

Abstract
Kinetochore proteins associate with centromeric DNA and spindle microtubules and play essential roles in chromosome segregation during mitosis. In this study, we uncovered a zebrafish mutant, stagnant and curly (stac), that carries the Tol2 transposon element inserted at the kinetochore protein H (cenph) locus. Mutant embryos exhibit discernible cell death as early as 20 hours postfertilization, extensive apoptosis, and upward curly tail during the pharyngula period and deform around 5 days postfertilization. The stac mutant phenotype can be rescued by cenph mRNA overexpression and mimicked by cenph knockdown with antisense morpholinos, suggesting the responsibility of cenph deficiency for stac mutants. We demonstrate that the intrinsic apoptosis pathway is hyperactivated in stac mutants and that p53 knockdown partially blocks excess apoptosis in stac mutants. Mitotic cells in stac mutants show chromosome missegregation and are usually arrested in G2/M phase. Furthermore, compared with wild type siblings, heterozygous stac fish develop invasive tumors at a dramatically reduced rate, suggesting a reduced cancer risk. Taken together, our findings uncover an essential role of cenph in mitosis and embryonic development and its association with tumor development.
Keywords: Apoptosis, Mitosis, p53, Tumor, Zebrafish, Cenph, Transposon
Introduction
Kinetochore is a structure formed at the outer surface of the centromere of a mitotic chromosome. In vertebrate cells, the kinetochore assembles as trilaminar ultrastructure on the centromere during prophase (1, 2) and acts as a bridge between the centromeric chromatin and spindle microtubules. The inner and outer kinetochore plates form the interface with the centromere and microtubules, respectively. Therefore, kinetochores play essential roles in sister chromatin adhesion and separation, connection of chromosome and microtubules, chromatid movement, and mitotic checkpoint control (3, 4).
In vertebrates, a kinetochore consists of a large number of proteins, many of which are highly conserved in invertebrates and plants (3). However, only a few of the vertebrate kinetochore proteins have been studied for their functions at the organism level. Cenpa, Cenpc, or Cenpe knock-out mice are embryonic lethal with severe chromosome instability and mitotic defects (5–8), whereas Cenpb knock-out mice are viable (9), which suggest that different kinetochore proteins have different functions in development. Furthermore, altered levels of some kinetochore proteins have been shown to affect cancer susceptibility in animals (10, 11).
Kinetochore protein H (Cenph) was originally identified as a protein specifically and constitutively localized in kinetochores throughout the cell cycle in mouse cells (12). Subsequent studies indicate that Cenph is colocalized with Cenpa and Cenpc in the inner kinetochores and is required for recruiting Cenpc and Cenp-50 (13–15). Cenph overexpression in human HCT116 cells and mouse 3T3 cells induces aneuploidy due to chromosome missegregation that may result from abnormal location of ectopic Cenph (16). Depletion of Cenph in cultured chicken or human cells also causes chromosome missegregation and cell death (13, 17). These reports support the notion that Cenph is essential for normal chromosome segregation during mitosis. However, it remains unknown how Cenph deficiency leads to cell death and whether it plays a role in vertebrate development. Nevertheless, up-regulation of CENPH expression in several types of human tumors suggests the involvement of CENPH in tumorigenesis (16, 18–22).
In an attempt to identify developmentally important genes during zebrafish embryogenesis, we performed a mutagenesis using a Tol2 transposon-based gene trapping approach (24, 25). One of the mutant lines, stagnant and curly (stac) line, carries an insertion of the Tol2 transposon element at the cenph locus. Massive cell death occurs in stac mutant embryos and as a result leads to embryonic lethality, whereas heterozygous fish are viable. Compared with wild type sibling embryos, we found that stac mutants have more mitotic cells with aberrant spindles and chromosome missegregation with an arrest of the cell cycle in G2/M phase. In stac mutants, the intrinsic apoptotic pathway components, including tp53/p53, mdm2, and bbc3/puma genes as well as caspases 3, 7, and 9, are overexpressed or hyperactivated. Upon induction with the carcinogen MNNG,2 stac heterozygous fish develop malignant tumors at a significantly reduced rate than the wild type sibling fish. Thus, these findings provide novel insights into developmental roles and clinical significance of Cenph.
EXPERIMENTAL PROCEDURES
Gene Trapping and Transposon Mutagenesis
The transposon-based gene trap vector T2BGS was modified from T2KSAG (23) by replacing the original splicing acceptor with a splicing acceptor in the first intron of the zebrafish bcl2 gene, which was a gift from Dr. Jian Zhang. Injection of transposon DNA and transposase mRNA, screening of transgenic fish, and identification of mutants were done essentially as described previously (24, 25).
Thermal Asymmetric Interlaced PCR, Genotyping, RT-PCR, and Real Time PCR
To identify the flanking sequences of the transposon insertion sites in stactsu055 mutant line, genomic DNA was extracted from GFP-positive stac embryos, and thermal asymmetric interlaced PCR was performed as described previously (24).
Homozygotes, heterozygotes, and wild type siblings were easily separated based on GFP expression level around 24 hpf. For accurate genotyping, genomic DNA was extracted from single embryos, and the regions in the vicinity of the transposon insertion were amplified by PCR with the combination of three specific primers as follows: cenph5 (5′-CGGCAGTTTCAGACAGGATTGGAATC-3′), T3-1 (5′-CTCAAGTACAATTTTAATGGAGTAC-3′), and cenph3 (5′-CCGGAGACACCAAAAACCTAATTTACATC-3′). The amplification conditions were as follows: 94 °C for 3 min; 35 cycles of denaturation at 94 °C for 45 s, annealing at 53 °C for 45 s, and extension at 72 °C for 45 s. An additional extension was executed at 72 °C for 7 min.
For RT-PCR, total RNA was isolated from individual embryos using the RNeasy mini kit (Qiagen), and the first-strand cDNA was synthesized by reverse transcription with MMLV reverse transcriptase (Promega). Specific primers used for RT-PCR were as follows: cphRT5 (5′-GGAACTGACCAATCAGAACTCTGAGATTTGTG-3′) and cphRT3 (5′-CCTTCTCCTGCTTCAACTCGTTTATCTCCTGC-3′) for cenph; GFPRT5 (5′-GTGCAGCTCGCCGACCACTACCAGCAGAAC-3′) and GFPRT3 (5′-GGTCACGAACTCCAGCAGGACCATGTGATC-3′) for GFP; and actinRT5 (5′-ATGGATGATGAAATTGCCGCAC-3′) and actinRT3 (5′-ACCATCACCAGAGTCCATCACG-3′) for βactin2. Reaction conditions were as follows: 94 °C for 3 min; 30 cycles of denaturation at 94 °C for 30 s, annealing at 53 °C for 30 s, and extension at 72 °C for 30 s. An additional extension was made at 72 °C for 7 min. PCR products were resolved by 2% agarose gel electrophoresis. Real time RT-PCR was performed as described previously (26).
Cloning of Cenph cDNA
The full-length coding sequence of cenph was amplified by RT-PCR with a pair of specific primers as follows: cph5RNA (5′-CGGAATTCCACCATGAGTTCCAGTAACGTTAATC-3′) and cph3RNA (5′-CCCTCGAGTTAGCTGGGTATGTGTTCCAGTTTC-3′), which were designed based on the sequence with an original name cenp-h like (GenBankTM accession number NM_001113609). The PCR product was cloned into the vector pXT7 for in vitro synthesis of mRNA and into pBluescript II KS for synthesis of in situ hybridization probe.
mRNA and Morpholino Microinjection
mRNA was synthesized in vitro from linearized plasmids using the mMessage Machine kit (Ambion). Two morpholinos, cenph-MO1 (5′-TCCAATCCTGTCTGAAACTGCCGCC-3′) and cenph-MO2 (5′-GTTACTGGAACTCATCTTTGTATGT-3′), were designed to target the 5′UTR of cenph. p53-MO (5′-GACCTCCTCTCCACTAAACTACGAT-3′) (27) was purchased from Gene Tools, LLC. Specific morpholino or mRNA was injected into one-cell embryos at the indicated doses.
Whole-mount in Situ Hybridization and Immunohistochemistry
Whole-mount in situ hybridization for detection of gene expression in embryos was done as described previously (25). For spindle and active caspase 3 immunofluorescence, embryos fixed in 4% paraformaldehyde were dehydrated in graded methanol at −20 °C for at least 30 min, followed by rehydration three times for 5 min each in PBST (1× PBS, 0.3% Triton X-100). The embryos were then permeabilized in acetone for 7 min at −20 °C. After washing three times with PBST, embryos were incubated in block solution (2% blocking reagent (Roche Applied Science), 10% fetal calf serum, 1% dimethyl sulfoxide in PBST) for 1 h at room temperature. Monoclonal mouse α-tubulin antibody (T6199, Sigma) or rabbit caspase 3 active form antibody (559565, BD Biosciences) was added at a concentration of 1:500. Embryos were incubated for binding of the primary antibody at 4 °C overnight, rinsed four times for 20 min each in PBST (1× PBS, 0.1% Triton X-100), and incubated in block solution (10% fetal calf serum, 1% dimethyl sulfoxide in PBST) for 1 h at room temperature. Rhodamine-conjugated goat anti-mouse secondary antibody or goat anti-rabbit secondary antibody (Jackson ImmunoResearch) was added at 1:50 dilution and incubated for 2 h. Next, embryos were rinsed four times for 20 min each in PBST. When necessary, DAPI (10 mg/ml) was added during the third wash at a 1:10,000 dilution. The stained embryos were directly observed by fluorescence microscopy or, for the spindle observation, were mounted on glass slides with antifade mounting medium (N-propyl gallate in glycerol) for observation under a Zeiss LSM710 laser scanning confocal microscope.
TUNEL and pH3 Staining
For detection of apoptotic cells in embryos, TUNEL assay was performed using the In Situ Cell Death Detection kit, TMR red (Roche Applied Science), according to the manufacturer's instruction with some modifications (28). The embryos were observed by fluorescence microscopy (Zeiss SteREO Discovery version 20). Staining of phosphohistone H3 in zebrafish embryos was performed using Ser-10 phosphohistone H3 antibody as described previously (28).
Flow Cytometry
Live zebrafish embryos, about 150 embryos for each group, were dechorionated at desired ages and homogenized with a handheld pestle in 1 ml of DMEM + 10% fetal bovine serum. The homogenates were passed through a 40-μm mesh filter into a 15-ml conical tube to obtain one-cell suspensions. The total volume was raised to 5 ml with PBS. The tube was spun at 1300 rpm for 2 min, and the pellet was resuspended in 500 μl of PBS and fixed in 500 μl of 100% cold ethanol dropwise while gently vortexing. The fixed cells were incubated at 4 °C for at least 20 min and precipitated by spinning at 1000 rpm for 2 min to remove ethanol. The pellet was resuspended in 500 μl of propidium iodide solution (0.1% sodium citrate, 0.05 mg/ml propidium iodide (Sigma), 100 μg/ml RNase A, 0.0002% Triton X-100). Then the mixture was incubated at room temperature in the dark for at least 30 min. The DNA content was determined by flow cytometry analysis with the BD FACS AriaII flow cytometer.
Transmission Electron Microscopy
Zebrafish embryos at 30 hpf were fixed by 2.5% glutaraldehyde (pH 7.2) overnight at 4 °C and then incised with sharp blade to get the desired tissues (the brain from the optic tectum to the 4th ventricle combined with partial spinal cord). The tissues were washed three times, for 10 min each, with PBS (pH 7.2) and osmicated in 1% osmium tetroxide for 1.5 h, followed by washing three times with PBS. The tissues were dehydrated in graded cold ethanol from 50 to 100% at an interval of 15 min and further dehydrated in series acetone. Then the tissues were embedded in the Spur resin and polymerized at 60 °C for 1 or 2 days. Blocks were cut to produce 70-nm-thick sagittal sections with Leica EM UC6 ultracut microtome. Sections were stained with lead citrate and imaged under a HITACHI H-7650 electron microscope.
Carcinogenesis
MNNG was freshly dissolved in 100% ethanol at a concentration of 2 mg/ml. Twenty one-day-old stac heterozygous fry and wild type siblings, which were derived from crosses between heterozygotes and pre-sorted based on GFP expression at 36 hpf, were exposed to MNNG under dark conditions at a final concentration of 2 μg/ml for 24 h. Fish were sacrificed 6 months after exposure, and serial step 5-μm-thick sagittal sections were made for each fish and examined for the existence of tumors by hematoxylin and eosin staining.
RESULTS
stac Embryos Express GFP Ubiquitously, and Homozygotes Are Embryonic Lethal
We identified a mutant line stactsu055, which was named for the stagnant and curly phenotype, by insertional mutagenesis using the Tol2 transposon-based gene trap vector T2BGS (23–25). In this line, the heterozygous females, when mated to wild type or heterozygous males, produced embryos all showing GFP expression at the one-cell stage (Fig. 1A), suggesting that the trapped gene is maternally expressed. Among embryos produced by pairwise heterozygous crosses, GFP expression (fluorescence) levels at or after 20 hpf could be categorized into the following three groups: weak, moderate, and strong (see Fig. 2D for examples); the difference should have resulted from zygotic expression of GFP, implying that the trapped gene is also zygotically expressed. We observed GFP expression in 1475 embryos at 30 hpf derived from 10 pairwise crosses of heterozygotes, and we found that the weak, moderate, or strong GFP group accounted for 25.56, 47.8, or 26.64%, respectively. We speculated, and later confirmed, that weak, moderate, and strong GFP represented wild type, heterozygous, and homozygous insertion, respectively. The observed GFP segregation according to the Mendel law of segregation, suggested that GFP in the stactsu055 line is expressed by a single locus. Notably, GFP is distributed ubiquitously before 20 hpf, and since then higher levels of expression appear mainly in the head and spinal cord (Fig. 1A).
FIGURE 1.

GFP expression pattern and phenotype of stac transgenic embryos. A, fluorescent images of stac embryos showing GFP expression at indicated stages. The genotypes of pictured embryos at early stages were not determined, whereas pictured embryos at 24–48 hpf were heterozygotes. B, morphology of stac mutant and wild type (WT) sibling embryos at indicated stages. The boxed trunk area was enlarged in the corner, and the head was enlarged in the right panel. Note that mutant embryos had a darker head, rough skin, and curly posterior trunk with stage-dependent degrees.
FIGURE 2.

Genomic organization and expression of the cenph locus in wild type and stac mutant embryos. A, genomic structure of the cenph locus and putative transcripts. Exons are numbered. The sequence at the transposon insertion site is shown in parentheses with black letters for cenph-derived sequence and red letters for Tol2 transposon-derived sequences. The binding positions and directions of primers used in B are indicated. UTR, untranslated region; (A)n, polyadenine tail. B, genotyping of individual embryos by PCR. Embryos collected from stac heterozygote intercrosses were separated based on GFP expression levels and phenotypes at 24 hpf. Three primers, cenph5, cenph3, and T3-1, were used together for PCR. The upper (a) and lower (b) bands represented the wild type and recombinant allele, respectively. M, molecular marker lane. C, transcripts of cenph and GFP expression were detected by RT-PCR in embryos of different genotypes and phenotypes. Primers cphRT5 and cphRT3 were used for cenph, and primers for the other genes were described under “Experimental Procedures.” Samples 1–3 represented wild type with weak GFP, heterozygote with moderate GFP, and homozygote with strong GFP, respectively. Sample C was a control with no RNA template. β-Actin served as a control. Note that changes in cenph and GFP transcription levels were opposite. D, expression level of cenph was quantified by real time PCR. Embryos were pre-sorted as mentioned above. Total RNA was extracted from 60 embryos for each group. The relatedness of GFP expression levels (at 20 hpf) with genotypes was exemplified on the right. WT, wild type sibling; Ht, heterozygote; stac, homozygous mutant. Error bars indicate S.D.
Embryos with strong GFP, which are stac mutants and homozygous for the transposon insertion, have normal morphology until about 20 hpf as do the wild type (weak GFP) or heterozygous (moderate GFP) siblings. Mutant embryos start to have discernible degeneration (cell death) in the head at 24 hpf (Fig. 1B). As development proceeds, mutant embryos show more severe degeneration particularly in the head and spinal cord with darker appearance due to higher opacity. The additional morphological changes include vague brain ventricle borders, coarse and rough skin, smaller eyes, and upward bent posterior trunk (Fig. 1B). Mutant embryos usually deform around 5 days postfertilization. In contrast, the wild type and heterozygous siblings develop normally and survive to adulthood, but the heterozygous adult fish appear smaller in size than wild type fish at comparable ages. The consistency between the mutant phenotype and the GFP level strongly suggests that the interruption of the locus by the transposon insertion resulting in GFP expression is responsible for the mutant phenotype.
cenph Locus in the stac Line Is Interrupted by a Transposon Insertion
To identify the transposon insertion position in the genome of the stac line, we performed thermal asymmetric interlaced PCR. The result revealed that the transposon element has been inserted into the second exon of the gene NP_001107081.1 in Ensembl on the 8th linkage group, which was originally named cenp-h like gene. This gene has an open reading frame of 702 bp, which encodes a putative peptide of 233 amino acids. The putative peptide shares an overall sequence similarity of 59 and 58% to the human and mouse centromere protein Cenph, respectively. Importantly, several genes located adjacent to the CENPH locus on human chromosome 5 have their counterparts in the vicinity of the zebrafish cenp-h like locus on chromosome 8 (data not shown). This information suggests that zebrafish Cenp-h like protein is in fact an orthologue of mammalian Cenph and should be renamed Cenph (GenBank accession number GU977276).
The zebrafish cenph locus consists of 10 exons and 9 introns. The insertion of the transposon element into the 2nd exon in the 5′UTR was expected to allow the transcription of GFP expression cassette under the control of the cenph promoter without interrupting the cenph coding region (Fig. 2A). The correlation between GFP expression levels and the interrupted cenph locus was tested by PCR-based genotyping using specific primers. Results showed that strong GFP embryos and moderate GFP embryos carry homozygous and heterozygous transposon insertion at the cenph locus, respectively, although embryos with weak GFP have no insertion (Fig. 2B). Thus, GFP expression in the stac line is driven by the cenph promoter.
Next, we asked whether the transposon insertion would affect the expression level of cenph in stac embryos. RT-PCR analysis disclosed that, compared with the wild type siblings (weak GFP), the heterozygous siblings at 24 or 30 hpf express cenph at a reduced level, whereas homozygotes express cenph at almost undetectable levels (Fig. 2C). Analysis by real time PCR indicated that heterozygous and homozygous embryos at 30 hpf retained ∼50 and 3.8% of cenph mRNA amount in the wild type embryos, respectively (Fig. 2D). It is clear that the transposon insertion in the stac line inhibits the expression of wild type cenph mRNA.
Interruption of the cenph Locus Accounts for the stac Mutant Phenotype
To further verify the relationship between the stac mutant phenotype and cenph disruption, we first performed a rescue experiment using in vitro synthesized cenph mRNA. Injection of cenph mRNA into wild type embryos did not produce visible abnormalities (data not shown). We then injected stac mutant embryos at the one-cell stage with different doses of cenph mRNA, and we observed morphology at several stages with focuses on cell death in the head (visible at 30 hpf) and curly posterior trunk (visible at 36 hpf). As shown in Fig. 3A, the mutant embryos injected with 50 pg of cenph mRNA had reduced degrees of cell death at 30 hpf and straight trunk before or at 36 hpf, which resembled wild type embryos, but the injected mutant embryos still showed curly trunk at 48 hpf and later stages. Increasing the amount of cenph mRNA led to a better rescue effect as observed at 48 and 72 hpf. Nevertheless, these results support the idea that the deficiency of Cenph accounts for the stac mutant phenotype.
FIGURE 3.

stac mutants are rescued by cenph overexpression and phenocopied by cenph knockdown. A, overexpression of cenph mRNA rescued the mutant phenotype. One-cell embryos from stac heterozygote intercrosses were injected with cenph mRNA. The homozygous mutant embryos were sorted out at 24 hpf by their strong GFP expression, and their morphology was observed at indicated stages. As indicated on the right lower corner was the ratio of embryos with presented morphology to the total number of injected embryos with strong GFP. B, spatiotemporal expression pattern of cenph in zebrafish wild type embryos was investigated by in situ hybridization at indicated stages. C, knockdown of cenph in wild type embryos mimicked the stac phenotype. One-cell wild type embryos were injected with cenph morpholinos alone or in combination with cenph mRNA. The number of affected embryos and the total number of injected embryos are indicated.
We inspected spatiotemporal expression of cenph in wild type embryos by whole-mount in situ hybridization. The cenph transcripts were detected in one-cell embryos and distributed evenly until midsegmentation (Fig. 3B). From the 20-somite stage to 30 hpf, cenph expression occurred at higher levels in the head and spinal cord, but at 36 hpf or later stages, its expression was retained in the head region only. The similarity between cenph expression pattern in wild type embryos and GFP expression pattern in stac embryos is another indication that the stac mutant phenotype arises from the interruption of the cenph locus.
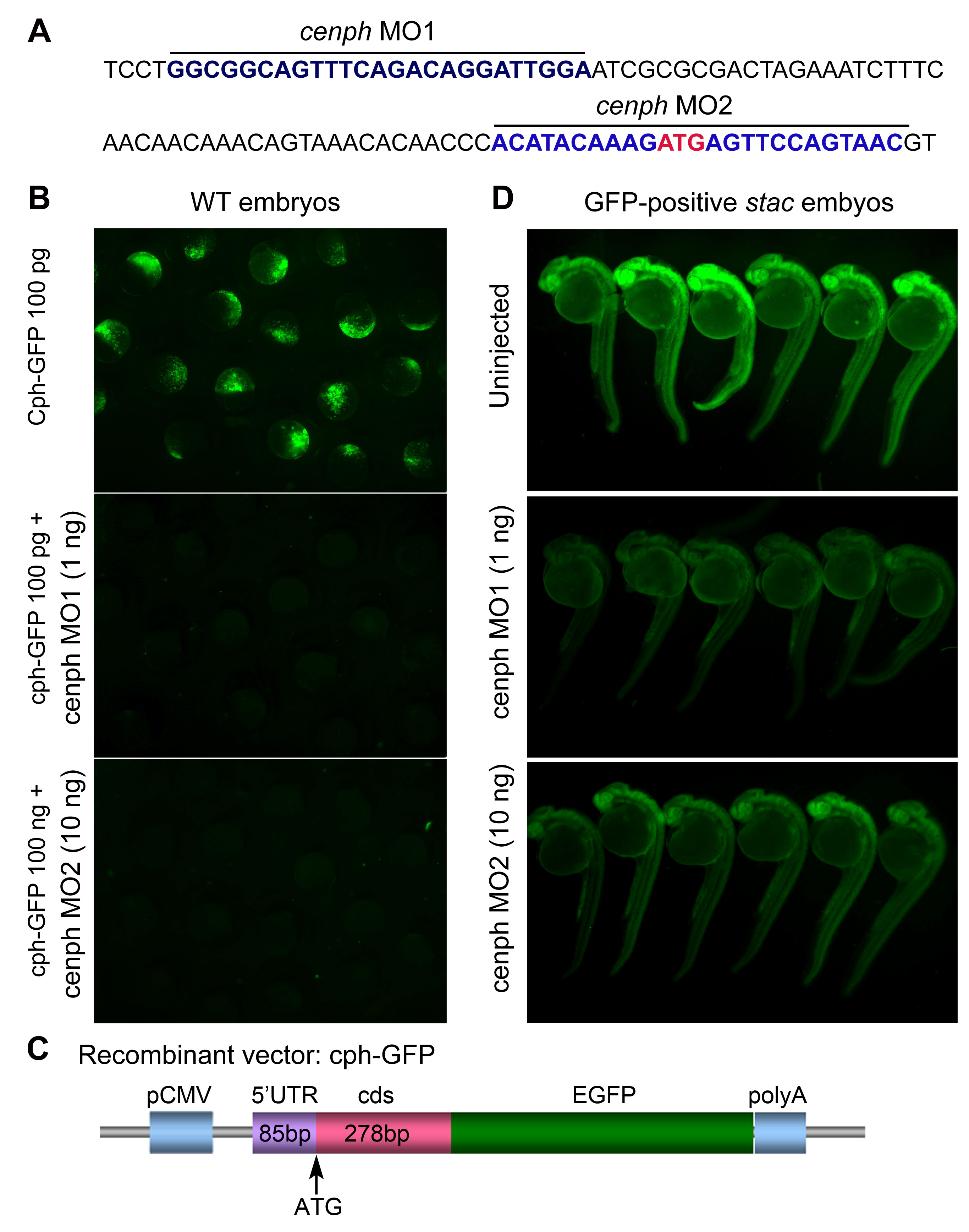
We took another strategy to confirm the relatedness of the Cenph deficiency to the stac mutant phenotype by inhibiting cenph expression in wild type embryos with antisense morpholinos. Two morpholinos, cenph-MO1 and cenph-MO2, were found to effectively block the expression of the reporter pcenph-5′UTR-GFP in which GFP was fused to 5′UTR and a 5′ part of the coding sequence of cenph and to reduce GFP expression in stac embryos (supplemental Fig. S1). Injection of 0.4 ng of cenph-MO1 and 4 ng of cenph-MO2 into one-cell wild type embryos resulted in abnormalities mimicking stac mutants, and the phenotypes were also rescued by coinjection of 100 pg of cenph mRNA (Fig. 3C). Taken together, the above data indicate that loss-of-function of cenph is responsible for the stac mutant phenotype.
Extensive Apoptosis Occurs in stac Mutant Embryos
To determine whether excess cell death in stac mutants belongs to apoptosis, we performed TUNEL assay that detects DNA fragmentation. We found that stac mutant embryos had many more TUNEL-positive cells, mainly in the brain and spinal cord, than in the wild type siblings at or after 24 hpf (Fig. 4A). Staining with acridine orange, a vital dye for detecting apoptotic cells with hypercondensed chromatin and apoptotic bodies, also detected more dying cells in stac mutants than in the wild type siblings (data not shown). By confocal microscopy of DAPI (diamidino-2-phenylindole)-stained embryonic nuclei, we found that, compared with the wild type siblings, stac mutants had more nuclei with superfluous apoptotic bodies and chromosomal condensation/fragmentation in the spinal cord and optic tectum of stac mutants (Fig. 4B). Electronic microscopy revealed that many cells in stac mutants displayed nuclear chromatin compaction and segregation, nuclear budding, and fragmentation (Fig. 4C), which are ultrastructural manifestations of apoptosis. Taken together, we conclude that the deficiency of Cenph in stac mutants induces widespread apoptosis.
FIGURE 4.

Excess apoptosis occurs in stac mutant embryos. A, DNA fragmentation was detected by TUNEL assay. Embryos were observed by fluorescence microscopy after staining at indicated stages. The boxed areas are enlarged in the corresponding right panels. Note that the mutants had more and stronger staining signals. B, chromosomes were stained by DAPI. The embryos were stained and observed by confocal fluorescence microscopy. The images were taken from the optic tectum and spinal cord regions. The areas indicated by arrowheads are enlarged in the corresponding lower panels. Note that in mutant embryos many nuclei had superfluous apoptotic bodies and chromosomal condensation/fragmentation. Scale bars, red is 40 μm; yellow is 10 μm. C, apoptotic cells were observed by transmission electron microscopy. Note that the nuclei of stac mutants showed chromatin compaction/segregation (top), budding (middle), or fragmentation (bottom). WT, wild type siblings of stac. Scale bar, 4 μm.
Intrinsic Apoptotic Pathway Is Triggered in stac Mutants
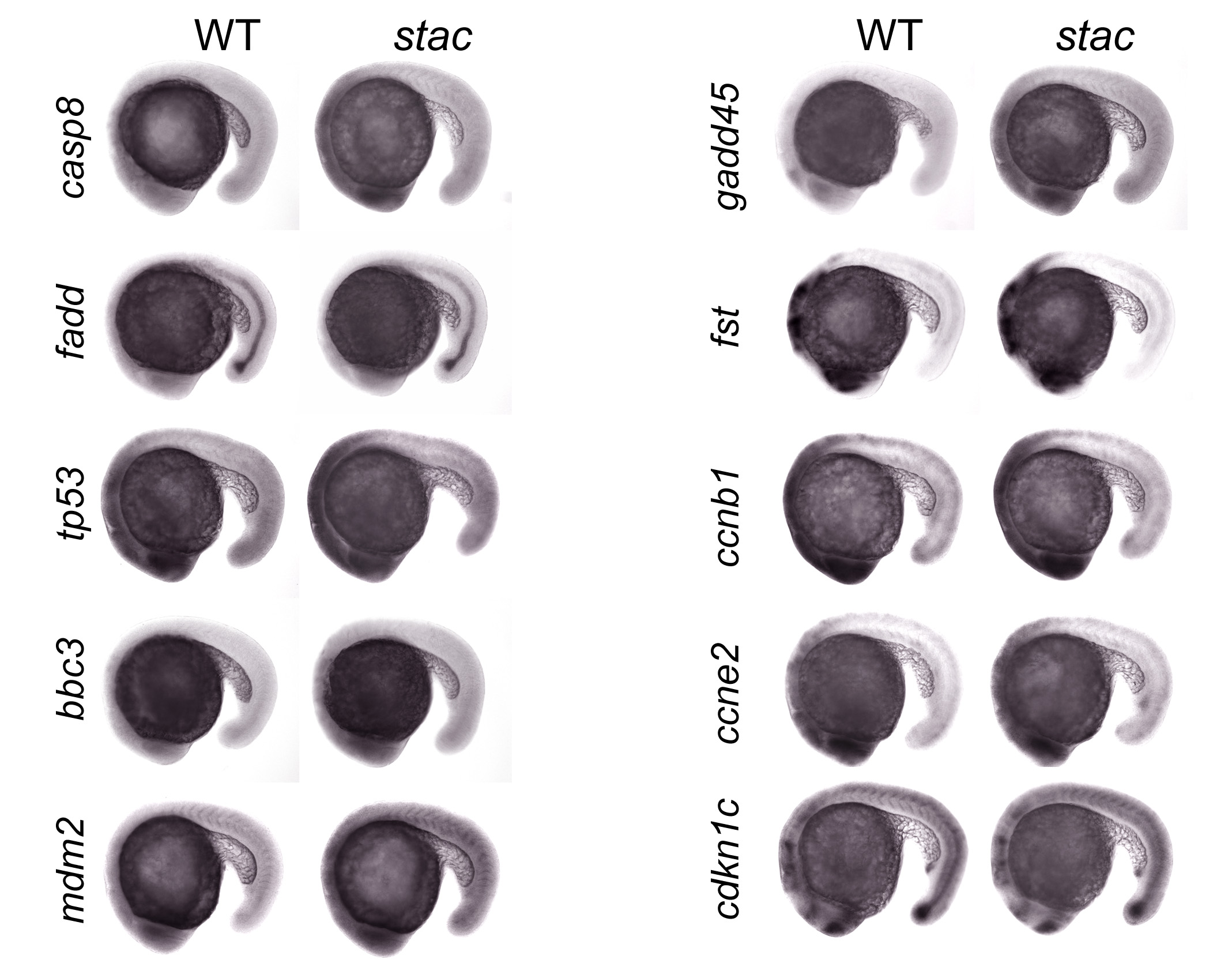
Like in human, both intrinsic and extrinsic apoptotic signaling pathways are involved in apoptosis in zebrafish (29, 30). We asked which apoptotic pathways are stimulated in stac mutants. We then did a microarray analysis using the Affymetrix GeneChip® Zebrafish Genome Array (NCBI GEO accession number GSE20707) and total RNA extracted from 24- or 30-hpf stac mutants or the wild type siblings. This analysis identified 49 up-regulated and 55 down-regulated genes in stac mutants (supplemental Tables S1 and S2 for known genes). We chose those involving apoptosis and cell cycle regulation, including tp53/p53, mdm2, fadd, bbc3/puma, gadd45al, follistatin (fst), caspase 8 (casp8), and cdkn1c/p57, for further validation by quantitative RT-PCR analysis and whole-mount in situ hybridization. Two other mitosis-related genes, cyclin B1 (ccnb1) and cyclin E2 (ccne2), were also analyzed, although they were not included in the array. The quantitative RT-PCR results indicated that in stac mutants bbc3, tp53, casp8, and mdm2 were significantly up-regulated at and after the 20-somite stage. The up-regulation of fst became apparent from 24 hpf onward; gadd45al was also up-regulated at 36 hpf. In contrast, ccne and cdkn1c were down-regulated from 24 hpf onward; however, the expression of fadd and ccnb1 appeared unchanged at examined stages (Fig. 5, A–C). The changes in the expression levels of these markers, as detected by in situ hybridization, showed a general tendency similar to the quantitative RT-PCR results (supplemental Figs. S2–S4). These data imply that both the intrinsic (mitochondria-dependent caspase 9 pathway) and extrinsic (death receptor-mediated caspase 8 pathway) apoptotic pathways might have been induced in stac mutant embryos and that the progression of the cell cycle of mutant cells may have been affected by down-regulation of genes such as ccne2 and cdkn1c.
FIGURE 5.

Apoptosis signaling pathway components are highly activated in stac mutants. A–C, quantification of apoptosis and cell cycle regulator expression levels by real time RT-PCR at different stages. The tested genes were selected from cDNA microarray assay. D, bioluminescent assay of caspase 8, 9, and 3/7 activities. Twenty stac and wild type (WT) sibling embryos at 30 hpf were treated with corresponding Caspase-Glo reagents (Promega), and luminescence was measured 2 and 3 h after treatment. The p value was 0.0847, 0.0273, or 0.0014 for 2-h treated caspase 8, 9, or 3/7 activity, respectively, and 0.0445, 0.0165, or 0.0004 for 3-h treatments. Significance of differences is as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001 (by Student's t test). Error bars indicate S.D. E, immunostaining of activated caspase 3 using an antibody against active caspase 3. F, excessive apoptosis in stac mutant embryos was inhibited by injection of p53 morpholino (p53-MO). One-cell embryos derived from stac heterozygote intercrosses were injected. The mutants were sorted based on GFP expression level around 24 hpf and were observed directly by bright field microscopy or observed by fluorescent microscopy after staining for apoptotic cells by TUNEL assay at indicated stages.
As the activation of caspases is a critical step of the extrinsic and intrinsic apoptotic pathways, we went on to measure the activities of caspase 3/7, caspase 8, and caspase 9 in embryonic cells at 30 hpf using the luminescent method (31). In stac mutant cells, the activities of caspase 3/7 and caspase 9 were increased significantly (Fig. 5D), compared with embryonic cells derived from the wild type siblings. The increase of the caspase 8 activity was less, but still statistically significant, if the incubation time was longer (3 h) (Fig. 5D). Thus, we hypothesize that the disruption of cenph expression in stac mutants primarily triggers the intrinsic but not extrinsic apoptotic pathway.
Because caspase 3 activation is a common essential step of the apoptotic pathways, we additionally examined the levels of activated caspase 3 by immunofluorescent staining. As shown in Fig. 5E, the stac mutants at 30 hpf showed stronger signals for active caspase 3 than the wild type siblings. This suggests that caspase 3-dependent apoptosis pathway is ignited in stac mutants.
Depletion of p53 Can Partially Rescue stac Mutants
Tp53/p53 is a key player of the intrinsic apoptotic pathway (32, 33). As the expression of p53 and several other components of the pathway is up-regulated in stac mutant embryos as demonstrated above, we expected that knockdown of p53 in stac mutants could inhibit massive apoptosis. To verify this, we injected different doses (5 or 10 ng) of p53 antisense morpholino (p53-MO) (27) into stac mutant embryos at the one-cell stage, followed by morphological observation and TUNEL assay at 30 hpf of development. As reported before (27), injection of p53-MO into wild type embryos did not cause any observable developmental defects (data not shown). In contrast, the cell death and the curly posterior trunk phenotype of stac mutant were partially inhibited by knockdown of p53 in a dose-dependent manner (Fig. 5F); this rescue effect was declined later, and as a result, the injected mutant embryos still developed the curly posterior trunk phenotype at 48 hpf. The TUNEL assay also detected varying degrees of decrease of signal for DNA fragmentation in the p53-MO-injected stac mutant embryos, which was dependent on the p53-MO doses (Fig. 5F). These results confirm that the p53 apoptosis pathway plays a role in cell death of stac mutant embryos. But the p53-independent apoptotic pathway may also contribute to the development of the stac mutant phenotype. On the other hand, the partial rescue effect by knocking down p53 could be ascribed to continuing dilution of the morpholino concentration during embryonic development.
Mitotic Cells in stac Mutants Are Arrested in G2/M Phase
Considering the important roles of Cenph and other centromere proteins on chromosome segregation and cell division (3, 4, 34, 35), we hypothesize that the deficiency of Cenph in the zebrafish stac mutants would lead to spindle conformation damage and mitotic arrest. To confirm this, we performed FACS-based cell cycle analysis of the DNA content of cells derived from stac mutant or wild type sibling embryos from 24 to 36 hpf. Results showed that the percentage (48.7%) of cells in G0/G1 phase in 24-hpf stac mutants was already lower than that (59.1%) in the wild type siblings, and the decrease became more apparent at 30 and 36 hpf (Fig. 6A). A much bigger difference between stac mutants and the wild type siblings was found in the proportion of the G2/M cells; the stac mutants at 36 hpf showed 3.43-fold increase compared with the wild type siblings, which suggests the accumulation of the G2/M cells in stac mutants. In contrast, the percentage of the S phase cells appeared unchanged between two groups of samples. Immunostaining for Ser-10-phosphorylated histone H3 (pH3), the mitosis marker, detected a dramatic increase of signals, particularly in the head and spinal cord, in stac mutants embryos at 24–36 hpf compared with the wild type siblings (Fig. 6B), which would suggest normal chromosome condensation but failure of chromosome segregation in the stac mutant cells. Taking these data together, we conclude that the mitotic cells in stac embryos can proceed through S and G0/G1 phases but are arrested in G2/M phase.
FIGURE 6.

stac mutant embryos have more abnormal mitotic cells. A, cell cycle analysis by flow cytometry. Cells from stac mutant or wild type (WT) sibling embryos at indicated stages were labeled with propidium iodide (PI), and DNA content was counted. The percentages of cells in different phases of the cell cycle are shown in bar graphs. Significance of differences is as follows: **, p < 0.01; ***, p < 0.001 (by Student's t test). Error bars indicate S.D. B, mitotic cells labeled with anti-pH3 antibody in stac mutant and wild type sibling embryos at indicated stages. The boxed area is enlarged at the right corner of each panel. Note that more mitotic cells are found in stac mutants. C and D, mitotic spindles and chromosome aggregation and segregation at metaphase and anaphase of mitotic cells in stac mutant and wild type sibling embryos. Immunohistochemistry was performed with anti-α-tubulin for spindles (red) and DAPI for chromosomes (blue) (C). Cells were picked up from the spinal cord. An abnormal mitotic cell in the anaphase from a mutant embryo was shown to have multipolar spindles (on the right lower corner of panel). Scale bar, 5 μm. The percentage of mitotic cells with aberrant spindles is shown in D.
Chromosome Missegregation Occurs in Mitotic Cells of stac Mutants
Our next question is why mitotic cells in stac mutant embryos are arrested in G2/M phase. Considering that Cenph is a component of the kinetochores (36), we would expect a failure of chromosome segregation due to the insufficiency of Cenph in stac mutant cells. To test this idea, we observed, by immunofluorescent confocal microscopy, microtubules/mitosis spindles and chromosomes of mitotic cells in embryos that were stained using α-tubulin antibody and DAPI. From the 20-somite stage to 36 hpf, we consistently identified a much higher proportion of abnormal mitotic cells in stac mutants than in the wild type siblings; and the difference became more dramatic at later stages (Fig. 6, C and D). Typically, the abnormal mitotic cells at the metaphase exhibited hypercondensation of chromosomes with failure to congregate in the metaphase plate, and the distorted (deformed, disordered and curved) organization of mitotic spindles; at anaphase, the abnormal cells showed aggregation or random dispersion of the chromosomes as well as disorganized and asymmetrical spindles (Fig. 6C). In a few extreme cases, we found mitotic cells with multipolar spindles (see an example in Fig. 6C). These results indicate that the deficiency of Cenph in stac mutants causes abnormal mitosis by affecting chromosome segregation and stability.
stac Heterozygous Adults Have Reduced Cancer Susceptibility in Response to Carcinogen Induction
Zebrafish has been successfully used as a cancer model system (37, 38). Because some of kinetochore proteins have been shown to be involved in cancer development (10), we extended our study to assess the effects of stac/cenph heterozygosity on the development of tumors. The stac heterozygous fish and wild type siblings at day 21, which were pre-sorted based on the GFP expression at 36 hpf, were treated with 2 μg/ml MNNG, a carcinogen that can induce different types of tumors (39). Six months later, fish were sacrificed, and serial cross-sections were made to examine the existence of tumors. Among 138 treated wild type sibling fish, we identified 9 fish carrying malignant tumors in the brain, esophagus, liver, spleen, retina, or lens (Fig. 7A), which showed hyperproliferation and in most cases invasion into the adjacent tissues/organs. The carcinogenesis rate (6.52%) in wild type sibling fish was comparable with those reported previously by other researchers (40, 41). In contrast, only 0.97% (1:103) of the treated stac heterozygous fish had retina hyperplasia. These data suggest that reduced levels of cenph due to loss of one allele suppress the development of invasive tumors. The reduction of cancer susceptibility in stac heterozygous fish may be related to the activation of the apoptotic pathways as seen in stac mutants (Fig. 5).
FIGURE 7.

stac heterozygous adults have a decreased susceptibility of carcinogenesis. stac heterozygous and wild type (WT) sibling fish at 21 days postfertilization were treated with the carcinogen MNNG and sectioned 6 months post-treatment. All pictures were taken from wild type embryos. A, malignant tumors were found in various tissues of wild type embryos. The malignant cells exhibited an increased nucleus/cytoplasm ratio, intensive and disorganized arrangement, and infiltration into adjacent tissues. The boxed areas are presented at a higher magnification on the right. Scale bars, black bar for lower or higher magnification, 200 μm; red bar for higher magnification, 100 μm. B, intestine adenomas showed large outgrowth of polyps, loss of goblet cells, and pseudostratification of nuclei. C, bar graphs showed the percentage of stac heterozygous (stac+/−) or wild type sibling (WT+/+) fish carrying tumors. The difference in malignant tumor rate between two groups was statistically significant (p = 0.046, Fisher's exact test), although the difference in intestine adenoma rate was statistically insignificant (p = 1).
We also paid attention to noninvasive neoplasms or aberrant hyperplasia in the treated fish. We found that comparable proportions of stac heterozygous fish and wild type sibling fish developed intestinal adenomas (Fig. 7, B and C). The intestinal adenomas manifested large outgrowth of polyps, loss of goblet cells, and pseudostratification of nuclei (Fig. 7B). It appears that stac haploinsufficiency in zebrafish has little impact on noninvasive neoplasms.
DISCUSSION
Cenph is a constitutive component of the centromeric inner kinetochores. Previous in vitro studies have disclosed that Cenph is required for normal chromosome segregation during mitosis. However, the importance of Cenph at the organism level has not been demonstrated. In this study, we identified a cenph-deficient zebrafish mutant, stac, in which the cenph locus is interrupted by the insertion of the Tol2 transposon element, and as a result, cenph expression is blocked. stac mutants are embryonic lethal due to widespread cell death via hyperactivation of the intrinsic apoptosis pathway. In stac mutants, mitotic cells show aberrant mitotic spindles and chromosome missegregation, resulting in the arrest of the cell cycle in G2/M phase. We also demonstrate that stac heterozygous adults are less susceptible to induction of the carcinogen MNNG for malignant tumor development. Thus, our study uncovers an essential role of Cenph in vertebrate development and its implications in tumorigenesis.
More apoptotic cells in stac mutant embryos than in wild type embryos are not obvious until about 20 hpf of development, being much later than the detectable time point (6 hpf during early gastrulation) of apoptosis in zebrafish embryos (42, 43). This phenomenon may be ascribed to the presence of maternal cenph transcripts and/or Cenph protein. Although stac mutant embryos injected with cenph mRNA survive a couple of days longer, they can never grow to adulthood, which suggests that constant supply of Cenph protein is necessary for life. Thus, it is not practical to obtain stac homozygous adult females by mRNA rescue or germ cell transfer. To study the requirement of maternal Cenph for the cleavage of blastomeres, new approaches should be taken to deplete maternal Cenph. Another interesting phenomenon is that more serious apoptosis occurs in the brain and spinal cord in stac mutants. It is likely that highly proliferating tissues require a large amount of Cenph.
Among known Cenp genes, Cenpa, Cenpb, Cenpc, or Cenpe null mice have been generated by site-specific targeting (5–9, 44). Homozygous mutants for Cenpa, Cenpc, or Cenpe all display mitotic defects and are embryonic lethal (5, 7, 8), indicating that these kinetochore genes are essential for embryonic development. In this study, we demonstrate for the first time an essential role of Cenph in vertebrate embryonic development using the zebrafish model. In contrast, Cenpb null mice develop normally; the mutant adults have reduced body weight and show age-dependent reproductive deterioration (9, 44). These reports suggest that different kinetochore genes/proteins have distinct roles in development, which may depend on their dynamic requirement for centromere architecture and function (4, 45). Each kinetochore in vertebrate cells consists of more than 80 proteins, most of which have not been studied for their function in development (3). Future effort should be made to address their developmental roles at the whole organism level.
In zebrafish stac mutants, many mitotic cells display disorganized spindles, chromosome hypercondensation, and missegregation, resembling those seen in chicken DT40 or human HEp-2 cells depleted of CENPH (13, 17), which suggests a conserved function of Cenph in mitosis. Compared with wild type sibling embryos, stac mutant embryos have a significantly higher fraction of mitotic cells in G2/M phase, suggesting mitotic arrest in mutants. The same phenomenon has also been observed in chicken DT40 cells depleted of CENPH (13). The observation that the cell cycle progresses normally in human HEp-2 cells depleted of CENPH by RNAi knockdown (17) may result from inefficient depletion of CENPH.
Previous studies have shown that the deficiency of kinetochore proteins usually leads to cell death following defective mitosis in cultured cells or organisms of vertebrates (for examples see Refs. 5, 7, 8, 13, 17, 46–48); however, the mechanisms underlying cell death are poorly understood. In this study, we demonstrate that in stac mutants several components of the intrinsic apoptotic pathway, including tp53/p53, mdm2, and bbc3/puma genes as well as caspases 3, 7, and 9, are expressed at higher levels or hyperactivated. Knockdown of p53 gene expression in stac mutants rescues the mutant phenotype only partially and temporarily. This suggests that p53-independent and even caspase-independent apoptosis pathways are involved in extensive cell death in zebrafish stac mutants. Another unsolved question is how the deficiency of kinetochore proteins or abnormal kinetochores triggers apoptosis. It is likely that microtubule/spindle damage due to abnormal kinetochores/centromeres causes activation of microtubule-associated apoptosis inducers or inactivation of apoptosis inhibitors. Another possibility is that mitotic arrest due to abnormal kinetochores will lead to DNA damage, and the latter activates related apoptosis pathways. Unfortunately, technical bottlenecks in fish have hindered our further studies.
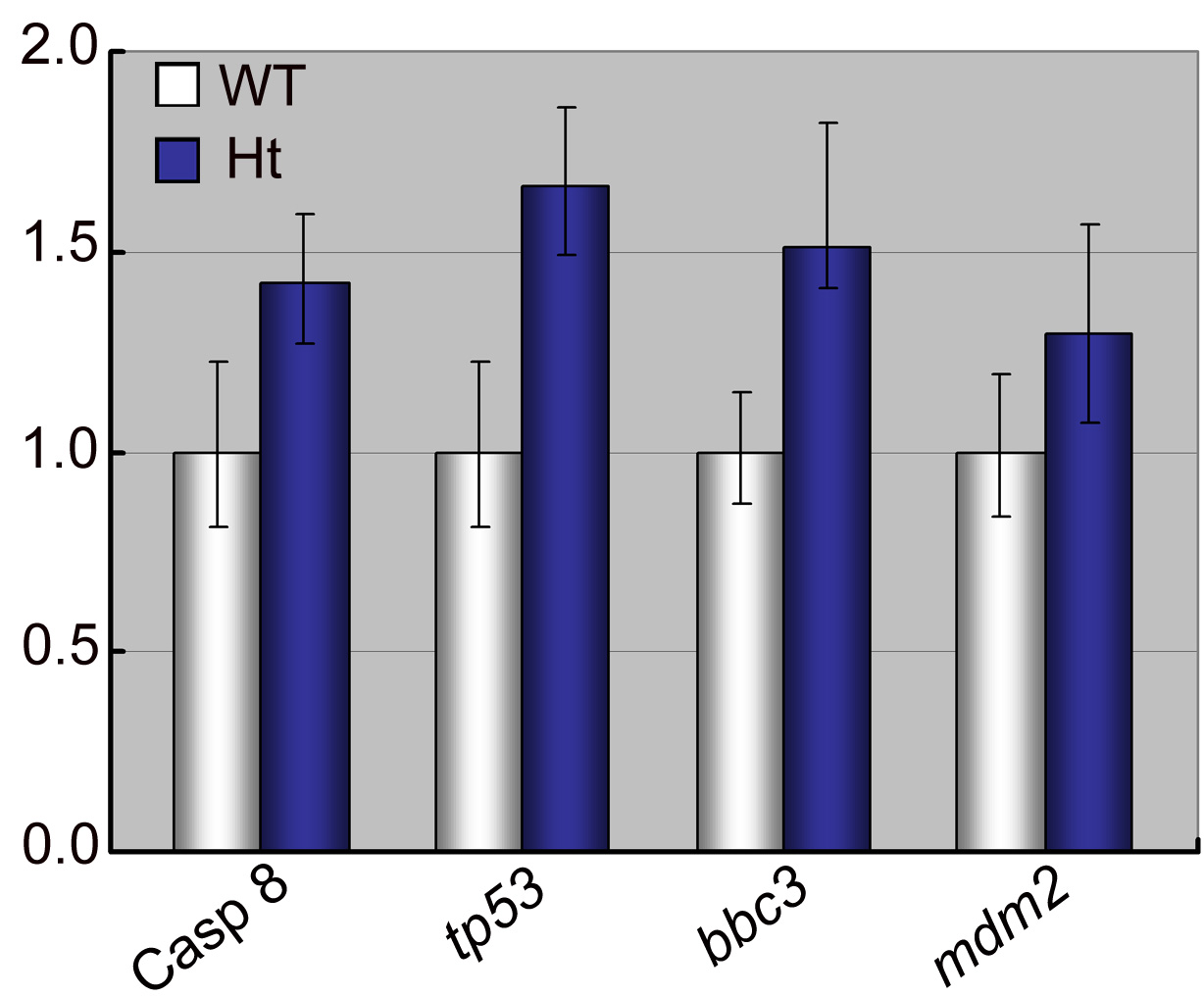
The genome instability caused by depletion of kinetochore or spindle checkpoint proteins could raise cancer susceptibility in most cases and inhibit the tumorigenesis in other contexts (10, 11). We demonstrate that haploinsufficiency of Cenph in stac heterozygous fish suppresses development of malignant tumors in several tissues upon chemical induction. This finding is similar to the previous report that Cenpe heterozygous mice have reduced tumor susceptibility to carcinogen stimulation (49). This possibly could be explained by the following: a higher level of apoptosis in the organisms triggered by dysfunctional kinetochores, which is supported by higher levels of casp8, tp53, bbc3, and mdm2 in stac heterozygous fish than in wild type fish (supplemental Fig. S5), may help destroy hyperproliferating cells. Interestingly, up-regulation of human CENPH expression has been reported in different types of cancers, including colorectal cancer (16), oral squamous cell carcinomas (18), esophageal (20), nasopharyngeal carcinomas (19), tongue cancer (22), and lung cancer (21). These findings suggest a potential significance of Cenph in tumor prognosis and therapy.
Supplementary Material
Acknowledgments
We thank Drs. Koichi Kawakami and Jian Zhang for Tol2 reagents. We are grateful to Drs. John H. Postlethwait, Kathleen L. Pfaff, Ming Li, Shuang Li, Ying Li, and members of the Meng Laboratory for helpful discussions or technical assistance.
This work was supported by Major Science Programs of China Grant 2006CB943401, National Basic Research Program of China Grant 2005CB522502, and the 863 Program Grant 2006AA02Z167.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) NM001113609 and GU977276.1.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. 1–5.
- MNNG
- N-methyl-N′-nitro-N-nitrosoguanidine
- hpf
- hours postfertilization.
REFERENCES
- 1.Brinkley B. R., Stubblefield E. (1966) Chromosoma 19, 28–43 [DOI] [PubMed] [Google Scholar]
- 2.McEwen B. F., Dong Y., VandenBeldt K. J. (2007) Methods Cell Biol. 79, 259–293 [DOI] [PubMed] [Google Scholar]
- 3.Cheeseman I. M., Desai A. (2008) Nat. Rev. Mol. Cell Biol. 9, 33–46 [DOI] [PubMed] [Google Scholar]
- 4.Santaguida S., Musacchio A. (2009) EMBO J. 28, 2511–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Howman E. V., Fowler K. J., Newson A. J., Redward S., MacDonald A. C., Kalitsis P., Choo K. H. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 1148–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalitsis P., Fowler K. J., Earle E., Griffiths B., Howman E., Newson A. J., Choo K. H. (2003) Chromosome Res. 11, 345–357 [DOI] [PubMed] [Google Scholar]
- 7.Kalitsis P., Fowler K. J., Earle E., Hill J., Choo K. H. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 1136–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Putkey F. R., Cramer T., Morphew M. K., Silk A. D., Johnson R. S., McIntosh J. R., Cleveland D. W. (2002) Dev. Cell 3, 351–365 [DOI] [PubMed] [Google Scholar]
- 9.Perez-Castro A. V., Shamanski F. L., Meneses J. J., Lovato T. L., Vogel K. G., Moyzis R. K., Pedersen R. (1998) Dev. Biol. 201, 135–143 [DOI] [PubMed] [Google Scholar]
- 10.Yuen K. W., Montpetit B., Hieter P. (2005) Curr. Opin. Cell Biol. 17, 576–582 [DOI] [PubMed] [Google Scholar]
- 11.Rao C. V., Yamada H. Y., Yao Y., Dai W. (2009) Carcinogenesis 30, 1469–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugata N., Munekata E., Todokoro K. (1999) J. Biol. Chem. 274, 27343–27346 [DOI] [PubMed] [Google Scholar]
- 13.Fukagawa T., Mikami Y., Nishihashi A., Regnier V., Haraguchi T., Hiraoka Y., Sugata N., Todokoro K., Brown W., Ikemura T. (2001) EMBO J. 20, 4603–4617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minoshima Y., Hori T., Okada M., Kimura H., Haraguchi T., Hiraoka Y., Bao Y. C., Kawashima T., Kitamura T., Fukagawa T. (2005) Mol. Cell. Biol. 25, 10315–10328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alonso A., Fritz B., Hasson D., Abrusan G., Cheung F., Yoda K., Radlwimmer B., Ladurner A. G., Warburton P. E. (2007) Genome Biol. 8, R148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomonaga T., Matsushita K., Ishibashi M., Nezu M., Shimada H., Ochiai T., Yoda K., Nomura F. (2005) Cancer Res. 65, 4683–4689 [DOI] [PubMed] [Google Scholar]
- 17.Orthaus S., Ohndorf S., Diekmann S. (2006) Biochem. Biophys. Res. Commun. 348, 36–46 [DOI] [PubMed] [Google Scholar]
- 18.Shigeishi H., Higashikawa K., Ono S., Mizuta K., Ninomiya Y., Yoneda S., Taki M., Kamata N. (2006) Oncol. Rep. 16, 1071–1075 [PubMed] [Google Scholar]
- 19.Liao W. T., Song L. B., Zhang H. Z., Zhang X., Zhang L., Liu W. L., Feng Y., Guo B. H., Mai H. Q., Cao S. M., Li M. Z., Qin H. D., Zeng Y. X., Zeng M. S. (2007) Clin. Cancer Res. 13, 508–514 [DOI] [PubMed] [Google Scholar]
- 20.Guo X. Z., Zhang G., Wang J. Y., Liu W. L., Wang F., Dong J. Q., Xu L. H., Cao J. Y., Song L. B., Zeng M. S. (2008) BMC Cancer 8, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao W. T., Wang X., Xu L. H., Kong Q. L., Yu C. P., Li M. Z., Shi L., Zeng M. S., Song L. B. (2009) Cancer 115, 1507–1517 [DOI] [PubMed] [Google Scholar]
- 22.Liao W. T., Yu C. P., Wu D. H., Zhang L., Xu L. H., Weng G. X., Zeng M. S., Song L. B., Li J. S. (2009) J. Exp. Clin. Cancer Res. 28, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawakami K., Takeda H., Kawakami N., Kobayashi M., Matsuda N., Mishina M. (2004) Dev. Cell 7, 133–144 [DOI] [PubMed] [Google Scholar]
- 24.Zhao L., Zhao X., Tian T., Lu Q., Skrbo-Larssen N., Wu D., Kuang Z., Zheng X., Han Y., Yang S., Zhang C., Meng A. (2008) Cardiovasc. Res. 80, 200–208 [DOI] [PubMed] [Google Scholar]
- 25.Tian T., Zhao L., Zhao X., Zhang M., Meng A. (2009) J. Genet. Genomics 36, 581–589 [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y., Li X., Qi J., Wang J., Liu X., Zhang H., Lin S. C., Meng A. (2009) J. Cell Sci. 122, 2197–2207 [DOI] [PubMed] [Google Scholar]
- 27.Robu M. E., Larson J. D., Nasevicius A., Beiraghi S., Brenner C., Farber S. A., Ekker S. C. (2007) PLoS Genet. 3, e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X., Jia S., Wang S., Wang Y., Meng A. (2009) Blood 114, 5464–5472 [DOI] [PubMed] [Google Scholar]
- 29.Eimon P. M., Kratz E., Varfolomeev E., Hymowitz S. G., Stern H., Zha J., Ashkenazi A. (2006) Cell Death Differ. 13, 1619–1630 [DOI] [PubMed] [Google Scholar]
- 30.Kratz E., Eimon P. M., Mukhyala K., Stern H., Zha J., Strasser A., Hart R., Ashkenazi A. (2006) Cell Death Differ. 13, 1631–1640 [DOI] [PubMed] [Google Scholar]
- 31.Geiger G. A., Parker S. E., Beothy A. P., Tucker J. A., Mullins M. C., Kao G. D. (2006) Cancer Res. 66, 8172–8181 [DOI] [PubMed] [Google Scholar]
- 32.Haupt S., Berger M., Goldberg Z., Haupt Y. (2003) J. Cell Sci. 116, 4077–4085 [DOI] [PubMed] [Google Scholar]
- 33.Chipuk J. E., Green D. R. (2006) Cell Death Differ. 13, 994–1002 [DOI] [PubMed] [Google Scholar]
- 34.Fukagawa T. (2008) Front. Biosci. 13, 2705–2713 [DOI] [PubMed] [Google Scholar]
- 35.Przewloka M. R., Glover D. M. (2009) Annu. Rev. Genet. 43, 439–465 [DOI] [PubMed] [Google Scholar]
- 36.Sugata N., Li S., Earnshaw W. C., Yen T. J., Yoda K., Masumoto H., Munekata E., Warburton P. E., Todokoro K. (2000) Hum. Mol. Genet. 9, 2919–2926 [DOI] [PubMed] [Google Scholar]
- 37.Amatruda J. F., Shepard J. L., Stern H. M., Zon L. I. (2002) Cancer Cell 1, 229–231 [DOI] [PubMed] [Google Scholar]
- 38.Feitsma H., Cuppen E. (2008) Mol. Cancer Res. 6, 685–694 [DOI] [PubMed] [Google Scholar]
- 39.Spitsbergen J. M., Tsai H. W., Reddy A., Miller T., Arbogast D., Hendricks J. D., Bailey G. S. (2000) Toxicol. Pathol. 28, 716–725 [DOI] [PubMed] [Google Scholar]
- 40.Shepard J. L., Amatruda J. F., Stern H. M., Subramanian A., Finkelstein D., Ziai J., Finley K. R., Pfaff K. L., Hersey C., Zhou Y., Barut B., Freedman M., Lee C., Spitsbergen J., Neuberg D., Weber G., Golub T. R., Glickman J. N., Kutok J. L., Aster J. C., Zon L. I. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13194–13199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shepard J. L., Amatruda J. F., Finkelstein D., Ziai J., Finley K. R., Stern H. M., Chiang K., Hersey C., Barut B., Freeman J. L., Lee C., Glickman J. N., Kutok J. L., Aster J. C., Zon L. I. (2007) Genes Dev. 21, 55–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hong J. R., Lin G. H., Lin C. J., Wang W. P., Lee C. C., Lin T. L., Wu J. L. (2004) Development 131, 5417–5427 [DOI] [PubMed] [Google Scholar]
- 43.Negron J. F., Lockshin R. A. (2004) Dev. Dyn. 231, 161–170 [DOI] [PubMed] [Google Scholar]
- 44.Fowler K. J., Hudson D. F., Salamonsen L. A., Edmondson S. R., Earle E., Sibson M. C., Choo K. H. (2000) Genome Res. 10, 30–41 [PMC free article] [PubMed] [Google Scholar]
- 45.Hemmerich P., Weidtkamp-Peters S., Hoischen C., Schmiedeberg L., Erliandri I., Diekmann S. (2008) J. Cell Biol. 180, 1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dobles M., Liberal V., Scott M. L., Benezra R., Sorger P. K. (2000) Cell 101, 635–645 [DOI] [PubMed] [Google Scholar]
- 47.Nishihashi A., Haraguchi T., Hiraoka Y., Ikemura T., Regnier V., Dodson H., Earnshaw W. C., Fukagawa T. (2002) Dev. Cell 2, 463–476 [DOI] [PubMed] [Google Scholar]
- 48.Tadeu A. M., Ribeiro S., Johnston J., Goldberg I., Gerloff D., Earnshaw W. C. (2008) EMBO J. 27, 2510–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weaver B. A., Silk A. D., Montagna C., Verdier-Pinard P., Cleveland D. W. (2007) Cancer Cell 11, 25–36 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}