conditions (respiration state) | H2O2 removal |
|---|---|
| nmol/min/mg protein | |
| None (state 1) | 1.0 ± 0.2 |
| Low O2, none | 1.2 ± 0.4 |
| Heat-inactivated mitochondria, none | 0.8 ± 0.04 |
| Malate/glutamate (state 2) | 11.1 ± 0.2 |
| Low O2, malate/glutamate | 7.0 ± 1.3 |
| Heat-inactivated mitochondria + malate/glutamate | 1.7 ± 0.2 |
| Malate/glutamate + ADP (state 3) | 11.8 ± 0.2 |
| Malate/glutamate + FCCP (uncoupled) | 9.0 ± 0.7 |
| Succinate | 8.3 ± 0.7 |
| Succinate + rotenone | 9.7 ± 0.5 |
To determine if similar responses occurred at biologically relevant O2 concentrations encountered by mitochondria, N2 gas was utilized to decrease O2 tension to ∼2% in the system for subsequent experiments. Under these low O2 conditions, state 1 H2O2 removal rates were unchanged. Malate/glutamate stimulation caused a significant increase in rates (7.0 ± 1.3 versus 1.2 ± 0.4; p < 0.05), indicating that respiration-dependent H2O2 removal occurs in brain mitochondria under these more physiological O2 conditions. However, state 2 rates were decreased by 37% compared with brain mitochondria under atmospheric (∼21%) O2 conditions (Table 1).
Identification of Enzymatic Systems Contributing to Brain Mitochondrial H2O2 Removal
The generation of mitochondrial H2O2 is thought to primarily occur through a combination of spontaneous and superoxide dismutase-catalyzed dismutation of O2˙̄ produced in the matrix, intermembrane space, and outer membrane (24). As a result, mitochondria possess multiple enzymatic systems for the detoxification of H2O2 that vary widely between different tissues. Because brain mitochondria are not believed to express catalase, GSH- and/or Trx-based peroxidases are anticipated to act as the major H2O2 detoxification system(s). Each of these enzymatic systems is dependent upon the action of a peroxidase (GPx or Prx) to directly react with H2O2, together with substrate (GSH or Trx), and a reductase (GR or TrxR) that uses NADPH to maintain these proteins in a reduced state. Pharmacological inhibition was used to assess the contributions of these mitochondrial systems toward H2O2 removal in the brain. As expected, inhibition of catalase via aminotriazole had no effect on mitochondrial H2O2 removal (Fig. 2). GPx inhibition or GSH oxidation via malathion or diamide, respectively, showed minimal changes in removal rates. Inhibition of GR by carmustine decreased H2O2 consumption by 25%, whereas concomitant inhibition of GR and GPx had no further effect. In contrast, inhibition of the Trx/Prx system potently and dramatically attenuated brain mitochondrial H2O2 removal. Specifically, inhibition of TrxR with auranofin proved the most effective by attenuating H2O2 removal by 80%. Additionally, rapid oxidation of Prx3 by phenethyl isothiocyanate (25) caused a 50% decrease in removal rate. These results suggest that the Trx/Prx system is the major contributor to net H2O2 removal in brain mitochondria, whereas GSH/GPx plays a lesser role.
FIGURE 2.

Pharmacological inhibition of mitochondrial H2O2 removal. Mitochondria were incubated in incubation buffer with H2O2 (3 μm) under malate/glutamate-supported conditions. Inhibitors were added where indicated in Fig. 1 as follows: aminotriazole (2 mm, catalase (Cat)), malathion (100 μm, GPx), diamide (50 μm, GSH oxidation), carmustine (100 μm, GR), auranofin (1 μm, TrxR), or phenethyl isothiocyanate (1 mm, Prx). H2O2 removal rates were measured following the addition of malate/glutamate and adjusted to baseline rates with each inhibitor. H2O2 removal rates are expressed mean ± S.E. (n = 3–6). Effects of each inhibitor are also shown as percent change from vehicle (DMSO) control as mean (n = 3–6). *, p < 0.05 versus vehicle (DMSO) control (one-way analysis of variance).
Because the greatest attenuation of brain mitochondrial H2O2 removal occurred with inhibition of TrxR by auranofin, rather than the GSH/GPx pathway previously identified for this process (11), we explored its specificity for these two peroxidase systems. Auranofin-dependent inhibition was tested using an enzymatic assay that measures the coupled reductase and peroxidase activities in response to H2O2. At 1 μm, a concentration with maximal effects on mitochondrial H2O2 removal, auranofin significantly inhibited TrxR/Prx activity with no effect on GR/GPx (Fig. 3A), indicating specificity for the former peroxidase system. We next wanted to confirm that changes in mitochondrial H2O2 removal correlated with TrxR inhibition. Plotting concentration-response curves of the ability of auranofin to attenuate H2O2 removal and inhibit TrxR activity revealed that the two processes closely correlated (Fig. 3B). 1-Chloro-2,4-dinitrobenzene (DNCB), another specific inhibitor of TrxR with a unique mechanism of action to that of auranofin (26), also significantly decreased H2O2 removal. In a manner similar to auranofin, the effects of DNCB on H2O2 removal correlated with inhibition of TrxR activity (Fig. 3B), although DNCB showed greater maximal effects on H2O2 removal. With nearly complete inhibition of TrxR, DNCB caused over 90% inhibition of H2O2 removal, compared with 80% with auranofin. This difference likely arises from DNCB-dependent induction of O2˙̄ generative activity of TrxR at high concentrations (26), as opposed to off-target effects on other H2O2 removal systems (i.e. GSH or non-enzymatic pathways). Collectively, these results demonstrate that observed decreases in brain mitochondrial H2O2 removal rates are the consequence of specific inhibition of the Trx/Prx system, namely TrxR. We can also conclude that TrxR-dependent pathways account for up to 80% of H2O2 removal in brain mitochondria.
FIGURE 3.

Decreased mitochondrial H2O2 removal correlates with TrxR activity inhibition. A, auranofin specificity was measured using coupled activities of TrxR/Prx or GR/GPx in mitochondrial preparations in the presence or absence of auranofin (1 μm). Activity (units/g of protein) is expressed as mean ± S.E. (n = 3). *, p < 0.05 versus vehicle (DMSO) control (one-way analysis of variance). B, H2O2 removal and TrxR activities were measured as described under “Experimental Procedures” with increasing concentrations of auranofin or DNCB. Data are expressed as mean ± S.E. (n = 3).
Effect of Metal Ions on Mitochondrial H2O2 Removal
The absence of EDTA from the incubation buffer decreases H2O2 removal rate to non-enzymatic levels (data not shown), indicating that trace contamination by metals ions prevents the enzymatic scavenging of H2O2 by brain mitochondria. Therefore, changes in mitochondrial H2O2 removal rates were measured in the presence of various metal compounds. Neuronal mitochondria have shown the ability to accumulate, store, and release Ca2+ but are also susceptible to disruptions of Ca2+ handling in pathological conditions such as excitotoxicity, ischemic injury, and neurodegeneration. Excessive Ca2+ levels can also lead to perturbation of mitochondrial membrane potential and opening of the permeability transition pore (27). As expected, Ca2+ inhibited mitochondrial H2O2 removal in a dose-dependent manner, with an IC50 value of 42 μm. Because the effects of Ca2+ on mitochondrial function are widespread with significant changes in the activities of multiple enzymes, including peroxidases, arsenic and copper were used as alternative metals to determine the relative contribution of the Trx/Prx and GSH/GPx systems.
As shown in Fig. 4A, sodium arsenite attenuated mitochondrial H2O2 removal by ∼25%. Arsenite also produced significant inhibition of TrxR/Prx activity (75%), whereas GR/GPx activity was unaffected. Copper also decreased rates of H2O2 removal (Fig. 4B). CuCl2 (50 μm) resulted in ∼75 and ∼50% decreases in GR/GPx and TrxR/Prx activities, respectively, and attenuated H2O2 removal rates by ∼25% compared with controls. Interestingly, increasing CuCl2 concentrations to 100 μm caused further decrease of H2O2 removal to only 15% of control rates. This decrease in H2O2 removal rates with increasing CuCl2 from 50 μm to 100 μm was associated with a further inhibition of TrxR/Prx activity to ∼25% compared with controls. However, GR/GPx activity showed no further decreases in response to 100 μm CuCl2. These results demonstrate that inhibition of H2O2 removal rates by metal ions occurs in association with decreased TrxR/Prx activity, providing further support for the thioredoxin system as the major contributor to mitochondrial H2O2 removal.
FIGURE 4.

Effect of arsenite or copper on mitochondrial H2O2 removal and reductase/peroxidase activity. Mitochondrial H2O2 removal (■, solid line), GR/GPx activity (▴, dashed line), and TrxR/Prx activity (▾, dotted line) were measured as described under “Experimental Procedures” in the presence of arsenite, NaAsO2 (A), or copper, CuCl2 (B). Data are expressed as percent control for each parameter in mean ± S.E. (n = 3).
Concentration Response of Brain Mitochondrial H2O2 Removal Mechanisms
Estimates for the physiological concentration of H2O2 in fluid and tissues vary from nanomolar to low micromolar levels (28, 29). Therefore, a kinetic analysis was utilized to determine if similar mechanisms contribute to brain mitochondrial H2O2 removal throughout this range. Using initial concentrations of exogenous H2O2 from 0.25 μm (which represents the lower limit of accurate detection for the polarographic methods employed here) to 3 μm, brain mitochondria supported with malate/glutamate showed concentration-dependent rates of H2O2 removal, as increased rates were observed with higher initial concentrations of exogenous H2O2 (Fig. 5). Furthermore, these rates were significantly decreased in the presence of auranofin by 50–90%. These results indicate that respiration- and Trx-dependent mechanisms of H2O2 removal in brain mitochondria occur over a range of physiologically relevant H2O2 concentrations.
FIGURE 5.
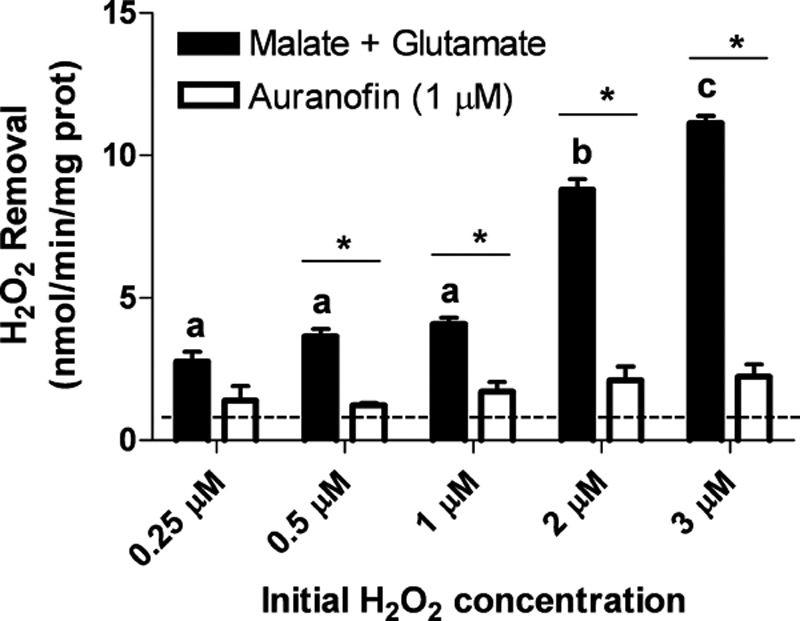
Trx-dependent H2O2 removal by mitochondria occurs in a concentration-dependent manner. H2O2 removal rates were measured in malate/glutamate-supported mitochondria with varying initial concentrations of exogenous H2O2 (0.25–3 μm). Auranofin was added at a final concentration of 1 μm to inhibit TrxR (open bars). The dashed line represents the baseline rate of H2O2 removal by unstimulated mitochondria. H2O2 removal rates are expressed as mean ± S.E. (n = 3). Bars with different letters represent significant differences between H2O2 removal rates of malate/glutamate-stimulated mitochondria (without auranofin) under varying H2O2 concentrations only. *, p < 0.05 between controls and auranofin treatment groups for each individual H2O2 concentration (one-way analysis of variance).
Mechanisms of H2O2 Removal in Liver-derived Mitochondria
To determine if the observed characteristics of H2O2 removal in brain mitochondria were applicable to other organs, mitochondria were also prepared from rat liver and subjected to similar experimental conditions (Table 2). Interestingly, liver-derived mitochondria showed nearly 5-fold higher maximal rates of H2O2 removal compared with those from brain. Additionally, these rates occured independent of the presence of malate/glutamate, suggesting that liver mitochondria do not depend upon a respiration-driven process to detoxify H2O2. This was confirmed by pharmacological studies, which showed that inhibition of Trx/Prx or GR/GPx systems via auranofin or carmustine, respectively, did not affect H2O2 removal rates. Instead, a 50% decrease in rates was seen with aminotriazole, confirming the role of catalase in the oxidative stress defense of liver mitochondria (13). These results suggest that respiration- and Trx/Prx-dependent mechanisms of H2O2 removal may be unique to the brain.
TABLE 2.
H2O2 removal rates by rat liver mitochondria
H2O2 removal rates were measured in liver mitochondria under the same conditions as brain. Rates of H2O2 removal by mitochondrial samples (0.1 mg/ml) in incubation buffer with H2O2 (3 μm) with respiration substrates and/or inhibitors as indicated. Rates are expressed as mean ± S.E. (n = 3–6).
| Substrate/conditions (respiration state) | H2O2 removal |
|---|---|
| nmol/min/mg protein | |
| No substrates (state 1) | 43.4 ± 5.7 |
| Malate/glutamate (state 2) | 49.5 ± 6.1 |
| Malate/glutamate + aminotriazole | 23.3 ± 4.0* |
| Malate/glutamate + auranofin | 45.1 ± 4.8 |
| Malate/glutamate + carmustine | 37.8 ± 8.1 |
*, p < 0.05 versus malate/glutamate (state 2) conditions (one-way analysis of variance).
Effect of TrxR Inhibition on Mitochondrial H2O2 Production
To examine the functional consequences of TrxR inhibition, we measured the net rate of H2O2 production from mitochondria following the addition of paraquat (PQ), a neurotoxicant linked with environmental causes of parkinsonism (30, 31). We have previously shown that mitochondria are a major source of PQ-induced H2O2 production in the brain (16). In contrast with the use of polarography to measure H2O2 removal in previous experiments, we utilized a well established fluorometric method to measure net H2O2 production from mitochondria. Auranofin (1 μm) alone caused a small but insignificant increase in net H2O2 production in control mitochondria supported with malate/glutamate (Fig. 6). PQ alone increased rates of H2O2 production, significantly at 10 μm (Fig. 6, open bars). Co-treatment with auranofin greatly exacerbated PQ-induced rates of H2O2 production by ∼5-fold (Fig. 6, closed bars). By undermining mitochondrial H2O2 removal systems, these results demonstrate the implications of TrxR disruption in susceptibility to environmental neurotoxicants.
FIGURE 6.
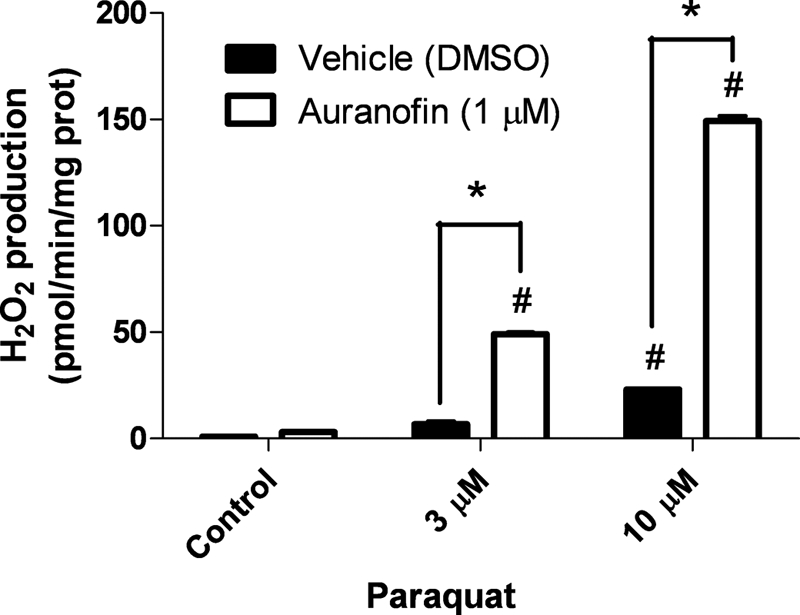
TrxR inhibition via auranofin exacerbates PQ-induced mitochondrial H2O2 production. H2O2 production was measured in malate/glutamate-supported isolated mitochondria with PQ via HRP-linked Amplex Red oxidation. Auranofin was added as indicated at 1 μm final concentration. H2O2 production rates are expressed as mean ± S.E. (n = 3–4). *, p < 0.05 versus vehicle (DMSO) control at same PQ concentration. #, p < 0.05 versus group control (one-way analysis of variance).
H2O2 Removal by Intact Dopaminergic Cells
To determine the biological relevance of H2O2 removal observed in isolated mitochondria, N27 dopaminergic cells were used to assess contributions of the Trx/Prx systems in a cell-based system. In D-PBS buffer containing glucose and pyruvate, N27 cells showed low rates of H2O2 removal (Fig. 7). In comparison with other common substrates (i.e. malate/glutamate, l-glutamine, and succinate), glucose and pyruvate supplementation caused maximal rates of H2O2 removal in N27 cells (data not shown). The addition of auranofin caused a significant 36% decrease in H2O2 removal by N27 cells (Fig. 7B). Furthermore, cellular TrxR activity was nearly completely abolished (95% decrease) with auranofin treatment. These data demonstrate that intact dopaminergic cells remove H2O2 in part via an auranofin-sensitive mechanism.
FIGURE 7.

H2O2 removal by N27 dopaminergic cells. A, representative polarographic traces of H2O2 removal in N27 dopaminergic cells. Cells were suspended in the incubation chamber with D-PBS containing glucose and pyruvate in the presence of vehicle (DMSO) or auranofin (500 nm). Exogenous H2O2 (3 μm) was added following baseline stabilization of the electrode. B, H2O2 removal rates in N27 cells in the presence of vehicle (DMSO) or auranofin (500 nm). H2O2 removal rates are expressed as mean ± S.E. (n = 3). *, p < 0.05 versus vehicle (DMSO) (unpaired t test).
DISCUSSION
Using a novel polarographic method in this study, we were able to assess an overlooked aspect of ROS metabolism, the ability of brain mitochondria to remove H2O2 under physiological conditions. In addition to examining the detailed mechanisms of H2O2 removal, we also measured the contributions of mitochondrial systems to H2O2 removal in the brain. Four major findings emerge from this work: 1) brain mitochondria are capable of scavenging exogenous H2O2 under biologically relevant conditions in a respiration-dependent manner, suggesting important roles for this organelle in antioxidant defense against both intra- and extra-mitochondrial ROS sources and as a regulator of cell signaling and redox-dependent processes; 2) in contrast to previous literature, we demonstrate that Trx/Prx is the major contributing enzyme system to respiration-dependent H2O2 removal in brain mitochondria, whereas GSH/GPx and non-enzymatic systems show only minor contributions; 3) inhibition of Trx-dependent antioxidant systems exacerbates H2O2 production by PQ, an environmental neurotoxicant; and 4) partial Trx-dependent removal of H2O2 occurs in intact dopaminergic neurons.
As a result of the overwhelming focus of studies in the literature on mitochondria as a source of H2O2 production, a multitude of reagents and techniques have developed to measure this aspect of ROS metabolism. Although mitochondrial metabolism of O2 and nitric oxide (NO) has been extensively studied, mitochondrial H2O2 removal has received little attention and remains a challenge to researchers. A limited number of studies have adapted fluorometric methods to assess H2O2 removal in mitochondrial systems (11, 13, 32). However, to our knowledge this is the first demonstration of the use of a polarographic method to quantitatively measure H2O2 removal by mitochondria in real time. In contrast to fluorometric methods, polarography does not require the addition of reagents (i.e. fluorescent probe, HRP) that may alter the system by generating artificial reactions that do not normally occur in the cellular environment. Polarography is also capable of measuring steady-state, dynamic changes in net H2O2 resulting from both production and/or removal in real-time (22), whereas fluorometry is typically limited to the measurement of a single aspect of H2O2 metabolism. In our studies, polarography revealed nearly 2-fold higher rates in the removal of H2O2 by respiring brain mitochondria in comparison with rates measured previously by fluorometric methods at similar initial H2O2 concentrations (state 2 = 11.1 versus 6.7 nmol/min/mg of protein, respectively (11)). This increased sensitivity likely results because polarography relies on the diffusion of H2O2 across a permeable membrane, whereas fluorometric detection is dependent on the reaction of the molecular probe with H2O2. As a result, fluorometry may show lower sensitivity as the probe competes with intrinsic removal systems (i.e. Trx/Prx) for reaction with H2O2. In sum, the polarographic method described here represents a reliable and sensitive means to quantitatively measure H2O2 removal in a physiological system.
The demonstration that H2O2 removal by brain mitochondria was respiration-dependent confirms previous work by Zoccarato et al. (11). In addition, our results show that respiration-driven enzymatic processes account for the vast majority (∼90%) of net H2O2 removal. However, as results from heat-inactivation experiments show, non-enzymatic processes also participate in H2O2 scavenging to a small degree (∼10%). Redox compounds contributing to non-enzymatic H2O2 removal may include GSH, NADH, or NADPH, tocopherols, ascorbic acid, and ubiquinone or cytochrome c of the respiratory chain. Furthermore, these same H2O2 detoxification mechanisms, including Trx/Prx dependence, were confirmed under conditions to simulate a biologically relevant environment using decreased O2 tension and a range of H2O2 concentrations.
The existence and dependence of brain mitochondria on linked respiration-supported and enzymatic mechanisms of H2O2 removal is intriguing. In contrast, H2O2 removal by liver-derived mitochondria did not display the same characteristics. The differences between H2O2 removal mechanisms in mitochondria from brain and liver may be related to the presence of catalase. This efficient and high capacity enzyme has been identified in mitochondria from liver (13), but not brain (14), which is consistent with the pharmacological inhibition we observed with aminotriazole (Fig. 2 and Table 2). The basis of the differences in H2O2 removal mechanisms in mitochondria from brain and other organs warrants further investigation.
The data in Table 1 show that supplementation with substrates and heat-inactivation experiments identify a role for both respiration- and enzymatic-dependent activities in mitochondrial H2O2 removal, respectively. Interestingly, we found that neither uncoupling via carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) or TCA cycle disruption nor respiratory chain inhibition individually attenuates mitochondrial H2O2 removal rates by >20%, indicating that enzymatic-dependent removal does not require actively respiring mitochondria. We speculate that H2O2 removal in brain mitochondria is highly dependent on the availability of reducing equivalents, particularly NADPH. The mitochondrial pool of NADPH is maintained in a reduced state by several systems, including mitochondrial membrane potential (ΔΨm)-dependent transhydrogenase and enzymes of the TCA cycle, notably NADP+-linked isocitrate dehydrogenase 2. Therefore, it appears that enzymatic-dependent H2O2 removal in brain mitochondria is acutely maintained by multiple systems capable of preserving the necessary reducing equivalents independent of respiration.
In the characterization of respiration-dependent H2O2 removal in brain mitochondria, Zoccarato et al. (11) concluded that the GR/GPx system accounted for the majority of mitochondrial peroxidase activity. Surprisingly, we found that Trx/Prx was the major peroxidase system involved in the respiration-dependent H2O2 removal process, whereas GSH/GPx showed only minimal contributions. Pharmacological inhibition of TrxR showed the greatest attenuation of brain mitochondrial H2O2 removal, which correlated with decreases in enzyme activity in a dose-dependent manner. It is important to recognize that previous work has demonstrated that auranofin-dependent TrxR inhibition does not stimulate the production of H2O2 from mitochondria, which could be a potential artifact during measurement of H2O2 removal. Additionally, we confirmed previous reports that auranofin-dependent inhibition is specific to the TrxR/Prx system with no effect on GR/GPx (33). It is interesting that TrxR plays such a critical role in regulating the activity of Prx, although the reductase lies upstream of the direct reaction with H2O2. Because TrxR is responsible for reducing Trx, and indirectly Prx, to their active states for H2O2 detoxification, these data highlight the critical step of maintaining and utilizing reducing equivalents (i.e. NADPH) in the mitochondria to drive H2O2 removal.
GSH and Trx are differentially oxidized by metal ions (34), a finding that we used to further examine the contributions of peroxidase systems to mitochondrial H2O2 removal in the brain. Zoccarato et al. concluded that brain mitochondrial H2O2 removal is largely dependent on GR/GPx based on results showing that Ca2+ could significantly inhibit H2O2 removal by intact mitochondria as well as the enzyme activities of GR and GPx in disrupted mitochondria (11). However, Ca2+ can also inhibit the activity of TrxR (35). As expected, Ca2+ significantly decreased mitochondrial H2O2 removal in our experiments, but we chose to utilize arsenite and copper to further explore the involvement of Trx- and GSH-based peroxidases. Arsenite showed specific inhibition of the TrxR/Prx system with no effect on GR/GPx peroxidase activity. With such drastic inhibition of TrxR/Prx, it was expected that arsenite should have greater effects on mitochondrial H2O2 removal than we observed (25% decrease compared with controls). However, the addition of arsenite caused an artificial change in baseline of the polarographic trace, which likely diminished actual measured rates of H2O2 removal. On the other hand, copper inhibited the peroxidase activity of both GR/GPx and TrxR/Prx, which consequently affected H2O2 removal. However, increasing copper concentrations to 100 μm showed further inhibition of TrxR/Prx activity and H2O2 removal while GR/GPx activity remained depressed to similar levels. The inhibition of H2O2 removal by metal ions correlated with inhibition of TrxR/Prx, providing further support for the antioxidant role of this system in brain mitochondria.
Taking all the experiments performed here into consideration, we estimate the following contributions toward H2O2 removal in brain mitochondria: non-enzymatic scavenging, 10%; GR/GSH/GPx, 10–20%; and TrxR/Trx/Prx, 70–80% (Scheme 1). Based on these quantifications, we can speculate as to the fate of H2O2 during mitochondrial detoxification. Given the similar mechanisms of GPx and Prx, H2O would be the major product. O2 evolution would not be expected, because catalase does not play a role in the process in brain mitochondria. During non-enzymatic scavenging, H2O2 may also react non-specifically with other small molecules/proteins resulting in alternative products.
SCHEME 1.

Proposed model of H2O2 removal in brain mitochondria (adapted from Murphy (24)). ROS, in the form of O2˙̄ and H2O2, are generated from multiple intra- and extramitochondrial sources. O2˙̄ is converted to H2O2 through the action of superoxide dismutase (SOD) enzymes and/or spontaneous dismutation. H2O2 can diffuse into the mitochondrial matrix where it is removed via three different routes at the following contributions: 1, Prx coupled with Trx and TrxR (70–80%); 2, GPx coupled to GSH and GR (10–20%); or 3, non-enzymatic scavenging through redox compounds (10%). Respiration substrates (malate/glutamate or succinate) provide energy in the form of reducing equivalents (NADPH), which are maintained by ΔΨm-dependent transhydrogenase and TCA cycle enzymes. NADPH is utilized by the reductases (TrxR and GR) of the peroxidase systems to reduce disulfide bonds formed in proteins during the detoxification of H2O2.
The demonstration, that N27 dopaminergic cells remove H2O2, albeit at low rates, in a manner that is partially sensitive to auranofin, suggests that a Trx/Prx-dependent mechanism is operative in intact cells under respiring conditions. In these cells, near complete inhibition of cellular TrxR activity was associated with a 36% decrease in rates of H2O2 removal. The inability of auranofin to inhibit cellular H2O2 removal to a greater extent despite its lack of specificity toward cytosolic versus mitochondrial isoforms of TrxR was unexpected. This may be related to additional cytosolic systems contributing to H2O2 removal in intact cells and the relatively low numbers of mitochondria in undifferentiated N27 cells. Given that near complete inhibition of TrxR in N27 cells showed only a moderate decrease in H2O2 removal rates, it seems likely that the effects on H2O2 metabolism resulting from specific inhibition of the mitochondrial Trx/Prx system may be difficult to reliably quantify in intact cells. In fact, siRNA-based approaches to knockdown expression of mitochondrial isoforms of TrxR, Trx, and/or Prx have been compromised by adaptation and up-regulation of other antioxidant pathways (36). Additionally, knockdown of TrxR alone is not sufficient to inhibit the antioxidant actions of other enzymes in the Trx/Prx pathway (37). Although these pharmacologically based experiments have their limitations regarding mitochondrial specificity, they demonstrate that intact dopaminergic cells remove H2O2 in an auranofin-sensitive (TrxR-dependent) manner.
The role for H2O2 is gaining recognition in redox signaling pathways that mediate a diverse set of physiological responses, including cell proliferation, differentiation, and migration, as well as pathological conditions, including oxidative stress, neurodegeneration, and cancer (5). Therefore, mitochondrial H2O2 removal by Trx/Prx may have significant implications in regulating these processes. Even slight changes in the activity of the Trx cycle enzymes may disrupt proper signaling under physiological conditions leading to a pathological state. The up-regulation of Trx system enzymes has been implicated in the progression of several cancers. As a result, Trx system inhibitors, such as auranofin, are in development as chemotherapeutic agents (26). However, the role of the Trx system in the brain and central nervous system has emerged more slowly, as studies examining the mitochondrial isoforms of these proteins are limited. Trx2, TrxR2, and Prx3 expression have all been localized to the brain with highest expression levels occurring in regions associated with high metabolic activity and ROS production (38–40). Although the role of these proteins in neurological disorders remain to be determined, decreased Prx3 expression was reported in cases of Alzheimer disease, Down syndrome, and Parkinson disease (41, 42). Furthermore, under conditions of selenium deficiency, TrxR activity was the least compromised of selenoproteins (TrxR1, TrxR2, GPx1, GPx4, and selenoproteinP) in the brain suggesting the importance of this antioxidant enzyme (43). In this study, we highlighted the functional consequences of mitochondrial TrxR inhibition. By disrupting TrxR activity and hence H2O2 removal, PQ-induced ROS production was greatly exacerbated. This finding alone warrants further investigation into the role of Trx/Prx in neurotoxicity and neurological disorders arising from mitochondrial dysfunction. Although we demonstrated that GSH/GPx provides only minimal contributions to H2O2 removal in the brain, the importance of this enzyme system in regulating cellular redox potential and free thiol levels as well as aspects of neurodegenerative disease should not be overlooked (44).
As suggested previously (11, 45), these results fuel speculation that mitochondria may serve as a “net sink for ROS,” which contrasts with the usual recognized role of organelles as a ROS producer. The importance of mitochondrial-derived oxidants, including H2O2, has been established in cell-signaling processes with other cellular components (5, 46). However, with the identification of mitochondria as potent ROS scavengers, these findings would imply a delicate balance between production and removal of H2O2 that determines the physiological versus pathological roles of the molecule. To date, studies examining the mechanisms of mitochondrial H2O2 removal have used methods measuring the clearance of an exogenous bolus of H2O2, which may not truly reflect mitochondrial capacity to detoxify H2O2-generated endogenously. Therefore, caution must be taken when making generalized conclusions regarding mitochondrial H2O2 metabolism based upon major findings of studies, including this one, which exclusively address aspects of H2O2 removal or production. The interplay between H2O2 production and removal processes in the mitochondria and the factors or conditions regulating these aspects of ROS metabolism warrant further investigation.
In conclusion, we have shown that mitochondrial H2O2 removal can be reliably measured via polarography and attributed to the actions of the Trx-based peroxidase system. Trx and/or Prx have previously been implicated in mitochondrial antioxidant defense, yet we quantitatively demonstrate for the first time that Trx/Prx is the major contributing system to H2O2 removal in brain mitochondria. These results also highlight the importance of mitochondrial Trx/Prx antioxidant defenses that should be considered in our understanding of processes regulated by ROS such as cell signaling and neurodegenerative disease progression.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1NS04748 and RO1NS039587 (to M. P.) and 1F31NS061438 (to D. A. D.) from NINDS. This work was also supported by an American Foundation for Pharmaceutical Education pre-doctoral fellowship (to D. A. D.).
- ROS
- reactive oxygen species
- Trx2
- thioredoxin 2
- TrxR
- thioredoxin reductase
- GPx
- glutathione peroxidase
- Prx3
- peroxiredoxin 3
- DNCB
- 1-chloro-2,4-dinitrobenzene
- PQ
- paraquat
- GR
- glutathione reductase
- GSH
- glutathione.
REFERENCES
- 1.Jensen P. K. (1966) Biochim. Biophys. Acta 122, 167–174 [DOI] [PubMed] [Google Scholar]
- 2.Loschen G., Flohé L., Chance B. (1971) FEBS Lett. 18, 261–264 [DOI] [PubMed] [Google Scholar]
- 3.Andersen J. K. (2004) Nat. Med. 10, (suppl.) S18–S25 [DOI] [PubMed] [Google Scholar]
- 4.Sastre J., Pallardó F. V., Viña J. (2003) Free Radic. Biol. Med. 35, 1–8 [DOI] [PubMed] [Google Scholar]
- 5.Rhee S. G. (2006) Science 312, 1882–1883 [DOI] [PubMed] [Google Scholar]
- 6.Nonn L., Williams R. R., Erickson R. P., Powis G. (2003) Mol. Cell. Biol. 23, 916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lebovitz R. M., Zhang H., Vogel H., Cartwright J., Jr., Dionne L., Lu N., Huang S., Matzuk M. M. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 9782–9787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinerfeld D., Traini M. D., Weinberger R. P., Cochran B., Doctrow S. R., Harry J., Melov S. (2004) J. Neurochem. 88, 657–667 [DOI] [PubMed] [Google Scholar]
- 9.Schriner S. E., Linford N. J., Martin G. M., Treuting P., Ogburn C. E., Emond M., Coskun P. E., Ladiges W., Wolf N., Van Remmen H., Wallace D. C., Rabinovitch P. S. (2005) Science 308, 1909–1911 [DOI] [PubMed] [Google Scholar]
- 10.Guidot D. M., Repine J. E., Kitlowski A. D., Flores S. C., Nelson S. K., Wright R. M., McCord J. M. (1995) J. Clin. Invest. 96, 1131–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zoccarato F., Cavallini L., Alexandre A. (2004) J. Biol. Chem. 279, 4166–4174 [DOI] [PubMed] [Google Scholar]
- 12.Radi R., Turrens J. F., Chang L. Y., Bush K. M., Crapo J. D., Freeman B. A. (1991) J. Biol. Chem. 266, 22028–22034 [PubMed] [Google Scholar]
- 13.Salvi M., Battaglia V., Brunati A. M., La Rocca N., Tibaldi E., Pietrangeli P., Marcocci L., Mondovì B., Rossi C. A., Toninello A. (2007) J. Biol. Chem. 282, 24407–24415 [DOI] [PubMed] [Google Scholar]
- 14.Sinet P. M., Heikkila R. E., Cohen G. (1980) J. Neurochem. 34, 1421–1428 [DOI] [PubMed] [Google Scholar]
- 15.Sims N. R., Anderson M. F. (2008) Nat. Protoc. 3, 1228–1239 [DOI] [PubMed] [Google Scholar]
- 16.Castello P. R., Drechsel D. A., Patel M. (2007) J. Biol. Chem. 282, 14186–14193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castello P. R., Drechsel D. A., Day B. J., Patel M. (2008) J. Pharmacol. Exp. Ther. 324, 970–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prasad K. N., Carvalho E., Kentroti S., Edwards-Prasad J., Freed C., Vernadakis A. (1994) In Vitro Cell Dev. Biol. Anim. 30A, 596–603 [DOI] [PubMed] [Google Scholar]
- 19.Arnér E. S., Zhong L., Holmgren A. (1999) Methods Enzymol. 300, 226–239 [DOI] [PubMed] [Google Scholar]
- 20.Wendel A. (1981) Methods Enzymol. 77, 325–333 [DOI] [PubMed] [Google Scholar]
- 21.Chae H. Z., Kang S. W., Rhee S. G. (1999) Methods Enzymol. 300, 219–226 [DOI] [PubMed] [Google Scholar]
- 22.Drechsel D. A., Patel M. (2009) Methods Enzymol. 456, 381–393 [DOI] [PubMed] [Google Scholar]
- 23.Liu Y., Fiskum G., Schubert D. (2002) J. Neurochem. 80, 780–787 [DOI] [PubMed] [Google Scholar]
- 24.Murphy M. P. (2009) Biochem. J. 417, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown K. K., Eriksson S. E., Arnér E. S., Hampton M. B. (2008) Free Radic. Biol. Med. 45, 494–502 [DOI] [PubMed] [Google Scholar]
- 26.Tonissen K. F., Di Trapani G. (2009) Mol. Nutr. Food Res. 53, 87–103 [DOI] [PubMed] [Google Scholar]
- 27.Nicholls D. G. (2009) Biochim. Biophys. Acta 1787, 1416–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boveris A., Cadenas E. (2000) IUBMB Life 50, 245–250 [DOI] [PubMed] [Google Scholar]
- 29.Halliwell B., Long L. H., Yee T. P., Lim S., Kelly R. (2004) Curr. Med. Chem. 11, 1085–1092 [DOI] [PubMed] [Google Scholar]
- 30.Hertzman C., Wiens M., Bowering D., Snow B., Calne D. (1990) Am. J. Ind. Med. 17, 349–355 [DOI] [PubMed] [Google Scholar]
- 31.Liou H. H., Tsai M. C., Chen C. J., Jeng J. S., Chang Y. C., Chen S. Y., Chen R. C. (1997) Neurology 48, 1583–1588 [DOI] [PubMed] [Google Scholar]
- 32.Chinta S. J., Rane A., Yadava N., Andersen J. K., Nicholls D. G., Polster B. M. (2009) Free Radic. Biol. Med. 46, 939–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rigobello M. P., Folda A., Baldoin M. C., Scutari G., Bindoli A. (2005) Free Radic. Res. 39, 687–695 [DOI] [PubMed] [Google Scholar]
- 34.Hansen J. M., Zhang H., Jones D. P. (2006) Free Radic. Biol. Med. 40, 138–145 [DOI] [PubMed] [Google Scholar]
- 35.Gitler C., Zarmi B., Kalef E., Meller R., Zor U., Goldman R. (2002) Biochem. Biophys. Res. Commun. 290, 624–628 [DOI] [PubMed] [Google Scholar]
- 36.Rogers K. E., Powis G. (2005) Proc. Am. Assoc. Cancer Res. 46, 454 [Google Scholar]
- 37.Watson W. H., Heilman J. M., Hughes L. L., Spielberger J. C. (2008) Biochem. Biophys. Res. Commun. 368, 832–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rybnikova E., Damdimopoulos A. E., Gustafsson J. A., Spyrou G., Pelto-Huikko M. (2000) Eur. J. Neurosci. 12, 1669–1678 [DOI] [PubMed] [Google Scholar]
- 39.Jurado J., Prieto-Alamo M. J., Madrid-Rísquez J., Pueyo C. (2003) J. Biol. Chem. 278, 45546–45554 [DOI] [PubMed] [Google Scholar]
- 40.Seo M. S., Kang S. W., Kim K., Baines I. C., Lee T. H., Rhee S. G. (2000) J. Biol. Chem. 275, 20346–20354 [DOI] [PubMed] [Google Scholar]
- 41.Kim S. H., Fountoulakis M., Cairns N., Lubec G. (2001) J. Neural Transm. Suppl., 223–235 [DOI] [PubMed] [Google Scholar]
- 42.Krapfenbauer K., Engidawork E., Cairns N., Fountoulakis M., Lubec G. (2003) Brain Res. 967, 152–160 [DOI] [PubMed] [Google Scholar]
- 43.Hill K. E., McCollum G. W., Boeglin M. E., Burk R. F. (1997) Biochem. Biophys. Res. Commun. 234, 293–295 [DOI] [PubMed] [Google Scholar]
- 44.Schulz J. B., Lindenau J., Seyfried J., Dichgans J. (2000) Eur. J. Biochem. 267, 4904–4911 [DOI] [PubMed] [Google Scholar]
- 45.Andreyev A. Y., Kushnareva Y. E., Starkov A. A. (2005) Biochemistry 70, 200–214 [DOI] [PubMed] [Google Scholar]
- 46.Nemoto S., Takeda K., Yu Z. X., Ferrans V. J., Finkel T. (2000) Mol. Cell. Biol. 20, 7311–7318 [DOI] [PMC free article] [PubMed] [Google Scholar]