

Abstract
The cAMP-dependent protein kinase (PKA) signaling pathway plays a crucial role in the pathogenesis of many NF-κB-related diseases. However, there have been controversial reports with regard to the PKA actions in the regulation of NF-κB activity. In this study, we have demonstrated the effect of PKA on NF-κB activity in view of AKIP1 action; and in 293 and HeLa cells, where the endogenous AKIP1 expression is minimal, PKA-activating agents inhibited the NF-κB-dependent reporter gene expression, blocked the interaction of PKAc and p65 subunit of NF-κB, and attenuated PKA-dependent phosphorylation of p65 on Ser-276. This inhibitory function of PKAc in NF-κB signaling was reversed by overexpression of AKIP1 in 293 cells. In the breast cancer cell line, MDA-MB231 cells and MCF7 cells, where the endogenous AKIP1 is abundant, the PKA signal was found to be synergized with NF-κB activation; PKA-activating agents enhanced NF-κB-dependent transcriptional activity and the interaction between p65 and PKAc and augmented the phosphorylation of p65 on Ser-276. After RNAi knockdown of AKIP1 in these breast cancer cells, we observed that PKA-activating agents antagonized NF-κB-dependent activation. Meanwhile, PKA inhibitor suppressed NF-κB-induced breast cancer cell proliferation and multiple NF-κB-dependent anti-apoptotic gene expression. It is likely that expression of AKIP1 determines the relationship between these two signal transduction pathways. These findings explained controversial results from various independent groups regarding the action of PKA signaling on the NF-κB activation cascade and suggested a possible therapeutic potential of PKA inhibitor in developing anti-cancer strategies.
Keywords: Gene Regulation, Gene Transcription, NF-kB Transcription Factor, Protein Kinase A (PKA), Protein-Protein Interactions, Transcription, AKIP1, Interacting Protein
Introduction
NF-κB is an inducible transcriptional factor for the expression of multiple genes involved in immunoinflammatory responses, cell proliferation, and survival, thus playing crucial roles in the pathogenesis of many diseases, including cancer, leukemia, and autoimmune diseases (1–4). NF-κB primarily exists as the heterodimer consisting of p65 and p50 in the cytoplasm and is sequestered by binding to its inhibitory protein IκB (5). Upon signaling, such as by tumor necrosis factor α (TNFα), IκB is phosphorylated and is followed by proteasome-mediated degradation, which liberates NF-κB to the nucleus thereby activating the target genes.
PKA exists in the cytoplasm as an inactivated tetramer holoenzyme composed of dimer catalytic subunits and dimer regulatory subunits, which dissociate upon elevation of cAMP (6–10). PKAc is also predominantly involved in the IκB-NF-κB complex in the cytoplasm, and IκB sequesters PKAc by masking its catalytic domain (11). Following a variety of extracellular stimuli such as TNFα, IκB is phosphorylated by IKK2 and degraded by the 26 S proteasome (12, 13). As a consequence, the p65/p50 heterodimer complex is liberated, and the catalytic center of PKAc is exposed, enabling activated PKAc to phosphorylate p65 at Ser-276 (14, 15). This PKA-dependent phosphorylation of p65 facilitates the recruitment of transcription coactivator CBP and DNA binding activity of p65; therefore, activation of PKA augments NF-κB-dependent gene expression, and PKAc signaling is considered to up-regulate NF-κB activation (14–17). With regard to the role of PKAc in NF-κB signaling, some claimed that cAMP-dependent PKA activation down-regulated NF-κB-dependent transcription by changing its DNA binding ability (18, 19), modifying the transactivation domain of p65 (20), or blocking the degradation of IκB proteins (21, 22).
In our previous report, we identified AKIP1 as a novel p65-interacting protein (23). AKIP1 was initially found in breast cancer cells (24) in which it facilitated the nuclear translocation of PKAc (25). We found that AKIP1 appears to serve as a molecular bridge between p65 and PKAc, promoting their interaction and subsequent p65 phosphorylation at Ser-276, thus enhancing NF-κB signaling (23). Because NF-κB is constitutively activated in some breast cancer cells, endowing them with resistance to apoptosis (26–28), we hypothesized that the effect of PKA in NF-κB cascade is associated with AKIP1 and could be modulated by AKIP1 in a cell type-dependent fashion.
In this study, we further investigate the role of PKA in regulating NF-κB-dependent transcription in various cell lines with different expression levels of endogenous AKIP1. We provide evidence that in minimal AKIP1-expressing cell lines, elevation of cAMP decreased p65-PKA binding and p65 phosphorylation at Ser-276, which eventually leads to down-regulation of the NF-κB-dependent transcription. In contrast, in breast cancer cell lines MDA-MB231 and MCF7 with high AKIP1 expression, the PKA-activating agents increased p65-PKA binding and its phosphorylation and up-regulated the NF-κB-dependent transcription, particularly in anti-apoptotic genes.
MATERIALS AND METHODS
Cell Culture and Transfection
293, HeLa, MCF7, and MDA-MB231 cells (ATCC) were cultured at 37 °C with 5% CO2 in Dulbecco's modified Eagle's medium with 10% heat-inactivated fetal bovine serum, 100 units of penicillin, and 100 μg/ml streptomycin. Cells were transfected using FuGENE 6 transfection reagent (Roche Applied Science) according to the manufacturer's recommendations.
Western Blotting
Cell lysates from 293, HeLa, MCF7, and MDA-MB231 cells were collected, and Western blotting assay was performed by using polyclonal antibody raised against AKIP1 (kindly provided by Professor S. Taylor) (25).
AKIP1 Knockdown by RNAi
The RNAi sequence of AKIP1 (UCC UCU UGG CCC UCU CCA GCA CUUC) was designed from exon 2 of AKIP1 (amino acids 155–179) to degrade all endogenous AKIP1, including its splicing variants, including AKIP1b and AKIP1c (23). These RNAi reagents for AKIP1 were purchased from Invitrogen. Transfection of AKIP1 RNAi was performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's recommendations.
Immunoprecipitation-Western Blotting (IP-WB)
After transfecting with or without FLAG-AKIP1, 293 cells were cultured with the cAMP analogue Bt2cAMP (3 mm) for 12 h and TNFα (1 ng/ml) for an additional 6 h. Immunoprecipitation was performed as described previously (23) by using either anti-p65 antibody or anti-CREB antibody (Santa Cruz Biotechnology), and incubated with protein A-Sepharose beads (Sigma). Bound proteins were eluted with an equal volume of SDS loading buffer at 100 °C for 3 min and fractionated on 10% SDS-PAGE. Western blotting was conducted using either anti-PKAc (Santa Cruz Biotechnology), anti-p65-phospho-Ser-276 (Cell Signaling), anti-CBP (Cell Signaling), or anti-CREB-phospho-Ser-133 (Cell Signaling) antibodies. MDA-MB231 cells with or without AKIP1 knockdown by RNAi (Invitrogen) were also used for the same IP-WB assays.
Transient Luciferase Assay
HeLa, 293, MCF7, and MDA-MB231 cells were cultured in 24-well plates, and FLAG-AKIP1 expression plasmid together with pGL-3κB-luc and pRL-TK plasmids were transfected with FuGENE 6 transfection reagent (Roche Applied Science). 24 h after transfection, cells were treated with Bt2cAMP or Br-cAMP (1 mm, 3 mm) for 12 h and subsequently stimulated with TNFα (1 ng/ml) for an additional 6 h. These cells were harvested for luciferase assay. Luciferase activity was normalized by the Renilla luciferase activity of each transfection to normalize the transfection efficiency.
ELISA
ELISAs were performed using commercial IL-8 ELISA kits (R & D Systems). Culture supernatants from 293 cells and MDA-MB231 cells with or without Bt2cAMP/TNFα treatment were collected for the IL-8 concentration determination. Assays were carried out in triplicate, and readings were compared with a standard curve obtained with human recombinant IL-8 provided in the kit.
WST-1 Assay
MDA-MB231 and 293 cells were seeded in 96-well plates, incubated for 24 h, treated with the PKA inhibitor H89 (1 and 3 μm) for 4 h, and further incubated with the IKK inhibitor BAY11-7082 (5, 10, and 20 μm) for an additional 20 h. WST-1 assay was performed by adding 10 μl of WST-1 reagent (Roche Applied Science) to each sample and incubated for 1 h at 37 °C. The absorbance of each sample was then measured at 450 nm.
SYBR Green Real Time PCR Analysis
293 cells were cultured in 6-well plates with or without AKIP1 transduction and were treated with or without Bt2cAMP (3 mm) and/or TNFα (1 ng/ml). Total RNA was isolated using TRIzol reagent (Invitrogen), and 0.3 μg total RNA of each sample was reverse-transcribed into cDNA. Real time PCR was carried out with quantitative PCR MasterMix Plus for SYBR Green using the instruction manual: incubation at 95 °C for 10 min followed by a 40-cycle amplification (15 s at 95 °C, 1 min at 60 °C, 45 s at 72 °C, and 15 s at 80 °C) for SYBR Green detection. Amplification was performed with the following primers: cyclin D1, sense 5′-CCC TCG GTG TCC TAC TTC AAA-3′ and antisense 5′-CAC CTC CTC CTC CTC CTC TTC-3′; bcl-2, sense 5′-AGA TGT CCA GGC AGC TGC AC-3′ and antisense 5′-CAC AGG GCG ATG TTG TCC AC-3′; bcl-xL, sense 5′- CAG AGC AAC CGG GAG CTG GT-3′ and antisense 5′-GAT CCA AGG CTC TAG GTG GTC-3′; survivin, sense 5′- GGC ATG GGT GCC CCG ACG TTG-3′ and antisense 5′-CAG AGG CCT CAA TCC ATG GCA-3′; xiap, sense 5′-TGT CCG ATG TGC AAC ACA GT-3′ and antisense 5′- CCA TGT CAG TAC ATG TAG GC-3′; and β-actin, sense CCA GGC ACC AGG GCG TGA TG-3′ and antisense 5′-CGG CCA GCC AGG TCC AGA CG-3′ (27). The expression of each gene was normalized by the corresponding amount of β-actin mRNA. The relative amounts of each product were calculated by the comparative CT (2−ΔΔCT) method described in User Bulletin 2 of the ABI Prism 7500 Fast Sequence Detection System (Applied Biosystems).
RESULTS
Expression of AKIP1 in Various Cell Lines
To elucidate the role of AKIP1, a newly identified binding partner of the p65 subunit of NF-κB, we first examined its expression in various cell lines. Endogenous AKIP1 levels were detected by Western blot assay using a specific anti-AKIP1 antibody (25). As shown in Fig. 1, expression of endogenous AKIP1 was very low in 293 and HeLa cells, whereas in breast cancer cells MDA-MB231 and MCF7, AKIP1 was abundantly expressed. This result was consistent with the previous report indicating AKIP1 is highly expressed in breast cancer cells (24). It is also important to note that the highest AKIP1 expression was observed in MDA-MB231, which was derived from metastasized breast cancer tissue and was considered highly malignant (29).
FIGURE 1.
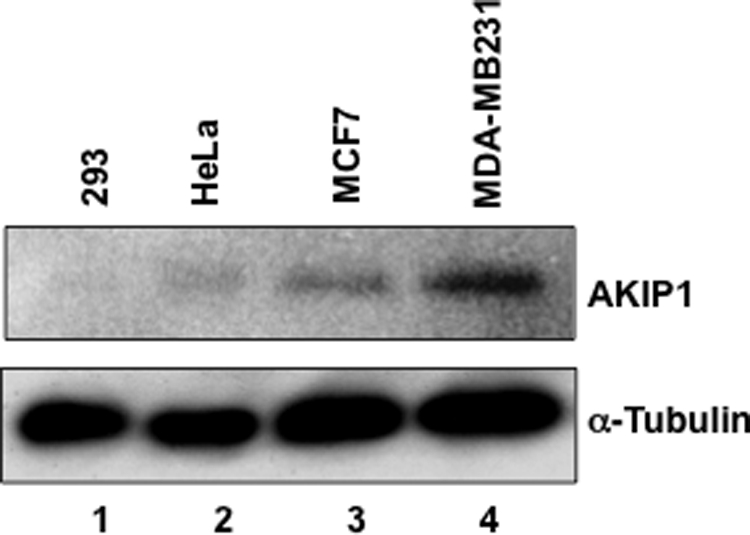
Expression of endogenous AKIP1 in various cells. 293, HeLa, MCF7, and MDA-MB231 cells were cultured and lysed for Western blotting using polyclonal antibody against AKIP1. Note the different expression levels of endogenous AKIP1 in these cell lines. The protein expression levels of α-tubulin indicate that the same amounts of cell lysates were loaded.
AKIP1 Regulates p65-PKAc Interaction and p65 Phosphorylation at Ser-276
Considering biological effects and regulation are mediated through protein-protein interaction, and it was established that PKAc is involved in regulating p65 phosphorylation at Ser-276, we attempted to explore the p65-PKAc interaction in various cell lines with different levels of AKIP1 expression. In Fig. 2A, 293 cells exhibiting low expression levels of endogenous AKIP1 were treated with the PKAc activator Bt2cAMP and NF-κB inducer TNFα. These cells were then subjected to immunoprecipitation with anti-p65 antibody to detect p65-PKAc binding through Western blotting using anti-PKAc antibody. As demonstrated here, treatment with Bt2cAMP enhanced the p65-PKAc interaction (Fig. 2A, upper panel, lane 2), suggesting cAMP could activate the PKAc-p65 interaction even without TNFα. Upon stimulation with TNFα, the interaction between p65 and PKAc was similarly enhanced (Fig. 2A, upper panel, lane 3). Intriguingly, treatment with both Bt2cAMP and TNFα at the same time led to a relatively weaker binding between p65 and PKA as compared with the treatment with TNFα alone (Fig. 2A, upper panel, compare lane 3 with 4), suggesting that the combination of PKA and NF-κB signaling could rather inhibit the p65-PKA interaction.
FIGURE 2.

Interaction of p65 and PKAc in 293 cells and MDA-MB231 cells. A, p65-PKAc interaction and p65 Ser-276 phosphorylation in wild type 293. Cells incubated with Bt2cAMP (3 mm) for 12 h and TNFα (1 ng/ml) for an additional 6 h were subjected to IP (p65)-WB (PKA) or (phospho-p65-Ser-276). B, effect of AKIP1 overexpression in 293 cells. The p65-PKAc interaction and p65 Ser-276 phosphorylation were examined in AKIP1-overexpressed 293 cells. 293 cells were transiently transfected with FLAG-AKIP1 and treated with Bt2cAMP and TNFα 24 h after the transfection. Then cells were subjected to IP-WB assay. C, p65-PKAc interaction and p65 Ser-276 phosphorylation in wild type MDA-MB231 cells. The cells were treated with Bt2cAMP and TNFα as above, and the cell lysates were subjected to IP-WB assay. D, effects of AKIP1 knockdown in MDA-MB231 cells. After transfecting RNAi-AKIP1 to MDA-MB231 cells, the p65-PKAc interaction and p65 Ser-276 phosphorylation were similarly examined. Equal amount of p65 was immunoprecipitated with anti-p65 antibody and subjected to the following Western blotting assays; 1:50 input of PKA expression was also indicated. The amounts of whole cell lysates were adjusted by examining the level of α-tubulin expression. The results were reproducibly observed at least three times, and the representative data are shown.
According to our previous study, AKIP1 could enhance NF-κB activation by promoting PKAc-dependent p65 phosphorylation at Ser-276 in most cell lines (23). Here, we examined whether there is a correlation between the extent of PKA-mediated p65 phosphorylation and the amount of AKIP1 expression. As shown in Fig. 2A (middle panel), when 293 cells were precipitated by anti-p65 antibody, and immunoblotted with anti-phospho-p65 (Ser-276) antibody, p65 phosphorylation was induced either by Bt2cAMP (lane 2) or TNFα (lane 3). However, when these cells were treated with both TNFα and Bt2cAMP, the extent of p65 phosphorylation and of p65-PKAc interaction was diminished (Fig. 2A, lane 4).
To examine the action of AKIP1 in regulating PKA signaling in the NF-κB activation pathway, we transfected the AKIP1-expressing plasmid in 293 cells, and the IP-WB assay was performed with p65 antibody followed by PKAc antibody. As shown in Fig. 2B (upper panel), p65-PKAc binding was moderately enhanced by Bt2cAMP (lane 2) and by TNFα (lane 3). Surprisingly, when AKIP1 was overexpressed, treatment of both Bt2cAMP and TNFα significantly increased p65-PKAc interaction (compare lane 4 of Fig. 2A with that of B, upper panels). Similarly, the extent of p65 (Ser-276) phosphorylation was further enhanced by both Bt2cAMP and TNFα. These results suggested that AKIP1 could reverse the inhibitory function of PKAc in antagonizing NF-κB cascade.
We investigated the effect of endogenous AKIP1 in regulating the enzymatic action of PKAc (Fig. 2, C and D). Either AKIP1-expressing breast cancer cell MDA-MB231 or AKIP1-knockdown MDA-MB231 cells were subjected to IP-WB assay with anti-p65 antibody followed by anti-PKAc antibody and phospho-p65 (Ser-276) antibody. Western blot assay revealed the significant degradation of endogenous AKIP1 protein after transfecting RNAi against AKIP1 in MDA-MB231 cells (data not shown). As shown in Fig. 2C (upper panel) with MDA-MB231 cells expressing endogenous AKIP1, either Bt2cAMP or TNFα could elevate the interaction between p65 and PKAc (lanes 2 and 3). Furthermore, upon the treatment with both Bt2cAMP and TNFα, p65-PKAc binding was up-regulated by PKAc activation (Fig. 2C, lane 4). Activation of PKAc induced an increased p65 phosphorylation at Ser-276 (Fig. 2C, middle panel, lane 2), and upon stimulation with TNFα, p65 phosphorylation was enhanced to the similar level (lane 3). When these cells were treated with both Bt2cAMP and TNFα, the p65 Ser-276 phosphorylation was further augmented (Fig. 2C, lane 4). These results mimic the observations with AKIP1-overexpressed 293 cells (Fig. 2B).
Fig. 2D indicated that in AKIP1-knockdown MDA-MB231 cells, PKAc activator inhibited both the extent of p65-PKA interaction and PKA-mediated p65 phosphorylation upon TNFα treatment, thus mimicking the property of 293 cells (Fig. 2A). Overall, these observations in Fig. 2 clearly indicate that AKIP1 could alter the p65-PKAc interaction and subsequent p65 phosphorylation; in the presence of AKIP1, PKAc activation promoted p65-PKAc binding and PKA-mediated p65 phosphorylation, whereas in the absence of AKIP1, PKAc activator blocked p65-PKAc interaction and p65 phosphorylation.
AKIP1 Facilitates the Recruitment of Transcriptional Coactivator CBP to p65
It is well established that PKA-mediated phosphorylation facilitates p65 to recruit its transcription coactivator, CBP (CREB-binding protein) (14, 17). Here, we examined whether CBP recruitment could be affected by AKIP1 expression. In Fig. 3A, after 293 cells were treated with Bt2cAMP and/or TNFα, cell lysates were prepared, and an equal amount of p65 was immunoprecipitated with anti-p65 and blotted with anti-CBP antibodies. The interaction between p65 and CBP was increased upon treatment of either PKA activator or TNFα, but it was not enhanced when cells were treated both with Bt2cAMP and TNFα at the same time (compare lane 4 with lanes 2 and 3), which is consistent with the results in Fig. 2. We then hypothesized that other PKA-mediated transcription factors might be involved and disrupt PKA-p65 interaction. CREB, which is another PKA-dependent transcriptional factor, was reported to compete with p65 in recruiting CBP upon PKA stimulation (15, 16, 30). As shown in Fig. 3B, the cell lysate was prepared from the same 293 cells and immunoprecipitated by anti-CREB antibody, and then an equal amount of immune complex was blotted with antibodies against CBP and the phosphorylated CREB at Ser-133. With treatment with either Bt2cAMP or TNFα, the CREB-CBP interaction was significantly augmented. However, in contrast to p65-CBP binding, the interaction between CREB and CBP was enhanced even by treatment with both Bt2cAMP and TNFα (compare Fig. 3, A and B, lanes 4, upper panel). Furthermore, CREB phosphorylation was also increased (Fig. 3B, lane 4), consistent with the previous work that CREB recruits CBP upon phosphorylation of CREB by PKA (31).
FIGURE 3.

Selective recruitment of coactivator protein CBP by AKIP1. A and B, 293 cells were treated with Bt2cAMP (3 mm) and TNFα (1 ng/ml). These cells were collected and immunoprecipitated with anti-p65 antibody (A) or anti-CREB antibody (B), and then an equal amount of immunocomplexes was blotted with anti-CBP antibody to detect the interaction of CBP with either p65 or CREB. The phosphorylated CREB at Ser-133 (S133) was also monitored (B). C and D, AKIP1-overexpressing 293 cells were treated Bt2cAMP and TNFα, and then IP-WB was similarly performed as in A and B. The 1:50 input of CBP expression was also indicated, and the α-tubulin expression levels were monitored as an internal control.
In contrast, in AKIP1-overexpressing 293 cells (Fig. 3C), the combination of Bt2cAMP and TNFα induced the extent of p65-CBP interaction (Fig. 3C, upper panel, lane 4); however, CREB-CBP binding and CREB phosphorylation were significantly decreased (Fig. 3D, lane 4). Taken together, these results demonstrated in Figs. 2 and 3 suggested that AKIP1 could modulate the relative preference for PKA-mediated phosphorylation and coactivator recruitment between p65 and CREB. AKIP1 attenuates the CREB phosphorylation and the subsequent binding to CBP, whereas AKIP1 appears to potentiate the recruitment of CBP to p65.
Regulation of NF-κB-dependent Transcription by AKIP1 upon PKA Signaling
The experiments described above indicated that AKIP1 appears to facilitate the PKA-dependent p65 phosphorylation and CBP recruitment upon TNFα induction. Thus, we examined whether AKIP1 could also regulate the effect of PKA in NF-κB-dependent transcriptional activity. As shown in Fig. 4A (left panel), in 293 cells (closed columns) where the expression of AKIP1 is limited, NF-κB-dependent gene expression was down-regulated by PKA activator Bt2cAMP in a dose-dependent manner. However, in MDA-MB231 cells (open columns), where AKIP1 is abundant, the NF-κB activation was up-regulated upon treatment with Bt2cAMP. We also tested another PKA activator Br-cAMP, and similar results were observed (data not shown). These results have demonstrated that AKIP1 determines the transcriptional activity of NF-κB through regulating the PKA signaling.
FIGURE 4.

Effects of Bt2cAMP on the NF-κB-dependent transcription and IL-8. A, AKIP1 switches the role of PKA signaling in NF-κB-dependent luciferase expression. Left panel, 293 (closed columns) and MDA-MB231 cells (open columns) were treated with Bt2cAMP (1 and 3 mm) for 12 h and TNFα (1 ng/ml) for an additional 6 h. Cells were lysed and subjected to luciferase assay and normalized with the Renilla luciferase activity as an internal control. The vertical axis indicates the relative luciferase activity in fold activation. Values are representative of triplicate experiments (mean ± S.D.). Right panel, 293 cells transfected with (open columns) or without FLAG-AKIP1 (closed columns) were treated with Bt2cAMP and TNFα and subjected to luciferase assay. Values are representative of triplicate experiments (mean ± S.D.). The inset in the right panel indicates the expanded representation of results in lanes 1–3 (the vertical axis indicates the fold activation). B, effects of AKIP1 on the NF-κB-dependent expression of IL-8. 293 and MDA-MB231 cells were cultured in 24-well plates and incubated with Bt2cAMP and TNFα. The concentration of IL-8 was measured by ELISA. The vertical axis indicates the secretion of IL-8 in the culture medium, and the values are representative of triplicate experiments (mean ± S.D.).
To further address the effect of AKIP1 in mediating PKA and NF-κB signaling, we performed luciferase assays in 293 cells with or without AKIP1 overexpression. As shown in Fig. 4A (right panel), in wild type 293 cells, Bt2cAMP-mediated PKAc activation significantly repressed NF-κB-dependent transcription (closed columns), as in Fig. 4A (left panel). In contrast, in the AKIP1-overexpressing 293 cells, PKA activation augmented the NF-κB-dependent transcription in a dose-dependent manner (Fig. 4A, open columns). These results together with those in Figs. 2 and 3 indicate that AKIP1 regulates the direction of cross-talk between PKA and NF-κB signaling presumably through the PKAc-mediated p65 phosphorylation and recruitment of the transcriptional coactivator CBP.
Considering IL-8 is one of the physiological target genes governed by NF-κB and is closely involved in cancer progression through promoting cancer cell proliferation (32) and angiogenesis (33, 34), we examined whether IL-8 expression is also regulated by AKIP1 through modifying the relationship between PKA and NF-κB signaling. As shown in Fig. 4B, 293 and MDA-MB231 cells were treated with Bt2cAMP and TNFα, and then the IL-8 concentration in cell culture supernatant was evaluated by sandwich-ELISA assay. In 293 cells, IL-8 production was triggered by TNFα stimulation, and in combination with Bt2cAMP, it was down-regulated in a dose-dependent manner (left panel). However, in MDA-MB231 cells, where the basic level of IL-8 is much higher than in 293 cells, the level of IL-8 production was further augmented by TNFα stimulation in concert with Bt2cAMP (Fig. 4B, right panel). Other PKA activators such as forskolin and Br-cAMP were also tested in these cell lines and MCF7 cells with similar results obtained (data not shown). These results indicated that NF-κB-dependent gene expression could be mediated by AKIP1 through PKA. This would all indicate that in the absence of AKIP1, PKA activation down-regulates NF-κB-dependent gene expression; however, in the presence of AKIP1, PKA activation up-regulates NF-κB-dependent gene expression.
PKA Inhibitor Represses the Proliferation of Breast Cancer Cells
Aberrant activation of NF-κB signaling causes proliferation of cancer cells (2, 3). Because PKA activation synergizes with NF-κB cascade in breast cancer cells (Fig. 4), we hypothesized that PKA inhibitor would also down-regulate proliferation of breast cancer cells through the regulatory cascade of NF-κB activation. Treatment with H89, a PKA inhibitor, decreased the growth of MDA-MB231 cells in a dose-dependent manner (Fig. 5A). In addition, when BAY11-7082, a specific inhibitor of IκB kinase (35, 36), was added, the inhibitory effects of H89 were further potentiated (Fig. 5B). However, this synergism by IKK and PKA inhibitors was not observed in 293 cells (Fig. 5C), presumably because AKIP1 expression is minimum, and neither NF-κB nor PKA signaling is constitutively activated in 293 cells. IKK inhibitor BAY11-7082 could only repress the basal level of NF-κB activation, and PKA inhibitor H89 failed to further synergize with BAY11-7082. These observations suggest a possibility that H89 may require either IKK or PKA signaling that is associated with AKIP1 expression.
FIGURE 5.

Effects of PKA inhibitor on the proliferation of MDA-MB231 cells. A, MDA-MB231 cells were cultured for 24 h, treated with PKA inhibitor H89 (1 and 3 μm) for 4 h, and then they were subjected to WST-1 assay to evaluate the cell viability. B, MDA-MB231 cells were treated with H89 (3 μm) for 4 h and further incubated with IKK inhibitor BAY11-7082 (5, 10, and 20 μm) for an additional 20 h. Then the proliferating cells were evaluated by WST-1 assay. Note the synergism between inhibition of PKA signaling and that of NF-κB activation signaling. C, 293 cells treated with H89 (3 μm) for 4 h and further incubated with IKK inhibitor BAY11-7082 (0.5, 2, 5, and 10 μm) for an additional 20 h, and WST-1 assay was performed.
AKIP1 Determines the Effect of PKA on NF-κB-dependent Expression of Anti-apoptotic Genes
Constitutive NF-κB activation induces expression of anti-apoptotic genes such as cyclin D1, bcl-2, bcl-xL, survivin, and xiap (26–28). We thus examined the effect of PKA activator Bt2cAMP on expression of these genes. Isolated total RNA from 293 cells with or without AKIP1 transfection and treatment of Bt2cAMP/TNFα were reverse-transcribed to cDNA. In wild type 293 cells (Fig. 6A), treatment of both TNFα and Bt2cAMP decreased the anti-apoptotic gene expression compared with TNFα-treated cells. However, among AKIP1-overexpressing 293 cells (Fig. 6B), double treatment of Bt2cAMP and TNFα enhanced the gene expression levels (lane 8). Consistent with our earlier results, AKIP1 appears to act as a major PKA determinant in the NF-κB activation cascade, and in cancer cells it promotes cell growth by up-regulating NF-κB and expression of its target genes.
FIGURE 6.

Expression of the NF-κB-dependent anti-apoptotic genes is regulated by AKIP1. 293 cells, transfected with AKIP1 or control vehicle vector, were treated with Bt2cAMP for 12 h and TNFα for an additional 6 h. The total RNA was prepared from cells, and cDNA was reverse-transcribed using 0.3 μg of total RNA of each sample. A–E, real time-PCR was performed in triplicate to analyze the relative expression levels of anti-apoptotic genes (cyclin D1, bcl-2, bcl-xL, survivin, and xiap); gene expression is expressed relative to β-actin (mean ± S.E. presented). F, semi-quantitative reverse transcription-PCR was performed with 0.3 μg for each sample.
DISCUSSION
In this study, we investigated the cross-talk between two distinct pathways, namely NF-κB and PKA cascades with AKIP1 acting as a common interacting protein partner and co-factor for their actions. We found that AKIP1 could regulate the mode of PKA signaling on NF-κB cascade based on its expression level. In cell lines where AKIP1 expression was low, PKA activator inhibited p65-PKA binding and p65 phosphorylation (Fig. 2), accompanied with down-regulation of NF-κB-dependent transcription (Fig. 4). On the contrary, in cells like MDA-MB231 or AKIP1-overexpressing 293 cells, the p65-PKA binding and subsequent p65 phosphorylation were enhanced by PKA signaling (Fig. 2), thus up-regulating NF-κB-dependent transcription (Fig. 4). We also obtained similar results with nonmetastatic MCF7 breast cancer cells (data not shown). It is noticeable that when AKIP1 was transduced into 293 cells, PKA activator could augment NF-κB activation, which highlighted the significance of AKIP1 as a determinant of synergism between two cascades. In the presence of AKIP1 expression, PKA signaling augmented expression of anti-apoptotic genes thus contributing to the NF-κB-dependent cell survival (Figs. 4 and 6). These findings give a clue to a long standing controversy regarding the action of PKA signaling in the NF-κB activation cascade (14–16, 18–22);: the synergism or antagonism between PKA and NF-κB signals appear to depend on cell type and the level of AKIP1 expression in particular.
It is well established that the recruitment of transcriptional coactivator CBP by p65 is an essential step for induction of its target genes by NF-κB (15, 30). Interestingly, PKA-mediated phosphorylation of p65 at Ser-276 promoted the interaction between p65 and CBP (Fig. 3). On the other hand, PKA also regulated the activity of CREB by phosphorylating on Ser-133 and promoting its binding to CBP (31). Thus, two transcription factors appear to compete with each other in recruiting CBP, whose amount is known to be limited in cells (37, 38). However, in the presence of AKIP1, such as in breast cancer cells, AKIP1 removes CBP from CREB and redirects it to p65, and AKIP1 seems to provide a molecular bridge for the interaction between PKAc and p65, inducing PKAc-mediated phosphorylation of p65 at Ser-276 thus endowing p65 with higher affinity to CBP (Figs. 2 and 3).
PKA activation is reported to positively regulate cancer progression through multiple cascades. For example, PKA activation induced osteolytic metastasis of MDA-MB231 breast cancer cells through ERK1/2-dependent pathway (39). In addition, inhibition of PKA signaling induced apoptosis via down-regulation of Bcl2 expression (40). Furthermore, the PKA inhibitor H89 could suppress breast cancer cell proliferation through blocking of the PKA-mediated STAT phosphorylation (41). It was reported that PKA-mediated phosphorylation of either steroid receptor coactivator (Src1) or estrogen receptor itself endowed breast cancer cells with resistance to tamoxifen (42, 43). Moreover, inhibition of PKA pathway by H89 could abolish the cAMP-mediated aromatase induction through up-regulating brca1 expression (44). Similar to our study, a number of previous reports support the possibility that PKA signaling contributes to the constitutive NF-κB activation (45–47). In this study, we demonstrated that NF-κB inhibitor BAY11-7082 repressed MDA-MB231 cell proliferation by blocking IKK, and PKA inhibitor H89 further augmented this inhibitory action (Fig. 5B). Thus, it is conceivable that PKA inhibitors would suppress the constitutive activation of NF-κB, potentiating the actions of NF-κB inhibitors. Potent inhibitors of PKA such as H89, PKI6-22 (23), and their derivatives could have potential therapeutic efficacy against cancer and leukemia. It is noticeable that PKA inhibitors could only block proliferation of breast cancer cells where AKIP1 is abundantly expressed but not in cells where it is absent (Fig. 5). Therefore, the expression level of AKIP1 may predict the clinical feasibility of PKA inhibitors in cancer treatment as a novel surrogate marker.
The findings demonstrated in this study not only support the involvement of AKIP1 in cancer progression but also indicate the critical role of AKIP1 in determining the effect of PKA signaling on the NF-κB activation cascade. AKIP1 could be used as a molecular determinant for designing tailor-made anti-NF-κB remedy. In AKIP1 high expressing cancer cells, where PKA signaling synergizes with NF-κB activation, the combination of IKK and PKA inhibitors would effectively repress cell growth and induce cancer cell death. In AKIP1-absent cells, activation of PKA cascade by up-regulating the intracellular cAMP level would promote cancer cell apoptosis by antagonizing with the NF-κB activation cascade.
Acknowledgment
We thank Dr. S. Taylor for kindly providing us the anti-AKIP1 antibody.
Footnotes
- IKK
- IκB kinase
- Bt2cAMP
- as dibutyryl cyclic AMP
- IP-WB
- immunoprecipitation-Western blotting
- CREB
- cAMP-response element-binding protein
- CBP
- CREB binding protein.
REFERENCES
- 1.Baeuerle P. A., Baichwal V. R. (1997) Adv. Immunol. 65, 111–137 [PubMed] [Google Scholar]
- 2.Baldwin A. S., Jr. (1996) Annu. Rev. Immunol. 14, 649–683 [DOI] [PubMed] [Google Scholar]
- 3.Okamoto T., Sakurada S., Yang J. P., Merin J. P. (1997) Curr. Top. Cell. Regul. 35, 149–161 [DOI] [PubMed] [Google Scholar]
- 4.Moynagh P. N. (2005) J. Cell Sci. 118, 4589–4592 [DOI] [PubMed] [Google Scholar]
- 5.Sun S. C., Ganchi P. A., Béraud C., Ballard D. W., Greene W. C. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 1346–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carnegie G. K., Scott J. D. (2003) Genes Dev. 17, 1557–1568 [DOI] [PubMed] [Google Scholar]
- 7.Taylor S. S., Buechler J. A., Yonemoto W. (1990) Annu. Rev. Biochem. 59, 971–1005 [DOI] [PubMed] [Google Scholar]
- 8.Scott J. D. (1991) Pharmacol. Ther. 50, 123–145 [DOI] [PubMed] [Google Scholar]
- 9.Francis S. H., Corbin J. D. (1994) Annu. Rev. Physiol. 56, 237–272 [DOI] [PubMed] [Google Scholar]
- 10.Walsh D. A., Van Patten S. M. (1994) FASEB J. 8, 1227–1236 [DOI] [PubMed] [Google Scholar]
- 11.Thompson J. E., Phillips R. J., Erdjument-Bromage H., Tempst P., Ghosh S. (1995) Cell 80, 573–582 [DOI] [PubMed] [Google Scholar]
- 12.Lallena M. J., Diaz-Meco M. T., Bren G., Payá C. V., Moscat J. (1999) Mol. Cell. Biol. 19, 2180–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiDonato J., Mercurio F., Rosette C., Wu-Li J., Suyang H., Ghosh S., Karin M. (1996) Mol. Cell. Biol. 16, 1295–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong H., SuYang H., Erdjument-Bromage H., Tempst P., Ghosh S. (1997) Cell 89, 413–424 [DOI] [PubMed] [Google Scholar]
- 15.Zhong H., Voll R. E., Ghosh S. (1998) Mol. Cell 1, 661–671 [DOI] [PubMed] [Google Scholar]
- 16.Hayashi T., Sekine T., Okamoto T. (1993) J. Biol. Chem. 268, 26790–26795 [PubMed] [Google Scholar]
- 17.Chen Lf., Fischle W., Verdin E., Greene W. C. (2001) Science 293, 1653–1657 [DOI] [PubMed] [Google Scholar]
- 18.Kim Y. S., Cho K. O., Lee H. J., Kim S. Y., Sato Y., Cho Y. J. (2006) J. Biol. Chem. 281, 39051–39061 [DOI] [PubMed] [Google Scholar]
- 19.Neumann M., Grieshammer T., Chuvpilo S., Kneitz B., Lohoff M., Schimpl A., Franza B. R., Jr., Serfling E. (1995) EMBO J. 14, 1991–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi N., Tetsuka T., Uranishi H., Okamoto T. (2002) Eur. J. Biochem. 269, 4559–4565 [DOI] [PubMed] [Google Scholar]
- 21.Majumdar S., Aggarwal B. B. (2003) Oncogene 22, 1206–1218 [DOI] [PubMed] [Google Scholar]
- 22.Minguet S., Huber M., Rosenkranz L., Schamel W. W., Reth M., Brummer T. (2005) Eur. J. Immunol. 35, 31–41 [DOI] [PubMed] [Google Scholar]
- 23.Gao N., Asamitsu K., Hibi Y., Ueno T., Okamoto T. (2008) J. Biol. Chem. 283, 7834–7843 [DOI] [PubMed] [Google Scholar]
- 24.Kitching R., Li H., Wong M. J., Kanaganayakam S., Kahn H., Seth A. (2003) Biochim. Biophys. Acta 1625, 116–121 [DOI] [PubMed] [Google Scholar]
- 25.Sastri M., Barraclough D. M., Carmichael P. T., Taylor S. S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 349–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kajino S., Suganuma M., Teranishi F., Takahashi N., Tetsuka T., Ohara H., Itoh M., Okamoto T. (2000) Oncogene 19, 2233–2239 [DOI] [PubMed] [Google Scholar]
- 27.Sanda T., Iida S., Ogura H., Asamitsu K., Murata T., Bacon K. B., Ueda R., Okamoto T. (2005) Clin. Cancer Res. 11, 1974–1982 [DOI] [PubMed] [Google Scholar]
- 28.Specht K., Haralambieva E., Bink K., Kremer M., Mandl-Weber S., Koch I., Tomer R., Hofler H., Schuuring E., Kluin P. M., Fend F., Quintanilla-Martinez L. (2004) Blood 104, 1120–1126 [DOI] [PubMed] [Google Scholar]
- 29.Bendre M. S., Gaddy-Kurten D., Mon-Foote T., Akel N. S., Skinner R. A., Nicholas R. W., Suva L. J. (2002) Cancer Res. 62, 5571–5579 [PubMed] [Google Scholar]
- 30.Perkins N. D., Felzien L. K., Betts J. C., Leung K., Beach D. H., Nabel G. J. (1997) Science 275, 523–527 [DOI] [PubMed] [Google Scholar]
- 31.De Cesare D., Fimia G. M., Sassone-Corsi P. (1999) Trends Biochem. Sci. 24, 281–285 [DOI] [PubMed] [Google Scholar]
- 32.Xie K. (2001) Cytokine Growth Factor Rev. 12, 375–391 [DOI] [PubMed] [Google Scholar]
- 33.Shono T., Ono M., Izumi H., Jimi S. I., Matsushima K., Okamoto T., Kohno K., Kuwano M. (1996) Mol. Cell. Biol. 16, 4231–4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin D., Galisteo R., Gutkind J. S. (2009) J. Biol. Chem. 284, 6038–6042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mori N., Yamada Y., Ikeda S., Yamasaki Y., Tsukasaki K., Tanaka Y., Tomonaga M., Yamamoto N., Fujii M. (2002) Blood 100, 1828–1834 [DOI] [PubMed] [Google Scholar]
- 36.Dai Y., Pei X. Y., Rahmani M., Conrad D. H., Dent P., Grant S. (2004) Blood 103, 2761–2770 [DOI] [PubMed] [Google Scholar]
- 37.Xu W., Chen H., Du K., Asahara H., Tini M., Emerson B. M., Montminy M., Evans R. M. (2001) Science 294, 2507–2511 [DOI] [PubMed] [Google Scholar]
- 38.Koh S. S., Li H., Lee Y. H., Widelitz R. B., Chuong C. M., Stallcup M. R. (2002) J. Biol. Chem. 277, 26031–26035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimo T., Kubota S., Yoshioka N., Ibaragi S., Isowa S., Eguchi T., Sasaki A., Takigawa M. (2006) J. Bone Miner. Res. 21, 1045–1059 [DOI] [PubMed] [Google Scholar]
- 40.Srivastava R. K., Srivastava A. R., Seth P., Agrawal S., Cho-Chung Y. S. (1999) Mol. Cell. Biochem. 195, 25–36 [DOI] [PubMed] [Google Scholar]
- 41.Xu H., Washington S., Verderame M. F., Manni A. (2008) Breast Cancer Res. Treat. 107, 63–70 [DOI] [PubMed] [Google Scholar]
- 42.Michalides R., Griekspoor A., Balkenende A., Verwoerd D., Janssen L., Jalink K., Floore A., Velds A., van't Veer L., Neefjes J. (2004) Cancer Cell 5, 597–605 [DOI] [PubMed] [Google Scholar]
- 43.Zwart W., Griekspoor A., Berno V., Lakeman K., Jalink K., Mancini M., Neefjes J., Michalides R. (2007) EMBO J. 26, 3534–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh S., Lu Y., Hu Y. (2008) Int. J. Biomed. Sci. 4, 260–265 [PMC free article] [PubMed] [Google Scholar]
- 45.Arun P., Brown M. S., Ehsanian R., Chen Z., Van Waes C. (2009) Clin. Cancer Res. 15, 5974–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu G. H., Qu J., Shen X. (2008) Biochim. Biophys. Acta 1783, 713–727 [DOI] [PubMed] [Google Scholar]
- 47.Ishinaga H., Jono H., Lim J. H., Kweon S. M., Xu H., Ha U. H., Xu H., Koga T., Yan C., Feng X. H., Chen L. F., Li J. D. (2007) EMBO J. 26, 1150–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]