

Abstract
In NCX proteins CBD1 and CBD2 domains are connected through a short linker (3 or 4 amino acids) forming a regulatory tandem (CBD12). Only three of the six CBD12 Ca2+-binding sites contribute to NCX regulation. Two of them are located on CBD1 (Kd = ∼0.2 μm), and one is on CBD2 (Kd = ∼5 μm). Here we analyze how the intrinsic properties of individual regulatory sites are affected by linker-dependent interactions in CBD12 (AD splice variant). The three sites of CBD12 and CBD1 + CBD2 have comparable Kd values but differ dramatically in their Ca2+ dissociation kinetics. CBD12 exhibits multiphasic kinetics for the dissociation of three Ca2+ ions (kr = 280 s−1, kf = 7 s−1, and ks = 0.4 s−1), whereas the dissociation of two Ca2+ ions from CBD1 (kf = 16 s−1) and one Ca2+ ion from CBD2 (kr = 125 s−1) is monophasic. Insertion of seven alanines into the linker (CBD12–7Ala) abolishes slow dissociation of Ca2+, whereas the kinetic and equilibrium properties of three Ca2+ sites of CBD12–7Ala and CBD1 + CBD2 are similar. Therefore, the linker-dependent interactions in CBD12 decelerate the Ca2+ on/off kinetics at a specific CBD1 site by 50–80-fold, thereby representing Ca2+ “occlusion” at CBD12. Notably, the kinetic and equilibrium properties of the remaining two sites of CBD12 are “linker-independent,” so their intrinsic properties are preserved in CBD12. In conclusion, the dynamic properties of three sites are specifically modified, conserved, diversified, and integrated by the linker in CBD12, thereby generating a wide range dynamic sensor.
Keywords: Calcium, Calcium-binding Proteins, Calcium Transport, Membrane, Sodium-Calcium Exchange, CBD1 and CBD2, Calcium Occlusion, Calcium Sensor, NCX, Splice Variants
Introduction
The Na+/Ca2+ exchanger proteins (NCX1–3) and their splice variants are expressed in a tissue-specific manner (1–3) and mediate Ca2+ entry/extrusion depending on various factors (4, 5). NCX is “autoregulated” by cytosolic Ca2+ at sites that do not directly participate in the ion translocation (6, 7). For example, the cardiac/neuronal NCX1 variants respond to 0.3–10 μm cytosolic Ca2+, whereas the kidney variant does not (8, 9). Allosteric regulation of NCX involves the interaction of cytosolic Ca2+ with the regulatory loop-f of NCX (10–14). Although the high affinity regulatory site of NCX1 was discovered 16 years ago (15), only recent studies identified two Ca2+ regulatory domains, CBD1 and CBD2, which form a tandem (CBD12) through a short linker (amino acids 501–504) (16). High resolution x-ray and NMR revealed C2-type protein folding in both CBDs (16–20) that display four Ca2+ sites (Ca1–Ca4) on CBD1 (17, 18, 20) and two Ca2+ sites (CaI and CaII) on CBD2 (16, 19). The NCX splice variants arise from a combination of 6 small exons (A, B, C, D, E, F), while the splice variant region is exclusively restricted to the CBD2 domain (2–4, 16, 19, 24). Excitable tissues, such as those of the brain (AD splice variant) and heart (ACDEF splice variant) contain exon A, whereas kidney, stomach, and skeletal muscle tissues comprise NCX with exon B (2–5).
Equilibrium binding studies have shown that isolated CBD1 protein contains two sites with high affinity for Ca2+ (Kd = 0.2–0.4 μm and nH = ∼2) and two low affinity sites (Kd > 20 μm), whereas CBD2 binds two Ca2+ ions with Kd values of 5–10 μm (21). Structural and mutational studies concluded that only three of the six Ca2+-binding sites of CBD12 (Ca3 and Ca4 on CBD1 and CaI on CBD2) actually contribute to [Ca2+]-dependent regulation of the intact NCX1 protein (22–26). The two high affinity sites of CBD1 (Ca3 and Ca4) may play a critical role in determining the affinity (K0.5 = ∼0.3 μm) for [Ca2+]-dependent regulation by cytosolic Ca2+ in cellular systems, whereas the primary site of CBD2 (CaI) may contribute to the Ca2+-dependent relief of Na+-dependent inactivation of intact NCX (23–25). Importantly, the segment that undergoes alternative splicing is located solely within the CBD2 domain, meaning that specific domain-domain interactions may shape the regulatory properties of primary Ca2+ sites at CBD1 (16–20, 24).
Fluorescence resonance energy transfer studies demonstrated that CBD domains of NCX can undergo significant conformational changes in the cell within the excitation-contraction coupling (22, 27), although dynamic aspects of relevant conformational transitions, as well as the contribution of each specific Ca2+ site to regulatory events, were not resolved. The crystal structure of CBD12 has not yet been determined, although low resolution small angle x-ray scattering studies have shown that Ca2+ interaction with CBD12 results in global reorientation of the two domains (24). The dynamic aspects of two-domain reorientation were not investigated, although the relevant mechanisms could be important for shaping the dynamic properties of Ca2+-dependent regulation in various isoforms/splice variants of NCX.
The Ca2+-dependent regulation of NCX during the action potential requires millisecond time scale of feedback regulation, whereas the slow modes of inactivation last for seconds (6–9, 12). A critical question arises: Which specific Ca2+ sites contribute to the rapid and slower modes of NCX regulation, and how is this related to regulation of distinct NCX isoforms/splice variants in different cell types? In cellular systems, 0.3–10 μm cytosolic Ca2+ can activate NCX1 within a few milliseconds, whereas lower Ca2+ levels (<0.3 μm) activate the exchanger more slowly (for review see Ref. 29). Moreover, the secondary regulation of NCX, referred to as Ca2+-dependent inactivation (I2) is a slow process (t0.5 = ∼3–7 s) (30–32) that considerably varies among NCX splice variants (8, 9). Interestingly, the heart and brain splice variants of NCX1 exhibit nearly identical affinities for [Ca2+]-dependent activation, whereas the response of the brain-expressed variant is 10-fold faster than that of the cardiac one (8, 9). Previously, we found that Ca2+ on/off rates of CBD domains resemble the properties of the rapid Ca2+ sensor that can properly respond to cytosolic Ca2+ changes during the action potential (21). However, these findings could not explain how CBDs contribute to slow inactivation of NCX.
In the current study we evaluate the role of linker-dependent interactions in shaping the equilibrium and kinetic properties of three regulatory Ca2+ sites in CBD12. For this purpose, the isolated preparations of CBD1, CBD2, CBD12, and CBD1 + CBD2 proteins and their corresponding mutants (CBD1-E454K and CBD12–7Ala) were analyzed by protocols developed in our laboratory (21). Here we demonstrate, for the first time, the slow mode of Ca2+ dissociation from CBD12, which appears to be the linker-dependent phenomenon. Our findings reveal that the linker-dependent interactions selectively diversify intrinsic properties of Ca2+ on/off kinetics at a specific site of CBD12 sensor, thereby yielding a wide range dynamic sensor capable of NCX regulation within a broad time interval ranging 4 orders of magnitude.
MATERIALS AND METHODS
Expression and Purification of CBD Proteins
The DNA constructs of CBD1, CBD2, and CBD12 (encoding residues 371–509, 501–657, and 371–657, respectively) of canine NCX1 (accession code P23685; AD splice variant) were cloned into a pET23b vector (generously provided by Dr. M. Hilge). Seven alanines were inserted between residues His501 and Ala502 of the CBD12 construct by a PCR-based method, generating the CBD12–7Ala construct. The insertion was confirmed by DNA sequencing. The DNA construct of CBD1-E454K was a generous gift from Dr. Kenneth D. Philipson and Debora A. Nicoll (David Geffen School of Medicine at UCLA). The vectors were expressed in Escherichia coli Rosetta2 (DE3) competent cells (Novagen) at 37 °C for 5 h by using 1 mm isopropyl 1-thio-β-d-galactopyranoside for induction as outlined previously (16, 21). The cells were disrupted twice in a microfluidizer (Microfluidics, Newton, MA), and after centrifugation the supernatant was loaded onto a nickel bead column (HIS-SelectTM, nickel affinity gel; Sigma) as previously described (21). Purified CBD preparations were washed with 10 mm EDTA and 20 mm β-mercaptoethanol using an Ultracel-3k (Millipore) device, and the EDTA was subsequently removed by washing the protein with decalcified buffer (10 mm Tris-HCl, 100 mm KCl, 20 mm β-mercaptoethanol) in five or six successive steps of ultrafiltration. Concentrated preparations of purified CBDs contain less than 100 nm free Ca2+ (as judged by a Fluo-32 assay), whereas the fractional occupation of CBD-binding sites by Ca2+ did not exceed 3% of the maximal binding capacity. The protein concentrations were determined at A280 nm. The extinction coefficients were estimated as ϵ = 2560 m−1 cm−11 for CBD1, ϵ = 5120 m−1 cm−1 for CBD2, and ϵ = 7680 m−1 cm−1 for CBD12. Purified protein preparations (>95%, as judged by SDS-PAGE) were concentrated to 300–1200 μm and stored at −80 °C. No changes in either Ca2+ binding affinity, stoichiometry, or off kinetics have been detected within at least 6 months of storage in deep freezing.
Equilibrium Binding of 45Ca2+ to Protein
All of the buffers for Ca2+ binding assays were prepared on the basis of decalcified water containing <0.1 ppm Ca2+ (LC-MS Chromasolv; Fluka). The decalcified buffer solutions contained less than 2 μm [Ca2+]free (21). To remove Ca2+, all glassware and tubes were washed with 0.1 m HCl, and the filters were pretreated with 5% NaHCO3 (33, 34). The 45Ca2+ binding experiments were done with an Ultracel-3k (4- or 15-ml sample compartment; 3-kDa cut-off; Millipore Corp.). The assay medium (1–2 ml) containing 10 μm protein was placed in the upper compartment of the Ultracel-3k concentrator, and the binding assay was performed as previously outlined (21). 1 μl of 5–6 mm 45Ca2+ stock solution (∼3 μCi) was added to the assay medium with protein, carefully mixed, and allowed to equilibrate. The sample was centrifuged at 1000 × g for 50 s, and 10–20-μl aliquots of filtrate and protein (upper chamber) were analyzed for sample radioactivity. Subsequently, nonradioactive CaCl2 was added serially to the protein sample, and after centrifugation, 10–20-μl aliquots of the filtrate and upper compartment were tested for sample radioactivity. The [Ca2+]free was measured as [Ca2+]free = [Ca2+]tot(a/b), where a and b represent the radioactivity in the ultrafiltrate and upper compartment, respectively. The CBD-bound Ca2+ was determined as [Ca2+-CBD] = [Ca2+]add + [Ca2+]res − [Ca2+]free (21, 33, 34). [Ca2+]add represents the externally added Ca2+, whereas [Ca2+]res is the residual (endogenous) Ca2+ in the solution. [Ca2+]res was determined by Fluo-3 or Fluo-4 as outlined before (21). The bound 45Ca2+/CBD (mol/mol) was plotted versus [Ca2+]free, and the data were fitted to an Adair or Hill equation using the GraFit software (Erithacus Software Ltd.) as described before (21). All other miscellaneous procedures were described elsewhere (38, 39).
Stopped Flow Experiments
In the stopped flow experiments, 150 μl of 10–40 μm CBD1, CBD2, CBD1 + CBD2, CBD12, or CBD12–7Ala in TK buffer (10 mm Tris-Cl, pH 7.2, and 100 mm KCl) containing 1–80 μm free Ca2+ (syringe A) were mixed (t = 10–30 ms) at 25 °C with 150 μl of TK buffer containing 200–600 μm Quin-2 (syringe B). The Quin-2 probe was utilized in this assay because under the given experimental conditions, this probe has an extremely high affinity toward Ca2+ (Kd = ∼50 nm), whereas the second order rate constant for Ca2+ binding (kon = ∼109 m−1 s−1) is diffusion-controlled. The Ca2+ off rates can be accurately measured because following the mixing of Ca2+-bound protein with Quin-2, the [Ca2+]free drops below 7–80 nm within <2 ms, thereby allowing the dissociation of Ca2+ from protein. Subsequently, Ca2+ dissociates from the protein complex (albeit on a slower time scale) and binds to Quin-2, resulting in a rise of fluorescence. The observed increase in fluorescence corresponds to the rate and the amount of Ca2+ dissociation from the complex. The stopped flow machine SFM-3 (BioLogic) was equipped with a two mixer/three syringe mixing system, a TC-100/15 cuvette (a mixing dead time of ∼2 ms), temperature controller, microprocessor, and single-wavelength monochromator (MOS-200). Quin-2 was excited at λex = 333 nm from a monochromator with a hydrogen-xenon lamp (150 W), and the emission was monitored at λem > 495 nm by using a long pass filter. Parameters (mixing time duration, flow rates, volume, signal sampling time, detection time length, etc.) for the desired stopped flow experiments were chosen with a MPF program (Bio-Logic). The data were analyzed with Bio-Kine 32 V4.45 (Bio-Logic) equipped with Pad-Laplace and Simplex analytical modes, allowing the automatic fitting to multi-exponential kinetics. Experimentally obtained traces were fitted to mono-exponential or multi-exponential curves by using the χ2 criteria to identify the best fit for a specific kinetic model of Ca2+ dissociation. All other details for data processing and analysis are described elsewhere (35–38, 40).
Statistics
The measured values are presented as the means ± S.E. Statistical analysis was performed by using the two-sample independent t test (Origin 7.0, Northampton MA).
RESULTS
To examine the effects of soluble CBD2 on the Kd values of the high affinity binding sites (Ca3 and Ca4) of CBD1, the 45Ca2+ titration curves of CBD1, CBD2, CBD1 + CBD2, and CBD12 proteins were analyzed by using the protein/ligand ultrafiltration approach (21, 33, 34). Only the AD splice variant of CBD2 and CBD12 was used in the present study. Adair analysis of 45Ca2+ binding curves reveals that the two high affinity sites (Ca3 and Ca4) of CBD1 (Kd = 0.2–0.6 μm) and the primary site (CaI) of CBD2 (Kd = 5–10 μm) have comparable values in the context of CBD1 + CBD2 and CBD12 (Fig. 1). These data suggest that linker-dependent interactions may decrease the Ca2+ binding affinities of the relevant three regulatory sites up to 3–4-fold, at most (because of the differences in protein preparations and the nature of the applied experimental procedures, these differences should be considered within or close to the experimental error). The isothermal titration calorimetry technique was utilized to detect any possible interaction between soluble CBD1 and CBD2 domains in solution at CBD concentrations of 20–50 μm concentrations, but no evidence was found for this type of protein-protein interaction (not shown).
FIGURE 1.

45Ca2+ binding curves of CBD1, CBD2, CBD12, and CBD1 + CBD2 proteins. The equilibrium binding of 45Ca2+ was measured by incubating 10 μm of the purified proteins with 45Ca2+ in 1 ml of assay medium containing 10 mm Tris-HCl, pH 7.2, and 100 mm KCl (TK buffer). The concentrations of Ca2+ were increased by adding stock solutions of nonradioactive Ca2+, and equilibrium binding was measured by ultrafiltration as described under “Materials and Methods.” The data are plotted as bound 45Ca2+/CBD protein (mol/mol) versus free concentrations of Ca2+. The curves were calculated according to the Adair equation with predefined number of binding sites as follows: CBD1 (four sites), CBD2 (two sites), CBD1 + CBD2 (six sites), and CBD12 (six sites). a, titration curves of CBD1 (□) and CBD2 (◇). b, titration curves of CBD1 + CBD2 (▵) and CBD12 (○). The Kd values derived from data fitting are summarized in Table 1.
The fluorescent probe Quin-2 was used in combination with the stopped flow technique for measuring the rate constants of Ca2+ dissociation from CBD1, CBD2, CBD12, CBD12–7Ala, or CBD1 + CBD2 by using the protocols developed in our laboratory (21, 35, 36). Before the stopped flow experiments, 10–20 μm of each protein analyzed were pre-equilibrated with Ca2+ containing buffer, and Ca2+ dissociation was then initiated by mixing Ca2+-bound proteins with 0.2–0.6 mm Quin-2. Remarkably, the Ca2+ dissociation from the low affinity sites (Kd > 20 μm) was so rapid (> 300 s−1), it could not be detected by the stopped flow technique. Approximately two Ca2+ ions dissociate from CBD1 (kf = 15 s−1), and one Ca2+ ion dissociates from CBD2 (kr = 125 s−1) showing monophasic kinetics, whereas under comparable experimental conditions the biphasic kinetics of Ca2+ dissociation from CBD12 is detected with kr = 280 s−1 and kf = 5 s−1 (Fig. 2a). Notably, the observed kinetics for biphasic Ca2+ dissociation from CBD12 cannot be completely resolved under the given experimental conditions, because the reaction does not reach saturation because of the extremely short signal detection time. To reliably resolve the slow phase of Ca2+ dissociation from CBD12, the signal detection duration was extended to 16–40 s, whereas final [Ca2+]free levels (after mixing) were kept as low as <7 nm, and rapid mixing parameters (mixing time, flow rates, data sampling time, detection duration, etc.) were adjusted to resolve the reactions with rate constants in the range of 0.1–80 s−1 (see below).
FIGURE 2.
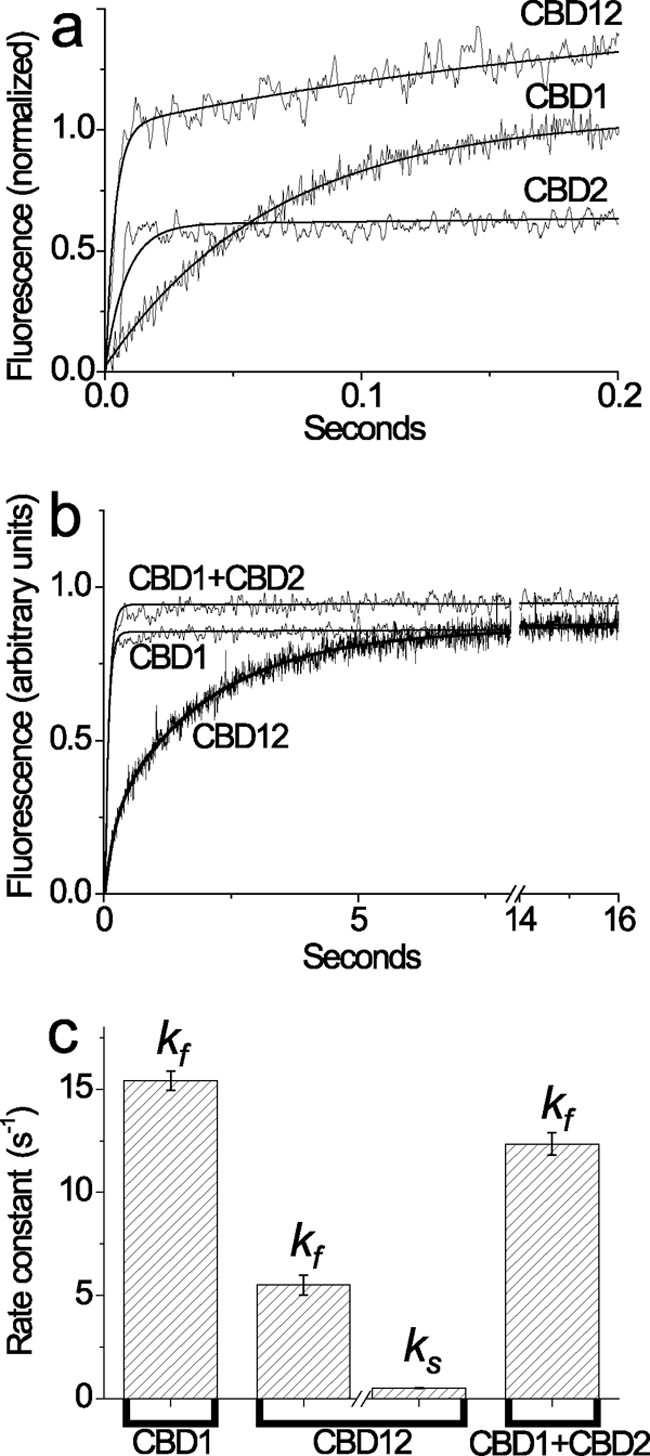
Stopped flow kinetics of Ca2+ dissociation from the high affinity sites of CBD1, CBD2, CBD12, and CBD1 + CBD2. All buffer solutions were prepared on the basis of 10 mm Tris-HCl, pH 7.2, 100 mm KCl (TK buffer) at 25 °C. a, syringe 1 contained 20 μm CBD12, CBD1, or CBD2 with 80 μm free Ca2+, and syringe 2 contained 153 μm Quin-2. 150-μl aliquots from syringe 1 were mixed with the identical volume of Quin-2 buffer in syringe 2. The representative trace of CBD12 was fitted to a double exponential curve with kr = 280 ± 32 s−1 and kf = 4.5 ± 0.19 s−1, whereas the traces of CBD1 and CBD2 were fitted to a single exponential curve with kf = 15.2 ± 0.15 s−1 and kr = 123.6 ± 4.42 s−1, respectively. b, syringe 1 contained either CBD1, CBD12, or CBD1 + CBD2 (10 μm of each protein) in TK buffer with 10 μm free Ca2+, and syringe 2 contained 200 μm Quin-2 buffer. 150-μl aliquots from syringe 1 were mixed with an identical volume of Quin-2 buffer from syringe 2 to decrease [Ca2+] below 7 nm (see “Materials and Methods”). The representative traces of CBD1 or CBD1 + CBD2 were fitted to a single exponential curve with kf = 14.8 ± 0.07 s−1 and kf = 13.1 ± 0.07 s−1, respectively. The representative trace of CBD12 was fitted to a double exponential curve with kf = 5.2 ± 0.08 s−1 and ks = 0.52 ± 0.001 s−1. c, the bars represent the statistics of experimentally observed kf and ks values (see above, a and b) obtained in four to seven independent experiments performed with three different preparations of CBD1, CBD2, CBD12, or CBD1 + CBD2 proteins. All of the measured values represent the means ± S.E.
Approximately two Ca2+ ions dissociate from high affinity sites of CBD1 and CBD1 + CBD2 preparations showing monophasic kinetics of kf = 13–16 s−1 (Fig. 2b), meaning that the interaction of CBD1 with CBD2 in solution does not alter koff. In sharp contrast to the CBD1 and CBD1 + CBD2 preparations, a slow mode of Ca2+ dissociation is observed in CBD12 preparations (Fig. 2b). These traces can be fitted either to monophasic dissociation of two Ca2+ ions with ks = 0.8 s−1 or to biphasic (sequential) dissociation of two Ca2+ ions with ks = 0.3–0.5 s−1 and kf = 7–10 s−1. The usage of the χ2 criteria consistently exhibits a better fit for biphasic (sequential) versus monophasic kinetics. Consistent results were obtained with three preparations of CBD12 tested in 37 independent experiments (Fig. 2c). Therefore, the CBD1-CBD2 linker decelerates the dissociation of one Ca2+ ion from CBD12 by 50–80-fold while exerting a rather small effect on the “fast” Ca2+ dissociation (kf). Notably, the Ca3-Ca4 sites retain reasonably high affinity properties in the context of CBD12 (Fig. 1b), despite the low values of koff (Fig. 2b) and kon (calculated as koff/Kd).
To resolve the effect of the medium/low affinity sites on the slow kinetics of Ca2+ dissociation from high affinity sites, CBD12 was incubated with 1–80 μm free Ca2+ to occupy two to six sites at fixed [Ca2+]free levels (Fig. 3a). Subsequently, the Ca2+-bound species of CBD12 were mixed with Quin-2 in the stopped flow apparatus to monitor Ca2+ off rates. The striking finding is that the slow kinetics of Ca2+ dissociation are observed even under conditions where only two high affinity sites of CBD12 are occupied by Ca2+ (Fig. 3b). Increasing occupation of medium/low affinity sites has no effect on slow kinetics (ks) of Ca2+ dissociation, although a 2–3-fold acceleration of kf may occur under these conditions (Fig. 3c). The effect on kf values could be even more prominent than the effects observed here, because the Ca2+ dissociation kinetics of low affinity sites (Kd > 20 μm) are so fast (>300 s−1) that the regulatory effect of the low affinity “secondary” sites cannot be properly characterized under the given experimental conditions.
FIGURE 3.
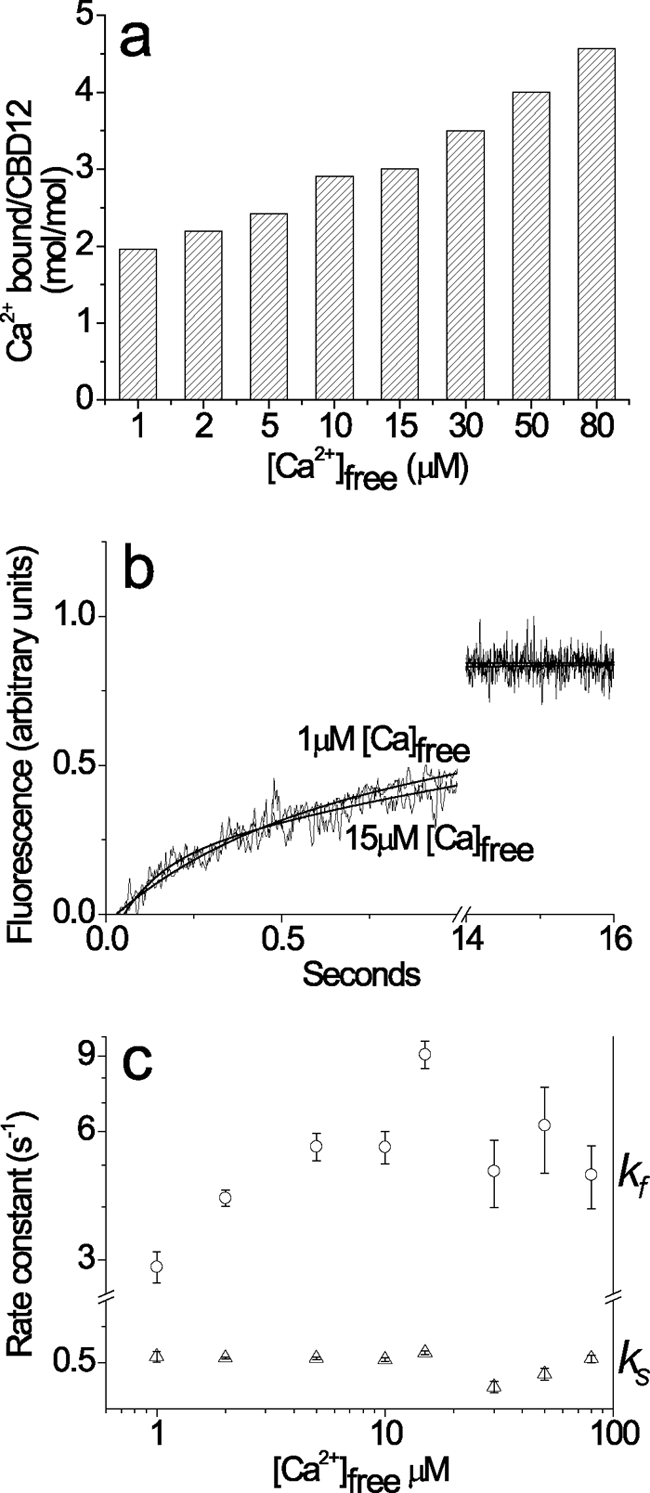
Ca2+ dissociation kinetics from CBD12, with two to six bound Ca2+ ions. Before performing the stopped flow experiment, the CBD12 protein was preincubated with various concentrations of Ca2+ to obtain the following fixed levels of [Ca2+]free = 1, 2, 5, 10, 15, 30, 50, and 80 μm. The amount of Ca2+ bound to CBD12 was measured independently at each fixed concentration of free Ca2+. All of the buffer solutions were prepared on the basis of 10 mm Tris-HCl, pH 7.2, 100 mm KCl (TK buffer). a, the occupancy of CBD12 Ca2+-binding sites (mol/mol) was estimated at varying [Ca2+]free by using the protein ultrafiltration method for assaying equilibrium 45Ca2+ binding (see Fig. 1). b, stopped flow experiments were performed under conditions described for Fig. 2. Syringe 1 contained 10 μm of CBD12 with varying Ca2+ concentrations. Syringe 2 contained 200 μm Quin-2 for [Ca2+]free < 30 μm or 600 μm Quin-2 for [Ca2+]free ≥ 30 μm. 150-μl aliquots from syringe 1 were mixed with identical volumes from syringe 2. In all of the experiments, the final [Ca2+]free after mixing were <10 nm. The trace for 1 μm free Ca2+ was fitted to a double exponential curve with kf = 2.76 ± 0.03 s−1 and ks = 0.53 ± 0.001 s−1. The trace for 15 μm Ca2+ was fitted to a double exponential curve with kf = 8.87 ± 0.32 s−1 and ks = 0.59 ± 0.002 s−1. c, the kf and ks values for Ca2+ dissociation from CBD12 were experimentally derived at the indicated [Ca2+]free values. The data were then plotted as the means ± S.E. Each point represents the average value obtained from four or five independent experiments.
For further characterization of the linker-dependent effects on the intrinsic properties of CBD12 Ca2+ sites, we prepared and analyzed the CBD12–7Ala protein, in which seven alanines were inserted into the CBD1-CBD2 linker between residues His501 and Ala502. The three relevant Ca2+ sites of CBD12–7Ala show 45Ca titration curves very similar to those of CBD1 + CBD2 or CBD12, suggesting that the Ca2+ binding affinities of Ca3-Ca4 (on CBD1) or CaI (on CBD2) are not considerably modified by the longer linker in CBD12–7Ala (Fig. 4a). Most strikingly, no slow dissociation of Ca2+ was observed in CBD12–7Ala while exhibiting typical Ca2+ off rates observed in CBD1 + CBD2 (Fig. 4b). This implies that with the longer linker, all three sites of CBD12–7Ala display kinetics and equilibrium properties similar to those observed in isolated CBD1 and CBD2 preparations. Therefore, CBD12 and CBD12–7Ala fundamentally differ in their Ca2+ dissociation kinetics, suggesting that the intact linker is capable of generating specific interdomain interactions responsible for decelerating on/off kinetics at a specific high-affinity site of CBD1 in intact CBD12.
FIGURE 4.

45Ca2+ titration curve and Ca2+ dissociation kinetics of the CBD12–7Ala mutant. The experimental conditions for the equilibrium 45Ca binding assay and stopped flow assay of Ca2+ dissociation were similar to those described in the legends of Fig. 1 and 2. a, the 45Ca2+ titration curves were calculated using the Adair equation, showing Cap = 6 (mol/mol) and Kd = 0.05, 0.1, 4, 6.5, 8.5, and 50 μm for CBD12–7Ala (□). The dashed lines depict typical titration curves of CBD1-WT and CBD12-WT. b, syringe 1 contained 10 μm CBD12–7Ala in TK buffer with 10 μm free Ca2+, and syringe 2 contained 200 μm Quin-2 in TK buffer. The representative trace of CBD12–7Ala was fitted to a single exponential curve with kf = 18.7 ± 0.08 s−1. The dashed lines represent typical traces observed for CBD1-WT and CBD12-WT.
To evaluate the role of the low affinity Ca1 site in regulating the affinity and/or Ca2+ off rates at high affinity sites of CBD1, the CBD1-E454K mutant was analyzed in terms of its Ca2+ titration curves (Fig. 5a) and Ca2+ dissociation kinetics (Fig. 5b). The rationale behind this is that although Glu454 is a part of the low affinity Ca1 site, it is known that the E454K mutant does not affect the [Ca2+]-dependent activation curve of intact NCX1 in the cellular system (25). The Ca2+ titration curve shows a maximal binding capacity of ∼2 Ca2+/CBD1-E454K (mol/mol) with a “normal” Kd of 0.24 μm and reduced cooperativity (nH = ∼1.3), whereas the wild type CBD1 shows a maximal binding capacity of ∼3 Ca2+/CBD1-WD (mol/mol) with Kd of 0.34 μm and high cooperativity of nH = ∼2.8 (Fig. 5a). The remarkable finding was that the Ca2+ off rate of CBD1-E454K (kf = 80 s−1) was much faster than that of wild type CBD1 (kf = 18 s−1) (Fig. 5b). Very similar data were obtained under conditions in which the stopped flow mixing was initiated at 10 or 80 μm free Ca2+ (not shown). Although the kon value was not directly measured in these experiments, it can be reliably quantified from the experimentally measured values of Kd and koff (kon = koff/Kd). Hence, the only reasonable explanation for the present results is that the E454K mutation accelerates both kon and koff proportionally, so the Ca2+ affinity (Kd) is not significantly altered. Therefore, at least one low affinity site (Ca1) of CBD1 may play an important role in determining the intrinsic on/off rates of Ca2+ interaction with Ca3-Ca4 sites while having a negligible (if any) effect on equilibrium binding at the same sites. Additional systematic research of relevant mutants is required to further elucidate these issues.
FIGURE 5.
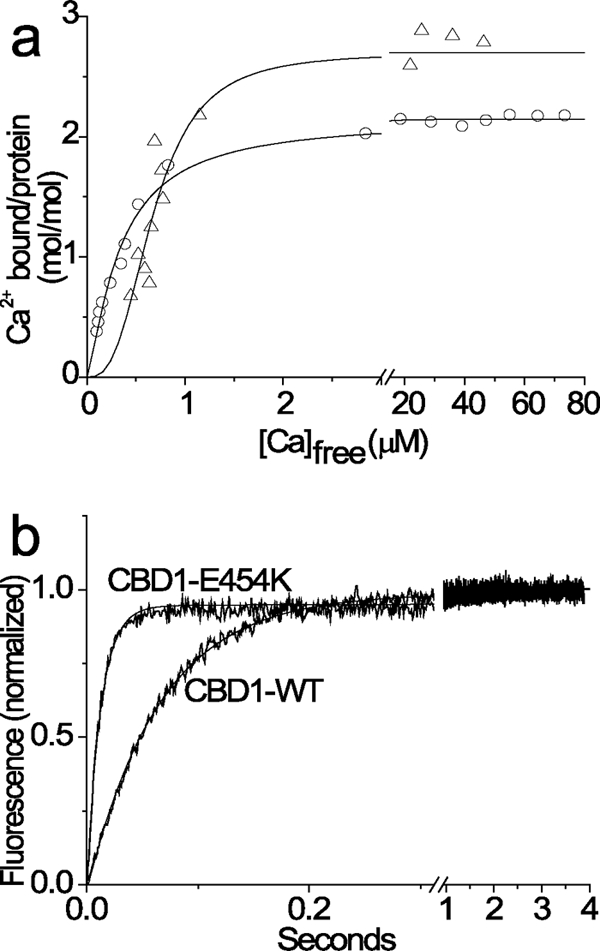
45Ca2+ titration curve and Ca2+ dissociation kinetics of the CBD1-E454K mutant. The experimental conditions for equilibrium 45Ca binding assay and stopped flow assay of Ca2+ dissociation were similar to those described in the legends of Fig. 1 and 2. a, the 45Ca titration curves were calculated by using the Hill equation, yielding Kd = 0.32 ± 0.11 μm, nH = 2.87 ± 0.72, and Cap = 2.70 ± 0.1 (mol/mol) for the wild type CBD1 (▵) and Kd = 0.25 ± 0.07 μm, nH = 1.28 ± 0.08, and Cap = 2.15 ± 0.02 (mol/mol) for the CBD1-E454K mutant (○). b, 10 μm of wild type or mutant CBD1 preparations were preincubated with 60 μm free Ca2+ and then mixed with Quin-2 in the stopped flow apparatus to initiate the Ca2+ dissociation (for details see “Materials and Methods” and Figs. 2 and 3). The data were fitted to a single exponential curve with values of kf = 18.0 ± 3.1 s−1 and kf = 80.2 ± 5.3 s−1 for the wild type CBD1 and CBD1-E454K mutant, respectively.
DISCUSSION
The present study was conducted to evaluate the contribution of linker-dependent interdomain interactions in shaping the affinity and kinetic properties of three regulatory Ca2+ sites in the brain splice variant CBD12 (AD). Our data reveal that (a) the linker slows down Ca2+ off rates at a specific Ca2+ site of CBD1 (by nearly 2 orders of magnitude), whereas the changes in Kd do not exceed 3–4-fold; (b) the linker does not significantly alter the intrinsic affinity and kinetic properties of the other two CBD12 regulatory sites (located on CBD1 and CBD2), thereby retaining their diverse kinetic properties in CBD12; and (c) the linker integrates the dynamic range of all three Ca2+ sites such that the dynamic range of Ca2+ sensing is expanded. The biological significance of the current findings is that linker-dependent interactions selectively retain, modify, and integrate dynamic properties of three regulatory sites of CBD12, thereby allowing effective regulation of NCX within a broad range time interval (10−3–10 s).
The Linker Dramatically Slows Ca2+ On/Off Rates at Specific High Affinity Site of CBD1 in CBD12
The striking finding is that CBD12 exhibits slow dissociation of Ca2+ (ks = 0.3–0.6 s−1), whereas all other protein preparations (CBD1, CBD2, CBD1 + CBD2, or CBD12–7Ala) lack this capacity (Figs. 2b and 4b). Moreover, the Ca2+ titration curve analysis shows that three regulatory sites of CBD1 + CBD2, CBD12, or CBD12–7Ala exhibit typical Kd values within 0.2–5 μm (Figs. 4a and 5a). However, CBD12 demonstrates a markedly distinct profile of Ca2+ dissociation kinetics differing from all the other proteins tested. Three Ca2+ ions dissociate from CBD12 with multiphasic kinetics (kr = 280 s−1, kf = 7 s−1 and ks = 0.4 s−1), whereas monophasic kinetics is observed for dissociation of two Ca2+ ions from CBD1 (kf = 16 s−1) and one Ca2+ ion from CBD2 (kr = 125 s−1) (Fig. 2). In agreement with the Ca2+ titration curves, the isothermal titration calorimetry measurements showed no evidence of interaction between soluble CBD1 with CBD2 in solution (not shown). Therefore, the slow dissociation of one Ca2+ ion from CBD12 represents specific interdomain interactions controlled by the linker. Most probably, the CBD1-CBD2 linker-dependent local interactions between the two CBD domains and/or linker-dependent conformational constraints at a specific CBD1 site (Ca3 or Ca4) specifically decelerate Ca2+ off rates from 16–18 s−1 to 0.2–0.4 s−1, whereas the Kd is retained within <0.6 μm. The only reasonable explanation for the obtained data is that the linker-dependent interdomain interactions decelerate the Ca2+ on/off rates by 50–80-fold at one specific CBD site, without greatly compromising the Ca2+ sensing affinity. The current findings are consistent with sequential dissociation of two Ca2+ ions from the Ca3-Ca4 sites, whereas the dissociation of the first (fast) Ca2+ ion is followed by dissociation of the second (“occluded”) Ca2+ ion, displaying characteristic slow kinetics (Fig. 6). Our findings are consistent with recent structural studies indicating that Ca2+ binding enhances the stability of the Ca2+ binding site of CBD1 near the hinge region while the overall structure of CBD1 remains largely unaffected (28). The underlying biological relevance is that the linker-dependent modification of Ca2+ on/off kinetics at a specific regulatory site can diversify and widen the dynamic range of Ca2+ sensing of CBD12 by nearly 2 orders of magnitude (while retaining a submicromolar range of Ca2+ affinity). Therefore, the linker-dependent interactions may extend the dynamic range of NCX regulation for up to 10–20 s.
FIGURE 6.

Ca2+ on/off kinetics in isolated preparations of CBD1, CBD2, CBD12, and CBD12–7Ala. Filled circles represent the Ca2+ occupied sites, whereas open circles depict the empty Ca2+ sites. The indicated koff values represent the typical empirical values. The kon values were calculated (kon = koff/Kd) from the Kd and koff values derived experimentally. The assigned terms rapid, fast, and slow represent the Ca2+ dissociation kinetics from the CaI, Ca3, and Ca4 sites, respectively.
Two Ca2+ Sites Retain Their Intrinsic Properties in CBD12 Context
The experimental fact is that two Ca2+ sites of CBD12 exhibit very similar kinetics and binding affinities in comparison with isolated preparations of CBD1 (kf = 7–18 s−1) and CBD2 (kr = 120–250 s−1) (Fig. 2). These data support the idea that the CBD1-CBD2 linker-dependent conformational modifications do not drastically influence (<3–4-fold) the kinetic and affinity properties of single Ca2+ sites located on CBD1 and CBD2. This conclusion has been further supported by analysis of the CBD12–7Ala mutant. The insertion of seven alanines into the linker (CBD12–7Ala) abolishes the slow dissociation of Ca2+, whereas all three sites of CBD12–7Ala still exhibit similar kinetics and affinity to isolated CBD1 and CBD2 (Figs. 4 and 5). A critical question that arises is what are the structure-activity relationships that determine “selective” modification of kinetic properties at one Ca2+ site on CBD1 while preserving the intrinsic properties of two other sites on CBD1 and CBD2? Because the crystal structure of CBD12 has not yet been determined, we are uncertain regarding the exact mechanisms involved. Nevertheless, the currently available x-ray structures (17, 19) of isolated CBD1 and CBD2 may provide some valuable and relevant clues on this issue. First of all, the primary site of CBD2 is at least 30 Å away from the linker, so it is not surprising that the linker-dependent structural transitions do not affect the intrinsic properties of this site. Second, the coordination chemistry of Ca2+ ligation of the Ca3 and Ca4 sites are considerably different in respect to the linker positioning (17), so these sites might be distinctly sensitive to the linker-dependent conformational transitions. Therefore, we postulate that in CBD12, the linker-dependent constraints alter on/off rates at specific Ca2+ site of CBD1 while retaining the intrinsic properties of the remaining two Ca2+ sites located on CBD1 and CBD2 intact (Fig. 6).
The Dynamic Properties of the Three CBD12 Regulatory Sites Are Diverse and Complementary
One important conclusion from the present work is that the CBD1-CBD2 linker not only specifically diversifies and preserves the dynamic properties of specific Ca2+ sites in CBD12 but also effectively integrates the dynamic properties of all three regulatory sites (Fig. 6). The rationale behind this is to provide a broad range dynamic sensor capable of dynamically regulating NCX proteins within a time scale spanning over 4 orders of magnitude (from milliseconds to seconds). The kinetically distinct Ca2+ sensors may represent a physical basis for differential Ca2+ sensing in various cellular compartments. More specifically, Ca2+ interactions with a “rapid” CBD2 site (the CaI site) can respond to transient swings of [Ca2+] that occur within less than 10 ms. This sensor may play an essential role in restricted areas of the cell (e.g. subsarcolemma space and dyadic cleft), where the [Ca2+] levels can transiently reach 300–600 μm levels within a few milliseconds, when the cell is depolarized (47–49). On the other hand, the fast CBD1 site is capable of responding to cytosolic [Ca2+] changes that take place within the duration of the action potential (10–200 ms) (Fig. 2a). This would be consistent with the notion that the NCX1 molecules are selectively co-localized and regulated within the cell (48). The dynamic range of CBD12 is further extended by the linker-dependent modulation of the “slow” site of CBD1, which may be involved in Ca2+-dependent events that last up to 10 s. This slow Ca2+ sensor may represent I1 and/or I2 inactivation modes of NCX observed in electrophysiological experiments (30–32).
Is the Linker Involved in Interdomain Signal Transmission?
The [Ca2+]-dependent activation curve of intact NCX1 (23, 25) is almost identical to Ca2+ titration curves obtained for isolated CBD1 (21), meaning that Ca3-Ca4 sites of CBD1 might determine the affinity for Ca2+-dependent activation. Insertion of the 7 alanine residues into the linker of the intact exchanger (NCX1–7Ala) decreases the affinity of [Ca2+]-dependent activation of NCX1–7Ala 15–30-fold, exhibiting a K0.5 of 5–10 μm (43). Two alternative explanations can be considered for interpretation of these results. Either the elongation of the linker results in decreased affinity of the Ca3-Ca4 sites on CBD1, or alternatively the primary site of CBD2 (CaI) becomes a predominant sensor for activation in NCX1–7Ala. In our CBD12–7Ala construct, two high affinity sites exhibit values typical of the Ca3-Ca4 sites, meaning that the long linker does not decrease the affinity of the two high affinity sites of CBD1 (Fig. 4). In light of the present considerations, we suggest that the long linker “disconnects” (uncouples) the interaction between the two CBD domains in NCX1–7Ala, so that the CaI site (Kd = 5–10 μm) of CBD2 (and not the Ca3-Ca4 sites of CBD1) becomes a “master” sensor for [Ca2+]-dependent activation of NCX1–7Ala. Currently, it is unclear how the kinetic aspects described here are related to the recently discovered Ca2+-dependent reorientation of two CBD domains in CBD12 (24). In any case, further research and application of alternative biophysical approaches (e.g. fluorescence resonance energy transfer) is required to resolve the dynamic aspects of linker-dependent domain-domain interactions. Artificial variations in the linker length could be a useful probe for identifying functionally relevant conformational transitions.
Low Affinity Sites of CBD12 Do Not Modulate Slow Dissociation of Ca2+
Mutational and structural studies have shown that two low affinity sites (Ca1 and Ca2) of CBD1 and one low affinity site of CBD2 (CaII) are not directly involved in [Ca2+]-dependent activation of intact NCX1 (23–26). On the other hand, a recently described model suggests that Ca2+ interaction with all four sites of CBD1 might play an important role in determining the electrostatic charge that drives the reorientation of the two CBD domains (24). Here we demonstrate that the slow mode of Ca2+ dissociation from CBD12 is not conditioned or modulated by Ca2+ occupation of medium/low affinity sites (Kd > 5 μm) of CBD12 (Fig. 3). Therefore, slow Ca2+ kinetics of CBD12 represents a “Ca2+-independent” mode, which is exclusively constrained by the linker. On the other hand, our data indicate that [Ca2+] > 2 μm can moderately accelerate (3–4-fold) the kf off rates (Fig. 3C), suggesting the possibility that occupation of CaI of CBD2 may accelerate Ca2+ dissociation from the “fast” CBD1 site. Taking into account the conformational changes associated with Ca2+ binding to CBD2 (42), it is more probable that Ca2+ binding to CBD2 controls the signal transmission from CBD2 to transmembrane segments rather than regulates the Ca2+ on/off rates at CBD1.
Glu454 contributes to Ca2+ ligation at the low affinity Ca1 site of CBD1 (16, 23–26), whereas substitution of Glu454 with lysine significantly stabilizes the protein folding of the CBD1-E454K mutant (25). Here we analyzed Ca2+ titration curves and Ca2+ off kinetics of CBD1-E454K and found that the replacement of the negative charge by the positive one in E454K does not have any considerable effect on the equilibrium binding of Ca2+ at Ca3-Ca4 sites (Fig. 5a). However, the CBD1-E454K mutant exhibits 5–7-fold faster kinetics for Ca2+ dissociation (kf = 80 s−1) in comparison with the wild type CBD1 (kf = 14–18 s−1) (Fig. 5b). Therefore, even in the absence of the CBD1-CBD2 linker, the low affinity site(s) of CBD1 can modulate on/off kinetics (but not the equilibrium binding) at the high affinity Ca2+ sites of CBD1. In light of the present findings, the electrostatic interactions of Ca2+ with the low affinity sites of CBD1 may shape the kinetics rather than the equilibrium properties of high affinity sensors on CBD1.
Interplay between the Kinetic and Equilibrium Properties of CBD12 Ca2+ Sensing
Our analyses clearly demonstrate that the kinetic diversity of three Ca2+ sites is much more prominent (koff = 0.4–250 s−1) than the affinity range (Kd = 0.2–5 μm) (Table 1). Although the observed Ca2+ on/off rates do not necessarily represent protein conformational transitions, dynamic diversity of the three regulatory sites may represent a physical scope for defining the kinetic range of Ca2+ sensing. In general, the interplay between the kinetic and affinity properties must be thoroughly balanced in Ca2+-sensing proteins in terms of structure-function relationships (44–46). Consistent with this principle, the Ca2+ off rates of two distinct domains could be very different even in the same protein (45, 46). For example, in calmodulin, the off rate for the EF-hands in the N-terminal lobe is over 100 times faster than that of the C-terminal domain (46). However, in contrast to calmodulin, the C2 domains (including CBDs) do not undergo concerted conformational changes upon Ca2+ binding (16–19, 41, 44–46, 50, 51). The remarkable feature of the CBD12 sensor is that the linker-dependent conformational constraints modify the Ca2+ on/off rates ∼100-fold at a specific regulatory site of CBD1 without greatly compromising the binding affinity (Figs. 1–4). Interestingly, linker-dependent interactions between two C2 domains are fundamentally different, as demonstrated in by Rabphilin-3A (51) and Synaptotagmin (41, 50), so the linker-related domain-domain interactions could substantially diverge in the course of evolution. It is currently unclear whether the linker-dependent effects described here represent exclusive properties of CBD12 or whether the other C2 proteins also possess similar structure-function relationships. This issue may be of general interest because of the vast amount of functionally important proteins (synaptotagmins, PKC, titin, fibronectin, neuronal cell adhesion factors, etc.) that sense Ca2+ through C2 folded domains, and ∼30% of them form two-domain structures (41, 44, 45, 50, 51). More dedicated kinetic analyses of two-domain C2-tandems are required to reach meaningful conclusions.
TABLE 1.
Microscopic Kd values of CBD1, CBD2, CBD12, and CBD1 + CBD2 proteins derived from the equilibrium 45Ca binding curves
The 45Ca titration curves of CBD1, CBD2, CBD12, and CBD1 + CBD2 proteins were performed as described in the legend to Fig. 1, and the lines were calculated to fit the experimental points according to the Adair equation until the “best fit” is obtained (judged by the χ2 weighting criteria). The maximal binding capacities were fixed for each protein, whereas the Kd values were allowed to fit the experimental data obtained from 45Ca titration curves. The Adair equations were written according to predefined models describing Ca2+ binding to specific protein containing four sites (CBD1), two sites (CBD2), or six sites (CBD12 or CBD1 + CBD2) by using the GraFit software (for details see “Materials and Methods”).
| Maximal binding capacity | Microscopic Kd values | |
|---|---|---|
| mol/mol | μm | |
| CBD1 | 4.0 | 0.03, 0.1, 6.8, 21 |
| CBD2 | 2.0 | 5.3, 22 |
| CBD12 | 6.0 | 0.2, 1.6, 4, 35, 60, 110 |
| CBD1 + CBD2 | 6.0 | 0.05, 0.1, 9, 12, 60, 80 |
Acknowledgments
We are grateful to Drs. K. D. Philipson and D. A. Nicoll for the gift of the DNA construct encoding the CBD1-E454K mutant.
This work was supported in part by Israeli Ministry of Health Grant 2010-3-6266, USA-Israeli Binational Research Grant 2009334, and Israel Science Foundation Grant 23/10.
- Fluo-3
- N-[2-[2[bis(carboxymethyl)amino]-5-(2,7dichloro-6-hydroxy-3-oxy-3H-xanthen-9-yl) henoxy]ethoxy]-4 methylphenyl]-N-(carboxymethyl) glycine
- Fluo-4
- 2-{[2-(2-{5-[bis(carboxymethyl)amino]-2-methylphenoxy}ethoxy)-4-(2,7-difluoro-6-hydroxy-3-oxo-3H-xanthen-9-yl)phenyl](carboxymethyl)amino}acetic acid
- Quin-2
- 2-{[2-bis(carboxymethyl) amino-5-methylphenoxy]methyl}-6-methoxy-8-bis(carboxymethyl)aminoquinoline.
REFERENCES
- 1.Philipson K. D., Nicoll D. A. (2000) Annu. Rev. Physiol. 62, 111–133 [DOI] [PubMed] [Google Scholar]
- 2.Kofuji P., Lederer W. J., Schulze D. H. (1994) J. Biol. Chem. 269, 5145–5149 [PubMed] [Google Scholar]
- 3.Lee S. L., Yu A. S., Lytton J. (1994) J. Biol. Chem. 269, 14849–14852 [PubMed] [Google Scholar]
- 4.Quednau B. D., Nicoll D. A., Philipson K. D. (1997) Am. J. Physiol. 272, C1250–C1261 [DOI] [PubMed] [Google Scholar]
- 5.Lytton J. (2007) Biochem. J. 406, 365–682 [DOI] [PubMed] [Google Scholar]
- 6.Blaustein M. P., Lederer W. J. (1999) Physiol. Rev. 79, 763–854 [DOI] [PubMed] [Google Scholar]
- 7.Khananshvili D. (1998) in Advances in the Molecular Cell Biology Life Sciences Program (Anderson J. P. ed) 23B, pp. 309–356, JAI Press, Inc., Greenwich, CT [Google Scholar]
- 8.Dyck C., Omelchenko A., Elias C. L., Quednau B. D., Philipson K. D., Hnatowich M., Hryshko L. V. (1999) J. Gen. Physiol. 114, 701–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn J., Elias C. L., Le H. D., Omelchenko A., Hryshko L. V., Lytton J. (2002) J. Biol. Chem. 277, 33957–33962 [DOI] [PubMed] [Google Scholar]
- 10.Matsuoka S., Nicoll D. A., Reilly R. F., Hilgemann D. W., Philipson K. D. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 3870–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levitsky D. O., Nicoll D. A., Philipson K. D. (1994) J. Biol. Chem. 269, 22847–22852 [PubMed] [Google Scholar]
- 12.Bers D. M. (2002) Nature 415, 198–205 [DOI] [PubMed] [Google Scholar]
- 13.Weber C. R., Ginsburg K. S., Philipson K. D., Shannon T. R., Bers D. M. (2001) J. Gen. Physiol. 117, 119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maack C., Ganesan A., Sidor A., O'Rourke B. (2005) Circ. Res. 96, 91–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiPolo R., Beaugé L. (2006) Physiol. Rev. 86, 155–203 [DOI] [PubMed] [Google Scholar]
- 16.Hilge M., Aelen J., Vuister G. W. (2006) Mol. Cell 22, 15–25 [DOI] [PubMed] [Google Scholar]
- 17.Nicoll D. A., Sawaya M. R., Kwon S., Cascio D., Philipson K. D., Abramson J. (2006) J. Biol. Chem. 281, 21577–21581 [DOI] [PubMed] [Google Scholar]
- 18.Johnson E., Bruschweiler-Li L., Showalter S. A., Vuister G. W., Zhang F., Brüschweiler R. (2008) J. Mol. Biol. 377, 945–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Besserer G. M., Ottolia M., Nicoll D. A., Chaptal V., Cascio D., Philipson K. D., Abramson J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 18467–18472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blaustein M. P., Charpentier T. H., Weber D. J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 18349–18350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyman L., Mikhasenko H., Hiller R., Khananshvili D. (2009) J. Biol. Chem. 284, 6185–6193 [DOI] [PubMed] [Google Scholar]
- 22.Ottolia M., Philipson K. D., John S. (2004) Biophys. J. 87, 899–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaptal V., Ottolia M., Mercado-Besserer G., Nicoll D. A., Philipson K. D., Abramson J. (2009) J. Biol. Chem. 284, 14688–14692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilge M., Aelen J., Foarce A., Perrakis A., Vuister G. W. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 14333–14338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ottolia M., Nicoll D. A., Philipson K. D. (2009) J. Biol. Chem. 284, 32735–32741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaptal V., Besserer G. M., Ottolia M., Nicoll D. A., Cascio D., Philipson K. D., Abramson J. (2007) Channels 1, 397–399 [DOI] [PubMed] [Google Scholar]
- 27.Wu M., Wang M., Nix J., Hryshko L. V., Zheng L. (2009) J. Mol. Biol. 387, 104–112 [DOI] [PubMed] [Google Scholar]
- 28.Wu M., Le H. D., Wang M., Yurkov V., Omelchenko A., Hnatowich M., Nix J., Hryshko L. V., Zheng L. (2010) J. Biol. Chem. 285, 2554–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reeves J. P., Condrescu M. (2008) Channels 2, 322–328 [DOI] [PubMed] [Google Scholar]
- 30.Hilgemann D. W., Matsuoka S., Nagel G. A., Collins A. (1992) J. Gen. Physiol. 100, 905–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hilgemann D. W., Collins A., Matsuoka S. (1992) J. Gen. Physiol. 100, 933–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hryshko L. V., Matsuoka S., Nicoll D. A., Weiss J. N., Schwarz E. M., Benzer S., Philipson K. D. (1996) J. Gen. Physiol. 108, 67–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ames J. B., Hendricks K. B., Strahl T., Huttner I. G., Hamasaki N., Thorner J. (2000) Biochemistry 39, 12149–12161 [DOI] [PubMed] [Google Scholar]
- 34.Wingard J. N., Chan J., Bosanac I., Haeseleer F., Palczewski K., Ikura M., Ames J. B. (2005) J. Biol. Chem. 281, 37461–37470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baazov D., Wang X., Khananshvili D. (1999) Biochemistry 38, 1435–1445 [DOI] [PubMed] [Google Scholar]
- 36.Khananshvili D., Baazov D., Weil-Maslansky E., Shaulov G., Mester B. (1996) Biochemistry 35, 15933–15940 [DOI] [PubMed] [Google Scholar]
- 37.Khananshvili D. (1988) Biochemistry 27, 8290–8296 [DOI] [PubMed] [Google Scholar]
- 38.Boyman L., Hiller R., Lederer W. J., Khananshvili D. (2008) Biochemistry 47, 6602–6611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khananshvili D., Weil-Maslansky E. (1994) Biochemistry 33, 312–319 [DOI] [PubMed] [Google Scholar]
- 40.Khananshvili D., Shaulov G., Weil-Maslansky E. (1995) Biochemistry 34, 10290–10297 [DOI] [PubMed] [Google Scholar]
- 41.Herrick D. Z., Kuo W., Huang H., Schwieters C. D., Ellena J. F., Cafiso D. S. (2009) J. Mol. Biol. 390, 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Breukels V., Vuister G. W. (2010) Proteins 78, 1813–1824 [DOI] [PubMed] [Google Scholar]
- 43.Ottolia M., Nicoll D. A., John S., Philipson K. D. (2010) Channels 4, 159–162 [DOI] [PubMed] [Google Scholar]
- 44.Burgoyne R. D. (2007) Nat. Rev. Neurosci. 8, 182–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lemmon M. A. (2008) Nat. Rev. Mol. Cell. Biol. 9, 99–111 [DOI] [PubMed] [Google Scholar]
- 46.Gifford J. L., Walsh M. P., Vogel H. J. (2007) Biochem. J. 405, 199–221 [DOI] [PubMed] [Google Scholar]
- 47.Guatimosim S., Dilly K., Santana L. F., Saleet, Jafri M., Sobie E. A., Lederer W. J. (2002) J. Mol. Cell. Cardiol. 34, 941–950 [DOI] [PubMed] [Google Scholar]
- 48.Sher A. A., Noble P. J., Hinch R., Gavaghan D. J., Noble D. (2008) Prog. Biophys. Mol. Biol. 96, 377–398 [DOI] [PubMed] [Google Scholar]
- 49.Kargacin G. J. (2003) J. Theor. Biol. 221, 245–258 [DOI] [PubMed] [Google Scholar]
- 50.Fuson K. L., Montes M., Robert J. J., Sutton R. B. (2007) Biochemistry 46, 13041–13048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Montaville P., Schlicker C., Leonov A., Zweckstetter M., Sheldrick G. M., Becker S. (2007) J. Biol. Chem. 282, 5015–5025 [DOI] [PubMed] [Google Scholar]