

Abstract
HbpS is an extracellular oligomeric protein, which has been shown to act in concert with the two-component system SenS-SenR during the sensing of redox stress. HbpS can bind and degrade heme under oxidative stress conditions, leading to a free iron ion. The liberated iron is subsequently coordinated on the protein surface. Furthermore, HbpS has been shown to modulate the phosphorylation state of the sensor kinase SenS as, in the absence of oxidative stress conditions, HbpS inhibits SenS autophosphorylation whereas the presence of heme or iron ions and redox-stressing agents enhances it. Using HbpS wild type and mutants as well as different biochemical and biophysical approaches, we show that iron-mediated oxidative stress induces both secondary structure and overall intrinsic conformational changes within HbpS. We demonstrate in addition that HbpS is oxidatively modified, leading to the generation of highly reactive carbonyl groups and tyrosine-tyrosine bonds. Further examination of the crystal structure and subsequent mutational analyses allowed the identification of the tyrosine residue participating in dityrosine formation, which occurs between two monomers within the octomeric assembly. Therefore, it is proposed that oxidative modifications causing structural and conformational changes are responsible for the control of SenS and hence of the HbpS-SenS-SenR signaling cascade.
Keywords: Bacterial Signal Transduction, Oxidative Stress, Protein Conformation, Protein Cross-linking, Reactive Oxygen Species (ROS), Streptomyces, Dityrosines, Iron-mediated Stress, Oxidative Modification, Redox Sensing
Introduction
Iron is the fourth most abundant element in the Earth's crust and is an essential trace mineral for nearly all known organisms. Under physiological conditions, it exists either in the reduced Fe2+ (ferrous) or in the oxidized Fe3+ (ferric) form. It plays a crucial role in many biological processes, as photosynthesis, N2 fixation, H2 production and consumption, respiration, oxygen transport, or gene regulation. Because of its redox potential ranging from −300 to +700 mV, iron is a versatile prosthetic component that can be incorporated into proteins either as a mono- or binuclear species, or in a more complex form as part of iron-sulfur clusters or heme groups (1–3).
In the presence of oxygen, iron ions frequently lead to the formation of redox stress by the generation of reactive oxygen species (ROS)2 such as the superoxide radical anion (O2˙̄), hydrogen peroxide (H2O2), and the hydroxyl radical (OH·). Oxidation of ferrous iron by molecular oxygen (Reaction 1) yields O2˙̄ that can undergo the dismutation reaction (Reaction 2) to form H2O2. Hydrogen peroxide in turn can react with the ferrous iron via the Fenton reaction (Reaction 3), generating a hydroxyl radical.
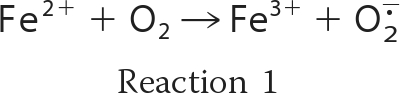 |
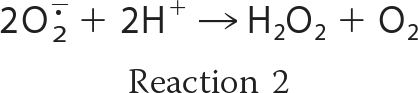 |
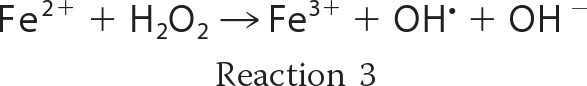 |
ROS can provoke the damage of DNA, lipids, and proteins (4–6). For instance, when proteins are exposed to ROS, they undergo a variety of oxidative modifications including: Nitration of aromatic amino acid residues, hydroxylation of aromatic groups, and aliphatic amino acid side-chains, sulfoxidation of methionine residues, and conversion of some amino acid residues to carbonyl derivatives. Oxidation can also induce the cleavage of the polypeptide chain and the formation of cross-linked protein derivatives (7, 8). These modifications can lead to functional changes of proteins that subsequently disturb the cellular metabolism. Thus, while bacteria and other organisms have to ensure that enough iron ions are present for the diverse biochemical reactions, they also have to avoid their harmful effects.
We have previously identified the two-component system SenS-SenR in the cellulose degrader Streptomyces reticuli (S. reticuli) and could demonstrate that it participates in the sensing of iron-mediated redox signals (9, 10). SenS is a histidine autokinase, and SenR its cognate response regulator, which together regulate the production of different redox active proteins such as the catalase-peroxidase CpeB, the regulator FurS and the heme-binding protein HbpS (11). Interestingly, we demonstrated that the extracellularly located HbpS interacts specifically with the membrane embedded histidine autokinase SenS. Further phosphorylation analyses revealed that under non-oxidative stress conditions high quantities of HbpS inhibit the autophosphorylation of SenS. However, HbpS significantly enhanced SenS autokinase activity in the presence of different redox-cycling compounds like heme, iron, DTT or H2O2 (10). This suggests that the switching of HbpS from its “inhibitor to activator” state of SenS autophosphorylation under oxidative stress conditions is controlled by conformational changes in HbpS.
Secretion of the oligomer-forming HbpS occurs in a Tat (twin-arginine translocation) dependent manner (12, 13). Homologs of HbpS exist in a number of ecologically relevant bacteria (including S. coelicolor A3(2), S. kasugaensis, Arthrobacter aurescens, Pseudomonas fluorescens, Pseudomonas putida, and Sphingomonas aromaticivorans) and medically relevant bacteria (including Vibrio cholera, Yersinia enterolitica, Leifsonia xyli, Acinetobacter baumani, Photorabdus luminiscens, and Bordetella avium) (11). Comparative physiological studies revealed that HbpS in S. reticuli provides an increased resistance against high concentrations of iron and heme (12, 10). We have previously reported the high resolution crystal structures (1.6 Å and 2.3 Å) of HbpS crystallized in the presence or absence of hemin (Fe3+-oxidized form of heme). A detailed examination of the HbpS crystal structure with a subsequent generation of mutants and biochemical and biophysical analyses allowed us to identify two residues (Ser-26 and His-28) that are essential for the oligomeric assembly of HbpS. Further studies demonstrated that this assembly is required for efficient interaction with SenS and modulation of its phosphorylation state (14).
Electron density analysis from the crystal structure obtained in the presence of hemin revealed the presence of a bound iron. Subsequently, spectroscopic and biochemical studies demonstrated that HbpS can bind and degrade heme through a non-enzymatic H2O2-dependent mechanism known as “coupled oxidation,” leading to free iron. The liberated iron is then coordinated on the surface of HbpS. This surface displayed iron is assumed to act as mediator of oxidative stress conditions leading to the start of the HbpS-SenS-SenR-signaling cascade (14, 11).
The aim of the present report is to gain deeper insight into the molecular mechanisms encompassing conformational changes within HbpS under iron-mediated oxidative stressing conditions. For this purpose, we have used mutant and wild-type (WT) HbpS, as well as different biochemical and biophysical techniques such as fluorescence resonance energy transfer (FRET), circular dichroism (CD) spectroscopy, fluorescence spectroscopy, SDS-PAGE, and immunoblotting. The existing crystal structure of HbpS was used to design experiments as well as to analyze the obtained data. We present data showing that amino acid side-chains in HbpS become oxidized upon iron-mediated oxidative stress. These modifications provoke secondary structure and overall conformational changes. Furthermore, using different S. reticuli strains and transcriptional studies the in vivo effect of iron-mediated oxidative stress on the expression of some genes belonging to the HbpS-SenS-SenR regulon was investigated.
MATERIALS AND METHODS
Bacterial Strains, Plasmids, Media, and Culture Conditions
The wild-type S. reticuli Tü54 (S. reticuli WT), S. reticuli hbpS mutant (12), and S. reticuli senS-senR mutant (9) as well as the Escherichia coli strains BL21(DE3)pLysS (Novagen), and DH5α (15) were used. The plasmid constructs pETHbpS (16) and pETHbpSHis28Ala (14) were taken to introduce specific mutations into the hbpS gene (see below). E. coli strains were grown in LB medium (17). Suspensions of S. reticuli spores were made as described elsewhere (18), and inoculated and propagated as described earlier (19).
Cleavage of DNA, Ligation, and Agarose Gel Electrophoresis
Plasmids were isolated from E. coli using a mini plasmid kit (Qiagen) and cleaved with various restriction enzymes according to the suppliers' (New Englad BioLabs) instructions. Ligation was performed with T4 ligase (Promega). Gel electrophoresis was carried out in 0.8–2% agarose gels using TBE buffer. Plasmids were used to transform E. coli DH5α by electroporation (20), or E. coli BL21 (DE3)pLys with the CaCl2 method as described (17).
Site-directed Mutagenesis
Point mutations in the hbpS gene on the plasmid pETHbpS or pETHbpSHis28Ala were introduced using either the overlap extension PCR technique that employs the two-step PCR method (21) or single PCR reactions. For the two-step PCR method overlapping mutagenic primers (MP; containing the desired mutation) as well as flanking primers (FP; designed to one end of the region of interest) were used. To obtain HbpSSer139C and HbpSHis28Ala-Ser139Cys following primers (5′ → 3′) were used: FPS139CFor1 (GACGGGGCGGGCCCGCAGTCGTACG), MPS139CRev1 (CGCCGCACGGAGCACCCGCGACGC), MPS139CFor2 (CCGTGCGGCG-ACCTGGACGAGCAGTAC) and FPS139C-Rev2 (CCCCTCAAGACCCGTTTAGAGGCCC); for HbpSTyr77Phe: FPY77PFor1 (GG-CGCCATGGCCGACACCACGGAG), MPY77-PRev1 (GACTCGAACGACTGCGGGCCCGCCCCG), MPY77FFor2 (GTCGTTCGAGTC-CGCGGAGCGCAAG) and FPY77FRev2 (GGGCAAGCTTGTGGCCGAGCACGG). To generate the HbpSTyr146Phe mutant, a single PCR step was performed using following primers FPY77PFor1 (see above) and MPY146FRev (GGCAAGCTTTCAGTGGCC-GAGCACGGCCGCGCCCGCCCGTGCGAACTGCTCGTCCAG). The numbering of the amino acids is in agreement with the sequence of the three-dimensional structure of HbpS (PDB ID: 3FPV). After PCRs, the amplicons were restricted and ligated. The ligation products were entered into E. coli DH5α. Subsequently, each of the obtained plasmid constructs was analyzed with restriction enzymes and by sequencing. The resulting correct plasmids were named pETHbpSSer139Cys, pETHbpSHis28Ala-Ser139Cys, pETHbpSTyr77Phe, and pETHbpSTyr146Phe, respectively. To overproduce the corresponding proteins, these plasmids were used to transform E. coli BL21 (DE3) pLysS as reported by Zou et al. (16).
Production, Purification, and Preparation of HbpS Samples
Protein production and purification were performed as described earlier (16), using Ni-NTA affinity chromatography, TEV protease cleavage and anion exchange chromatography over a DEAE-Sepharose column. The homogeneity of the His tag-free HbpS protein solutions (WT, H28A, S139C, H28A-S139C, Y77F, and Y146F, respectively) was checked by SDS-PAGE and by mass spectrometry analysis (ESI LC-MS). The concentration of purified HbpS proteins was calculated from their absorbance at 280 nm, assuming an ϵ280 of 8250 m−1 cm−1 (molecular mass = 15,498 Da) or using the Bradford method (22). Both methods yielded almost identical values.
To obtain oxidative stress samples, HbpS (25–50 μm) proteins were incubated with FeCl2 (500 μm) and DTT (5 mm) overnight at 30 °C. The presence of DTT in the reaction mixtures retains the iron ions in their ferrous (II) form, which under aerobic conditions can react with the spontaneously existing H2O2 via the Fenton reaction, leading to the formation of hydroxyl radicals (see introduction). In the absence of DTT and under O2-rich conditions, the iron ferrous form can be rapidly oxidized to the ferric (III) form, which is not able to act as a substrate for the Fenton reaction. For fluorescence measurements, samples were subsequently incubated with an excess of EDTA (10 mm) for 1 h to remove the iron ions. Prior to measurement, the reaction mixtures were purified using a PD10 gel filtration column previously equilibrated and eluted with the indicated buffer. Modifications of the sample preparation procedure will be appropriately stated.
Fluorescence Energy Transfer (FRET)
Labeling with IAEDANS
The labeling of the single cysteine in HbpSSer139C (S139C) and HbpSHis28A-Ser139C (H28A-Ser139C), respectively, was performed by reaction with the thiol-reactive fluorescent dye IAEDANS (Molecular Probes) in PBS buffer (pH 7.4) after treatment of the protein with 5 mm TCEP (Sigma). The reaction was carried out at 30 °C, in a light-proof vessel for 16 h. The labeled protein was subsequently purified and buffer-exchanged with buffer A (20 mm Tris-HCl, pH 7, 150 mm NaCl and 10% glycerol) using a PD10 (Sephadex G-25 medium) column (GE Healthcare). The protein solution was concentrated with a Microcon YM-3 Centrifugal Filter Unit (Milipore). All spectral measurements for unlabeled S139C proteins (WT and H28A, respectively) were performed in the presence of 2 mm DTT.
Steady-state Fluorescence Measurements
Fluorescence measurements were performed using a Jasco FP-6500 fluorimeter. Tryptophan was excited with a wavelength of 295 nm. The cell path-length was 1 cm and emission bandwidths were 5 nm. All samples contained 2 μm of HbpS proteins. Tryptophan at position 90 in HbpS was used as a natural fluorophore whereas IAEDANS at position S139C (Ser139Cys-IAEDANS) acted as an acceptor for FRET. Comparison between the fluorescence intensity of the donor in the absence and in the presence of the acceptor enabled the estimation of the donor-acceptor distance. Fluorescence energy transfer by dipolar interaction is described by the Förster rate equation: E = R06/(r6 + R06), where E is the energy transfer efficiency, R0 is the Förster or critical transfer distance, at which the energy transfer rate is equal to the decay rate, i.e. 50% fluorescence energy transfer efficiency, and r is the donor-acceptor distance. In the case of the IAEDANS/Trp pair, the value of R0 is 22.8 Å (23). On the basis of this value, the value of r (distance between Trp-90 and incorporated IAEDANS) was calculated for various states.
CD Spectroscopy
CD measurements of HbpS were recorded with a Jasco J-810A spectropolarimeter. The spectra in the far-UV region (180–250 nm), in which the absorbing group is principally the peptide bond, were obtained at room temperature in a quartz cell (0.01 cm optical path length) at a scanning speed of 50 nm/min with a sampling interval of 1 nm. To improve the signal/noise ratio the scans were averaged (n = 16). The samples contained native or treated HbpS (25 μm) in a 20 mm sodium phosphate buffer (0.5 m NaH2PO4/0.5 m Na2HPO4, pH 7.0). To analyze the effects of iron-mediated stress, the proteins were preincubated with FeCl2 (0.5 or 1 mm) in combination with or without DTT (5 mm). Molar ellipticity values were calculated by using a value of 15,498 Da for the mean residue weight of HbpS. Prediction of the secondary structure was derived by deconvoluting the data sets by a neural approach (24), and the CDpro package (25–27).
SDS-PAGE and Western Blotting
HbpS solutions were treated with the indicated concentrations of DTT and/or iron ions (FeCl2) in buffer A. In parallel, control reactions containing additional recombinant catalase from bovine liver or an excess of additional H2O2 (both from Sigma) were performed. After overnight incubation, the reactions were terminated by addition of loading buffer and analyzed on 12% or 15% SDS-polyacrylamide gels. After SDS-PAGE gels were stained with Coomassie Blue or transferred to a membrane and treated with anti-HbpS antibody as described (12).
Fluorescence Detection of Dityrosines
The appearance of dityrosines was monitored as previously described (28) with modifications. HbpS solutions (WT, H28A, Y77F, and Y146F, respectively) were prepared in buffer A and reacted with the indicated concentrations of DTT and/or iron ions at 30 °C. After overnight incubation, the reaction mixtures were treated with an excess of EDTA for 1 h. The samples were then purified with a PD10 gel filtration column previously equilibrated and eluted with 200 mm sodium bicarbonate buffer (pH 9.4). After excitation at 315 nm, the fluorescence intensities of the resulting mixtures were measured from 325 to 500 nm using a Jasco FP-6500 fluorescence spectrophotometer (Analytik Jena).
Detection of Carbonyl Groups
To detect protein carbonyl groups, which are introduced into amino acid side-chains during protein oxidation, the OxyBlot oxidation kit (Chemicon International) was used. Ten microliters of treated (FeCl2 + DTT) or untreated HbpS samples were mixed for 10 min with an equal volume of 12% SDS. 20 μl of 1× DNPH (dinitrophenylhydrazine) were added to the reaction mixture to perform dinitrophenyl (DNP) derivatization of the carbonyl groups. After 15 min of incubation at room temperature, the reaction was stopped by the addition of 15 μl of neutralization buffer. Aliquots of the samples were directly loaded to 12% SDS-polyacrylamide gels. After SDS-PAGE, proteins on the gel were either stained with Coomassie Blue or transferred to a membrane, on which the DNP groups were identified using supplied polyclonal anti-DNP antibodies. Anti-rabbit IgG coupled with alkaline phosphatase (Sigma) was used as secondary antibody.
Measurement of the Autokinase Activity of SenS
To perform in vitro phosphorylation assays the histidine autokinase SenS was first isolated as described previously (10). To analyze the effect of HbpS proteins (WT, S139C, Y77F, and Y146F) on the autokinase activity of SenS, aliquots (5 μm) of treated (with 500 μm FeCl2 and 5 mm DTT) HbpS proteins were added to SenS (5 μm) in phosphorylation buffer and incubated for 10 min at room temperature. Subsequently, 0.05 μCi of [γ-32P]ATP was added to the reaction mixture and further incubated at 30 °C for 10 min, and directly spotted onto a polyvinylidene fluoride (PVDF) membrane (Millipore). Signals detection and quantification was done using a PhosphorImager system and the ImageQuant 5.2 software as previously described (10, 14). Control samples of SenS were incubated in the presence of 0.05 μCi of [γ-32P]ATP alone to establish a baseline activity for SenS autokinase activity. The values presented indicate the area-integrated radioactivity of the sample divided by the area-integrated intensity of the control sample.
RNA Isolation and Analysis of Transcripts
To obtain well-grown mycelia, S. reticuli spores were inoculated in complete medium and cultivated as previously described (19). The cultures were subsequently washed four times with minimal medium (MM) and thereafter cultivated in MM containing crystalline cellulose (1% final concentration) in the absence or presence of 1 mm FeCl2 previously treated with 10 mm DTT. After 1 h of incubation, the genomic RNA was isolated using guanidinthiocyanate as described earlier (19). Isolated RNAs were treated with DNase using the TURBO DNA-freeTM kit (Applied Biosystems) to eliminate remain traces of DNA. The quality of the RNA was controlled on a 2% agarose gel and by PCR. RNA concentrations were estimated by UV spectrometry. To detect the mRNA expression levels of cpeB, hbpS, and the gene encoding the 16 S rRNA the OneStep RT-PCR Kit (Qiagen) was used. For this purpose 1 μg of the DNA-free genomic RNA was used as a template combined with specific oligonucleotides. To obtain the cpeB-cDNA following primers (5′ → 3′) were used: PCpeBFor (GACGCGATCGTCACAGACGCG-AAG) and PCpeBRev (GCAGGTCGGGCCC-AGGAGATGCTCTG); for the hbpS-cDNA: PHbpSFor (GAGACCATGGCCGACACCACG-GAG) and PHbpSRev (GGGCAAGCTTGTG-GCCGAGCACGG); and for the cDNA of the 16 S rRNA gene: P16sRNAFor (ACAAGCCT-GGAAACGGGGT) and P16sRNAFor (CACC-AGGAATTCCGATCT). The “Rev” primers acted as primers for the reverse transcription. The oligonucleotides used for amplification of the 16 S rRNA gene from Streptomyces species were previously reported (29).
RESULTS
Fluorescence Energy Transfer in HbpS
The FRET technique allows the estimation of the distance between two fluorophores (30–32). As relatively small perturbations of inter-fluorophore distances can change the intensity of FRET signals, this technique provides evidence of protein conformational changes. Usually, cysteine residues are efficient targets to covalently attach thiol-reactive fluorescence dyes. Interestingly, HbpS does not possess any cysteine residues. This property as well as the availability of the three-dimensional structure of HbpS allowed the choice of the serine residue at position 139 (Ser-139) for the insertion of a cysteine residue. Ser-139 was selected because of its high solvent accessibility as well as its location close to the end of the C terminus (Fig. 1). Another interesting feature of HbpS is the fact that it possesses only one tryptophan residue (at position 90, Trp-90) (Fig. 1), which also displays high solvent accessibility. Trp-90 was used as a donor (excitation wavelength 295 nm). As the acceptor dye IAEDANS (emission wavelength ∼480 nm) was selected. IAEDANS was covalently attached to designed HbpSSer139Cys and HbpSHis28A-Ser139Cys mutant proteins. In vitro phosphorylation assays revealed that like the wild-type protein the HbpSSer139Cys mutant induces a 12-fold increase of SenS autophosphorylation under iron-mediated oxidative stressing conditions, indicating that the exchange of Ser-139 to Cys has no effect on the function of the protein.
FIGURE 1.

An image of the structure of one HbpS subunit showing the location of His-28 (H28), Tyr-77 (Y77), Trp-90 (W90), Lys-108(K108), Phe-115 (F115), Ser-139 (S139), and Tyr-146 (Y146). The figure was produced using NOC.
A fluorescence energy transfer efficiency (EFRET) of ∼50% was observed in the native state of Ser139Cys-IAEDANS samples (Fig. 2 and Table 1). According to the crystal structure, the distance between the side-chains of Trp-90 and Ser-139 within one subunit of the octomeric HbpS is ∼22 Å. As HbpS displays eight monomers subunits further interactions between different Trp-90 and Ser-139C from different monomer subunits within the octomer could be expected. The distances are ranging from 10 to 54 Å. Using a simplified model of one donor/acceptor pair, the distance estimated from steady-state FRET in S139C as well as in H28A-S139C was 21 and 21.7 Å, respectively. Further distances were estimated after treatment of the proteins under different conditions (Table 1). Upon the exposure of Ser139Cys-IAEDANS proteins to redox-stressing conditions (FeCl2 and DTT), the calculated distance between Trp-90 and Ser139Cys-IAEDANS was estimated at 16.2 Å, which represents an increase of EFRET to ∼64%. Under the same oxidative stressing conditions, the His28Ala-Ser139Cys-IAEDANS mutant showed no significant change in the EFRET, compared with its native state; the estimated distance remains almost identical. Similarly, the previous incubation of the samples either with FeCl2 or DTT alone did not provoke significant changes in the EFRET (Table 1). The FRET results indicate that WT-Ser139Cys but not the H28A-S139C mutant undergoes a conformational change upon exposure to oxidative stress, resulting in an increase in efficiency of energy transfer. As the H28A mutant has lost the ability to form octomers (14), the presented data indicate that the oligomeric assembly in HbpS plays an important role during conformational changes.
FIGURE 2.

Fluorescence energy transfer. Two micromolar of unlabeled (solid line) WT-Ser139Cys (WT), and His28Ala-Ser139Cys (H28A) as well as the corresponding ones labeled with IAEDANS (dotted line) were excited at 295 nm. Substantial energy transfer occurs as shown by the large emission intensity from the IAEDANS group in the vicinity of 485 nm.
TABLE 1.
Calculated distances and fluorescence energy transfer efficiency (EFRET) from steady-state FRET in S139C proteins
| Distance | EFRET | |||
|---|---|---|---|---|
| Å | Å | % | % | |
| Condition | WT | H28A | WT | H28A |
| Native | 21 | 21.7 | ∼50 | ∼50 |
| DTT | 21 | 21.6 | ∼50 | ∼50 |
| FeCl2 | 20 | 22 | ∼50 | ∼50 |
| FeCl2 + DTT | 16.2 | 22 | ∼64 | ∼50 |
Oxidative Stress Induces Changes in HbpS Secondary Structure
As CD spectroscopy is a well-established technique to investigate the secondary structure of proteins (33, 34), we analyzed the secondary structures of HbpS to examine possible conformational changes under oxidative stressing conditions. CD spectra of untreated and treated (either with 5 mm DTT or 500 μm FeCl2, respectively) HbpS proteins showed similar shapes of curves with a band at 192 nm and a shoulder between 211–222 nm (Fig. 3A). In the presence of iron ions (FeCl2) and DTT, the amount of α-helical structures seem to be lowered (Fig. 3B). These observations could be verified by a structure analysis in which the fraction of α-helices was decreased from 42% to 33% (using the method I) after treatment with 0.5 mm FeCl2 in combination with DTT. An enhanced decrease of the α-helices was observed (from 42% to 20%) in the presence of 1 mm FeCl2 in combination with DTT. In both cases the percentages of the other secondary structures (β-sheets, random coils, and turns) showed a concomitant increase (Table 2). When HbpS was pre-incubated with FeCl2 alone, a lower decrease (from 42 to 39%) in α-helical structures could be observed. Verification of secondary structures using the other methods (II to IV) revealed an overall similar trend (Table 2). The validity of the CD data is underlined by the fact that the obtained secondary structures (using the method I) for untreated HbpS (α-helices: 42%, β-sheets: 15%, random coils: 24% and turns: 19%) are in agreement with those gained from the crystal structure (α-helices: 44%, β-sheets: 18%, random coils: 21% and turns: 18%).
FIGURE 3.

CD spectroscopy. A, CD spectra of native (line 1) and preincubated with 0.5 mm FeCl2 (line 2), or 5 mm DTT (line 3), respectively, HbpS proteins were recorded with a Jasco-J-810A spectropolarimeter as described under “Materials and Methods.” B, secondary structure changes upon iron-mediated oxidative stress were monitored, using native (line 1) and treated with 5 mm DTT in combination with FeCl2 (0.5 mm, line 2; 1 mm, line 3) HbpS proteins.
TABLE 2.
Calculated amounts of secondary structures of HbpS under different conditions
Four different algorithms were used (see “Materials and Methods”).
| Condition | Method | α-Helices | β-Sheet | Turn | Random coil |
|---|---|---|---|---|---|
| Native | I | 42 | 15 | 19 | 24 |
| II | 43 | 15 | 19 | 23 | |
| III | 44 | 15 | 20 | 22 | |
| IV | 42 | 13 | 18 | 27 | |
| DTT | I | 42 | 15 | 20 | 24 |
| II | 41 | 16 | 21 | 23 | |
| III | 44 | 15 | 20 | 21 | |
| IV | 39 | 14 | 18 | 29 | |
| FeCl2 | I | 39 | 15 | 20 | 26 |
| II | 40 | 16 | 19 | 25 | |
| III | 39 | 17 | 19 | 25 | |
| IV | 39 | 15 | 17 | 30 | |
| FeCl2 (0.5 mm) + DTT | I | 33 | 16 | 22 | 29 |
| II | 42 | 17 | 21 | 30 | |
| III | 37 | 18 | 18 | 27 | |
| IV | 32 | 14 | 20 | 34 | |
| FeCl2 (1 mm) + DTT | I | 20 | 25 | 22 | 33 |
| II | 26 | 18 | 23 | 33 | |
| III | 19 | 26 | 21 | 35 | |
| IV | 16 | 27 | 28 | 30 |
Iron-mediated Cross-linking of HbpS
In addition to the altered spectral properties of HbpS under oxidative stressing conditions, we could observe the formation of SDS-insensitive HbpS dimers and other high order oligomers upon SDS-PAGE and Western blot analysis. The untreated sample showed a protein band with a molecular weight of ∼16 kDa representing the monomer form of the protein (15.5 kDa). A dimeric form with low signal intensity can also be observed (Fig. 4, A and B, lane 1). Treatment of HbpS with increasing concentrations (100, 250, and 500 μm) of iron ions (FeCl2) and DTT led to the formation of SDS-resistant dimeric and tetrameric forms (Fig. 4, A and B, lanes 2–4). Under higher concentrations of iron ions a higher oligomeric form could be seen migrating between 175 und 80 kDa (Fig. 4, A and B, lanes 3–4), we have identified this as the native octomer, with expected molecular mass of 124 kDa. These cross-linking events cannot be attributed to disulfide (S-S) bridges, as HbpS does not possess a cysteine residue. Interestingly, when a bovine recombinant catalase (Sigma) was previously added to the reaction mixture to remove any H2O2 present and thus inhibit the formation of hydroxyl radicals via the Fenton reaction, the generation of cross-linked species was blocked (Fig. 4, A and B, lane 5). However, the addition of H2O2 (1 mm) for 15 min to the treated (with FeCl2 and DTT) sample led to the appearance of degraded HbpS products (Fig. 4, A and B, lane 6), indicating a role of increased formation of hydroxyl radicals. To investigate the influence of H2O2 in more detail, the treated (with FeCl2 and DTT) HbpS sample was incubated with 1 mm H2O2 for different times (10, 30, 60, and 120 min). A concomitant appearance of HbpS degradation products could be observed increasing with time. At the same time the apparent levels of cross-linked species slightly decreased (Fig. 4, C and D).
FIGURE 4.

Iron-mediated cross-linking of HbpS. HbpS WT (A and B) and HbpSHis28Ala (G and H) proteins were treated with FeCl2 (100 μm, lane 2; 250 μm, lane 3; 500 μm, lanes 4–6) and 5 mm DTT. Recombinant catalase was added before start of treatment (lane 5). Additional H2O2 was added after treatment (lane 6). All treated samples as well as the native one (lane 1) were subjected to 12% SDS-PAGE. C and D, WT-treated sample (with FeCl2 and DTT; lane 2) was further incubated with H2O2 for different times (10 min, lane 3; 30 min, lane 4; 60 min, lane 5; 120 min lane 6). All treated samples as well as the native one (lane 1) were subjected to 15% SDS-PAGE. E and F, HbpS was treated with FeCl2 (lane 1), FeCl3 (lane 2), or DTT (lane 3). Aliquots of each of these samples were incubated with H2O2 (H2O2 + FeCl2, lane 4; H2O2 + FeCl3, lane 5; H2O2 +DTT, lane 6). All samples were subjected to 12% SDS-PAGE. After electrophoresis, proteins were analyzed by Coomassie staining (A, C, E, and G) or by immunoblotting using anti-HbpS antibodies (B, D, F, and H). Arrows indicate positions for the monomeric (1mer), dimeric (2mer), tetrameric (4mer), or octameric (8mer) HbpS forms. The degradation products are also indicated (Deg). The approximate sizes of prestained protein markers are also shown (lane 7).
Incubation of HbpS with FeCl2 or FeCl3 orDTT alone did not induce the formation of cross-linked HbpS forms. Addition of H2O2 to such treated samples also did not show any effect (Fig. 4, E and F), implicating that the ferrous form needed for the Fenton reaction is not available. As an additional control, H28A sample reactions were run in parallel. Here, only slight bands representing a dimeric form could be observed (Fig. 4, G and H).
Oxidation of HbpS Leads to Dityrosine Formation
It is well known that hydroxyl radicals promote considerable protein-protein cross-linking by the formation of Cys-Cys (S-S) or Tyr-Tyr (dityrosine) bridges (7). Dityrosine is a covalently bound diphenol, which can be produced under oxidative stress conditions by a mechanism encompassing the formation of tyrosyl radicals, diradical reaction, and finally enolization. They can be detected by their characteristic ultraviolet fluorescence (28, 35–37). Fluorescence measurements were performed to assess the formation of dityrosines involved in the generation of cross-linked HbpS derivatives. Contrary to an untreated HbpS WT sample, those treated with DTT and FeCl2 showed a fluorescence emission spectrum with a maximum around 410 nm when exposed to 315 nm light, consistent with the formation of dityrosine cross-links (Fig. 5).
FIGURE 5.

Detection of dityrosines. The fluorescence emission spectra of native (solid line) and treated (dotted line) HbpS proteins (5 μm) are shown. The excitation wavelength was set at 315 nm. The emission maximun of Tyr-Tyr bonds is achieved at ∼410 nm.
As HbpS possesses two tyrosine residues (Tyr-77 and Tyr-146) that could be involved in the formation of dityrosine cross-links, the corresponding codons were individually exchanged by for those of phenylalanine. Subsequently, fluorescence emission spectra of the corresponding HbpS mutants were recorded. Under oxidative stress conditions Y146F displayed almost an identical dityrosine signal to the WT protein, while the Y77F mutant did not show this signal (Fig. 5), leading to the conclusion that Tyr-77 plays an essential role in dityrosine formation. It could be assumed that a pre-requisite for such Tyr-Tyr bonds is the physical vicinity between two tyrosines from at least two different protein monomers. The clearly lower dityrosine signal intensity displayed by similarly treated His28Ala samples (Fig. 5) corroborates this assumption, as this mutant is monomeric in vitro (14).
To test the functionality of the tyrosine mutants in terms of modulation of SenS autophosphorylation, in vitro phosphorylation assays were performed. Similar to the wild-type protein, the presence of the HbpSTyr146Phe mutant led to an identical increase (12-fold) in SenS autophosphorylation upon iron-mediated oxidative stress. Under the same conditions, the HbpSTyr77Phe mutant provoked an 8-fold increase of SenS autophosphorylation, indicating that Tyr-77 plays a role in the function of HbpS.
Introduction of Carbonyl Groups into HbpS during Oxidative Stress
Metal-catalyzed protein oxidation can also lead to the generation of protein derivatives possessing highly reactive carbonyl groups (aldehydes and ketones), which in turn can be involved in the formation of intra- or inter-protein cross-linking events (8). The observed cross-linked HbpS forms (Fig. 4) were analyzed for the presence of carbonyl groups using the OxyBlot kit as described under “Materials and Methods.” Contrary to the untreated sample, the treated ones showed an apparently similar degree of oxidation (Fig. 6). The monomeric, dimeric, as well as tetrameric HbpS forms observed after Coomassie staining (Fig. 6A) immunoreacted with an anti-DNP antibody (Fig. 6B), indicating that the iron-catalyzed oxidation of HbpS led to the introduction of carbonyl groups into its amino acid side-chains and to the subsequent generation of cross-linked species.
FIGURE 6.

Carbonylation of HbpS amino acid side-chains. Untreated (lane 1) or treated with FeCl2 (100 μm, lane 2; 250 μm, lane 3; 500 μm, lane 4) and 5 mm DTT HbpS WT proteins were subjected to DNP derivatization as described under “Materials and Methods.” Reaction products were loaded, without previous boiling, onto 12% SDS-polyacrylamide gels. After electrophoresis, proteins were analyzed by Coomassie staining (A) or by detecting derivatized DNP groups by immunoblotting using anti-DNP antibodies (B). Arrows indicate positions for the monomeric (1mer), dimeric (2mer), or tetrameric (4mer) HbpS forms. The approximate sizes of prestained protein markers are also shown (lane 5).
Physiological Studies
As HbpS as well as SenS-SenR is involved in the transcriptional regulation of cpeB and hbpS (9, 12), the expression of cpeB and hbpS in S. reticuli strains (wild-type, hbpS- and senS-senR-disruption mutants) upon iron-mediated oxidative stress was analyzed by RT-PCR. cDNAs corresponding to the cpeB and hbpS genes could be detected only in the stressed WT strain (Fig. 7), indicating that for the sensing of iron-mediated stress leading to the transcriptional activation of cpeB and hbpS a functional HbpS-SenS-SenR system is required. As a control the levels of transcripts of the gene for the 16 S rRNA were analyzed. In all cases, the level of this transcript remained equal (Fig. 7).
FIGURE 7.

Transcriptional analyses. S. reticuli wild-type (WT), hbpS disruption mutant (Δh), and senS-senR disruption mutant (Δs-r) were cultivated in the absence (−) or presence (+) of FeCl2 previously treated with DTT. After isolation of total RNAs, RT-PCRs were performed to amplify cDNAs corresponding to cpeB (cpeB-cDNA), hbpS (hbpS-cDNA) and the gene for the 16 S rRNA (16s rRNA-cDNA). 5 μl of each RT-PCR product were electrophoresed on an agarose gel.
DISCUSSION
In this study, we have analyzed the structural and conformational changes of the oligomeric forming protein HbpS under iron-mediated redox-stressing conditions by a variety of approaches. To probe a change in protein conformation, we applied the FRET technique. The presence of only one tryptophan (Trp-90) residue, which has been used as a natural fluorophore, and the absence of cysteine residues in HbpS provided an opportunity to create mutants and specifically place a fluorophore (IAEDANS) in a selected part (S139C) of the protein. Upon exposure of HbpS to oxidative stress an increased FRET efficiency within the WT-S139C protein could be observed. This is a clear indication of motion of HbpS subunits, leading to an overall conformational change dependent upon correct assembly of HbpS. These assumptions are supported by the observation that the FRET efficiency of the HbpS mutant H28A, which is monomeric in vitro, did not change under identical oxidative stress conditions. The distance between Trp-90 and Ser-139 within one subunit of the octomeric crystal structure matches the distance calculated from the FRET efficiency. However, because HbpS is an octomer, we have to consider seven possible additional donor-acceptor distances that could contribute to the overall FRET efficiency. The exact distance distributions within the octomeric assembly merit therefore further studies. For instance, the combination of site-directed spin labeling with electron paramagnetic resonance measurements and molecular modeling could be useful for this purpose (38). Recently, distance fingerprints were obtained within the cytoplasmic domain of the octomeric Wza protein by using of the pulsed electron-electron double resonance spectroscopy (39).
The conformational changes measured by FRET are accompanied by an altered proportion of secondary structure elements in HbpS. Measurement of HbpS CD spectra, recorded in the far-UV region (180–250 nm), combined with structure analysis using four different methods revealed that the fraction of α-helices is substantially decreased under oxidative stress conditions. This decrease is significantly enhanced (from 42 to 20%) upon addition of higher concentrations of iron ions. At the same time the percentages of β-sheets (from 15 to 25%) were also increased. Thus, HbpS seems to have the ability to interconvert between different forms of secondary structures under changing redox-stressing conditions. The transition of α-helices into β-sheet structures leading to oligomerization and aggregation in certain proteins has been associated with fatal diseases, such as Alzheimer disease and prion disease (40–42). Taking the CD and FRET data together, we can conclude that iron-mediated oxidative stress provokes significant structural and conformational changes in HbpS.
Further analyses revealed that iron-catalyzed oxidative stress led to the formation of SDS-insensitive HbpS oligomers, which have been probably formed by cross-linking processes. Because reduced iron, as a precursor of the Fenton reaction, can generate ROS, we assumed that these cross-linked derivatives were formed by oxidative modifications of amino acid side-chains in HbpS. The building of intra- or inter-protein cross-linked derivatives has been shown to be induced by several different mechanisms, as for example interaction of two tyrosine radicals, direct interaction of two carbon-centered radicals, oxidation of cysteine sulfhydryl groups, or interactions of the carbonyl groups of oxidized proteins with the primary amino groups of lysine residues in the same or a different protein, respectively (8). A metal-catalyzed carbonylation with concomitant cross-linking and degradation was also observed for the Bradyrhizobium japonicum Irr (iron response regulator) protein, which is involved in the regulation of iron transport as well as of heme biosynthesis genes (3, 43). It was concluded that the metal-catalyzed oxidation with subsequent degradation of Irr represents the molecular basis for the modulation of the activity of this regulator. Many common proteases are also involved in protein turnover, as they degrade oxidized proteins more rapidly than unoxidized forms (7). We showed that oxidation of HbpS induces the generation of carbonyl groups, which in part might participate in the formation of cross-linked derivatives. In the presence of exogenously added H2O2 a rapid degradation of HbpS has also been observed. We propose that turnover of HbpS is controlled by both oxidation and degradation processes.
The iron-mediated oxidation of HbpS has been also shown to catalyze the formation of dityrosines, which could be detected by their characteristic fluorescence with a maximum around 410 nm. Dityrosine has been proposed as a biomarker of organism oxidative stress status or of cumulative exposure of proteins to oxidation both in vitro and in vivo (44). Dityrosine cross-linking products have been shown for example in the heme-binding protein neuroglobin upon oxidative stress (28). In general, the formation of dityrosines requires the physical proximity of two tyrosyl radicals to form intra- or intermolecular protein cross-links (45, 46). HbpS has two tyrosine residues (Tyr-77 and Tyr-146) in a single monomer. The distance between these within the monomer is ∼21 Å; the distances between the other possible combinations of the Tyr77-Tyr146 pair within the octomer range from 24 to 50 Å. Further distances from different combinations of Tyr146-Tyr146 pairs within the octomer range from 12 to 54 Å. As all these inter-distances are relatively long, it can be assumed that these tyrosine pairs (Tyr77-Tyr146 or Tyr146-Tyr146, respectively) cannot be involved in the formation of dityrosines in HbpS. Whereas the distance between neighboring Tyr-77 residues is 4.8 Å in the available crystal structure (Fig. 8A and B). Indeed, when Tyr-77 was mutated to Phe the characteristic dityrosine fluorescence upon oxidative stress could no longer be observed. As expected, HbpSTyr146Phe proteins display an almost identical dityrosine spectrum compared with the WT sample under oxidative stress conditions. Furthermore, the H28A mutants have been shown to display considerably lower intensity of dityrosine fluorescence. As this mutant does not form stable oligomers, the physical proximity of two Tyr-77 is not present. The traces of fluorescence can be explained as a result from weakly formed cross-linked derivatives, which were observed upon iron-mediated oxidative stress (Fig. 4). We have previously demonstrated that the oligomeric assembly of HbpS is required for its interaction with the sensor kinase SenS (14); based on the data presented here, we concluded in addition that this assembly plays a central role during oxidation processes and in the sensing of oxidative stress.
FIGURE 8.

A, image showing the interacting subunits H (in blue) and F (in red) within the octomeric HbpS. The position of two adjacent Tyr-77 is marked with a box. B, image showing the arrangement of Tyr-77 on the subunit H (blue) and Tyr-77 on the subunit F (red) within the HbpS octomer. The distance (4.76 Å) between the two residues is indicated. The figures were produced using NOC. C, two EXXE (red) motifs within the HbpS sequence are indicated. Tyr-77 is also indicated (↓).
An important consideration during metal-catalyzed oxidation of proteins is the relationship between metal-binding sites and targets of ROS attack. Locally generated oxygen species may react at specific sites within or in the proximal vicinity of the metal-binding site (7). We have previously shown that lysine residues at position 108 in the octomeric HbpS coordinate an iron ion. In the vicinity (4 to 6 Å) of Lys-108 the aromatic amino acids Trp-90 and Phe-115 are located (Fig. 1). As nitration or hydroxylation of aromatic groups lead to the formation of reactive carbonyl derivatives (8), we could expect that interactions of two carbon-centered radicals or of the carbonyl groups with the primary amino groups of Lys-108 induce the formation of cross-links. Nevertheless, we cannot exclude the formation of other carbonyl derivatives at the surrounding regions. Tyr-77 is located in close proximity (10–15 Å) to this putative reaction center including Lys-108, Trp-90, and Phe-115. It can also not be excluded that iron binds somewhere else on the protein, at least transiently. Further sequence analysis in HbpS allowed the identification of two EXXE motifs (Fig. 8C), which have been postulated as iron-binding sites in the Salmonella PmrB and the Saccharomyces Ftr1p proteins (47, 48). One of these motifs encompasses the glutamate residues at position 78 and 81. As Tyr-77 is located in the direct vicinity of this motif, it could be expected that this putative iron-binding motif is involved in the formation of dityrosines upon iron-mediated oxidative stress. Further mutational analyses and biophysical measurements should clarify this aspect.
Previous physiological studies have demonstrated that HbpS together with the two-component system SenS-SenR regulates its own as well as the transcription of genes encoding for different redox proteins, including the redox regulator FurS or the mycelium-associated catalase-peroxidase CpeB (9, 12). Further biochemical analyses have shown that the autokinase SenS modulates the DNA-binding activity of the response regulator SenR. The unphosphorylated form of SenR binds to specific sites upstream of the furS-cpeB operon, leading to its transcriptional repression. Once SenR has been phosphorylated, it loses the ability to bind to this operator, releasing repression of the furS-cpeB transcription. Further specific sites within the regulatory region of hbpS were recognized by phosphorylated SenR (SenR∼P). It was additionally deduced that SenR∼P acts as an activator of the transcription of hbpS (49). These data indicate that SenR∼P positively regulates the expression of cpeB as well as hbpS. Additional data showed that HbpS interacts specifically with SenS, leading to the modulation of its phosphorylation. Under non oxidative stress conditions HbpS is able to repress the autophosphorylation of SenS whereas upon iron-mediated oxidative stress this phosphorylation is highly enhanced (49). SenS can in turn transfer the phosphate group to SenR, which positively regulates the transcription of furS-cpeB and hbpS. Transcriptional analyses presented here showed that upon iron-mediated oxidative stress the expression of cpeB as well as of hbpS is highly activated. Moreover, comparative analyses revealed that functional HbpS and SenS-SenR proteins are required for this in vivo activation. Consequently, once CpeB has been produced and secreted outside of the mycelia of S. reticuli the stressor H2O2 can be degraded. In this manner freshly synthesized HbpS proteins can be protected from oxidation and can further repress SenS autophosphorylation, switching off the HbpS-SenS-SenR signaling cascade.
In summary, we have here shown that upon iron-mediated oxidative stress HbpS becomes oxidized, leading to the generation of carbonyl groups, which in concert with the formation of dityrosine bonds enable the formation of cross-linked derivatives. As a consequence overall structural and conformational changes occur. These changes are expected to be responsible for the activation/inhibition of the signaling cascade, in which the three-component HbpS-SenS-SenR system is involved. In this context, the characterization of conformational changes within the sensor kinase SenS during the interaction with HbpS upon signal-sensing merit further study. The application of in vivo FRET techniques using reporter proteins could be interesting tools for clarifying these aspects.
Acknowledgments
We thank Dr. M. R. Groves (EMBL, Hamburg) for critical and proofreading of the manuscript, to apl. Prof. Dr. -Ing. R. Wagner (Biophysics, University of Osnabrueck) for facilitating equipment for UV-, Vis-, CD-, and fluorescence spectroscopy, and Dr. L. Becker (Biophysics, University of Osnabrueck) for advice and assistance in CD spectroscopy. We also thank I. Wedderhoff, M. Büttner, and I. Hormann (all three Applied Genetics of the Microorganisms, University of Osnabrueck) for some cloning experiments and initial transcriptional studies.
This work was supported by Grant OR 224/1–3 from the Deutsche Forschungsgemeinschaft (DFG).
- ROS
- reactive oxygen species
- H2O2
- hydrogen peroxide
- IAEDANS
- 5-[[2-[(iodoacetyl)amino]-ethyl]amino]naphthalene-1-sulfonic acid
- TCEP
- Tris(2-carboxyethyl)phosphine.
REFERENCES
- 1.Andrews S. C., Robinson A. K., Rodríguez-Quiñones F. (2003) FEMS Microbiol. Rev. 27, 215–237 [DOI] [PubMed] [Google Scholar]
- 2.Baker H. M., Anderson B. F., Baker E. N. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 3579–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudolph G., Hennecke H., Fischer H. M. (2006) FEMS Microbiol. Rev. 30, 631–648 [DOI] [PubMed] [Google Scholar]
- 4.Halliwell B., Gutteridge J. M. (1984) Biochem. J. 219, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim K., Rhee S. G., Stadtman E. R. (1985) J. Biol. Chem. 260, 15394–15397 [PubMed] [Google Scholar]
- 6.Touati D. (2000) Arch. Biochem. Biophys. 373, 1–6 [DOI] [PubMed] [Google Scholar]
- 7.Stadtman E. R. (1992) Science 257, 1220–1224 [DOI] [PubMed] [Google Scholar]
- 8.Stadtman E. R., Levine R. L. (2003) Amino Acids 25, 207–218 [DOI] [PubMed] [Google Scholar]
- 9.Ortiz de Orué Lucana D., Zou P., Nierhaus M., Schrempf H. (2005) Microbiology 151, 3603–3614 [DOI] [PubMed] [Google Scholar]
- 10.Bogel G., Schrempf H., Ortiz de Orué Lucana D. (2009) Amino Acids 37, 681–691 [DOI] [PubMed] [Google Scholar]
- 11.Ortiz de Orué Lucana D., Groves M. R. (2009) Amino Acids 37, 479–486 [DOI] [PubMed] [Google Scholar]
- 12.Ortiz de Orué Lucana D., Schaa T., Schrempf H. (2004) Microbiology 150, 2575–2585 [DOI] [PubMed] [Google Scholar]
- 13.Berks B. C., Sargent F., Palmer T. (2000) Mol. Microbiol. 35, 260–274 [DOI] [PubMed] [Google Scholar]
- 14.Ortiz de Orué Lucana D., Bogel G., Zou P., Groves M. R. (2009) J. Mol. Biol. 386, 1108–1122 [DOI] [PubMed] [Google Scholar]
- 15.Villarejo M., Zamenhof P. J., Zabin I. (1972) J. Biol. Chem. 247, 2212–2216 [PubMed] [Google Scholar]
- 16.Zou P., Groves M. R., Viale-Bouroncle S. D., Ortiz de Orué Lucana D. (2008) Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 64, 386–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: a Laboratory Manual, 2 Ed.Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 18.Hopwood D. A., Bibb M. J., Chater K. F., Kieser T., Bruton C. J., Kieser H. M., Lydiate D. J., Smith C. P., Ward J. M., Schrempf H. (1985) Genetic Manipulation of Streptomyces: a Laboratory Manual, Norwich, John Innes Foundation [Google Scholar]
- 19.Ortiz de Orué Lucana D., Schrempf H. (2000) Mol. Gen. Genet. 264, 341–353 [DOI] [PubMed] [Google Scholar]
- 20.Dower W. J., Miller J. F., Ragsdale C. W. (1988) Nucleic Acids Res. 16, 6127–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J., Russell W. (2006) Condensed Protocols from Molecular Cloning: Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 22.Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 23.Wu P., Brand L. (1994) Anal. Biochem. 218, 1–13 [DOI] [PubMed] [Google Scholar]
- 24.Böhm G., Muhr R., Jaenicke R. (1992) Protein Eng. 5, 191–195 [DOI] [PubMed] [Google Scholar]
- 25.Sreerema N., Woody R. W. (1993) Anal. Biochem. 209, 32–44 [DOI] [PubMed] [Google Scholar]
- 26.Sreerema N., Woody R. W. (2000) Anal. Biochem. 287, 52–260 [DOI] [PubMed] [Google Scholar]
- 27.Sreerema N., Woody R. W. (2003) Science 12, 384–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lardinois O. M., Tomer K. B., Mason R. P., Deterding L. J. (2008) Biochemistry 47, 10440–10448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rintala H., Nevalainen A., Rönkä E., Suutari M. (2001) Mol. Cell Probes 15, 337–347 [DOI] [PubMed] [Google Scholar]
- 30.Nishimura C., Riley R., Eastman P., Fink A. L. (2000) J. Mol. Biol. 299, 1133–1146 [DOI] [PubMed] [Google Scholar]
- 31.Selvin P. R. (2000) Nat. Struct. Biol. 7, 730–734 [DOI] [PubMed] [Google Scholar]
- 32.Heyduk T. (2002) Curr. Opin. Biotechnol. 13, 292–296 [DOI] [PubMed] [Google Scholar]
- 33.Kelly S. M., Price N. C. (2000) Curr. Protein Pept. Sci. 1, 349–384 [DOI] [PubMed] [Google Scholar]
- 34.Richardson L. G. L., Jelokhani-Niaraki M., Smith M. D. (2009) BMC Biochemistry 10, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kungl A. J., Visser A. J., Kauffmann H. F., Breitenbach M. (1994) Biophys. J. 67, 309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giulivi C., Davies K. (2001) J. Biol. Chem. 276, 24129–24136 [DOI] [PubMed] [Google Scholar]
- 37.Malencik D. A., Anderson S. R. (2003) Amino Acids 25, 233–247 [DOI] [PubMed] [Google Scholar]
- 38.Steinhoff H. J. (2004) Biol. Chem. 385, 913–920 [DOI] [PubMed] [Google Scholar]
- 39.Hagelueken G., Ingledew W. J., Huang H., Petrovic-Stojanovska B., Whitfield C., ElMkami H., Schiemann O., Naismith J. H. (2009) Angew. Chem. 121, 2948–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly J. W. (1998) Curr. Opin. Struct. Biol. 8, 101–106 [DOI] [PubMed] [Google Scholar]
- 41.Ding F., Dokholyan N. V., Buldyrev S. V., Stanley H. E., Shakhnovich E. I. (2002) J. Mol. Biol. 324, 851–857 [DOI] [PubMed] [Google Scholar]
- 42.Ding F., Borreguero J. M., Buldyrev S. V., Stanley H. E., Dokholyan N. V. (2003) Proteins 53, 220–228 [DOI] [PubMed] [Google Scholar]
- 43.Yang J., Panek H. R., O'Brian M. R. (2006) Mol. Microbiol. 60, 209–218 [DOI] [PubMed] [Google Scholar]
- 44.Dalle-Donne I., Rossi R., Colombo R., Giustarini D., Milzani A. (2006) Clin. Chem. 52, 601–623 [DOI] [PubMed] [Google Scholar]
- 45.Davies K. J. (1987) J. Biol. Chem. 262, 9895–9901 [PubMed] [Google Scholar]
- 46.DiMarco T., Giulivi C. (2007) Mass. Spectrom. Rev. 26, 108–120 [DOI] [PubMed] [Google Scholar]
- 47.Wösten M. M., Kox L. F., Chamnongpol S., Soncini F. C., Groisman E. A. (2000) Cell 103, 113–125 [DOI] [PubMed] [Google Scholar]
- 48.Severance S., Chakraborty S., Kosman D. J. (2004) Biochem. J. 380, 487–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bogel G., Schrempf H., Ortiz de Orué Lucana D. (2007) FEBS J. 274, 3900–3913 [DOI] [PubMed] [Google Scholar]