

Abstract
Helicobacter pylori, a pathogen that colonizes the human stomach, requires the nickel-containing metalloenzymes urease and NiFe-hydrogenase to survive this low pH environment. The maturation of both enzymes depends on the metallochaperone, HypA. HypA contains two metal sites, an intrinsic zinc site and a low-affinity nickel binding site. X-ray absorption spectroscopy (XAS) shows that the structure of the intrinsic zinc site of HypA is dynamic, and able to sense both nickel loading and pH changes. At pH 6.3, an internal pH that occurs during acid shock, the zinc site undergoes unprecedented ligand substitutions to convert from a Zn(Cys)4 site to a Zn(His)2(Cys)2 site. NMR spectroscopy shows that binding of Ni(II) to HypA results in paramagnetic broadening of resonances near the N-terminus. NOEs between the β-CH2 protons of Zn cysteinyl ligands are consistent with a strand-swapped HypA dimer. Addition of nickel causes resonances from zinc binding motif and other regions to double, indicating more than one conformation can exist in solution. Although the structure of the high-spin, 5–6 coordinate Ni(II) site is relatively unaffected by pH, the nickel binding stoichiometry is decreased from one per monomer to one per dimer at pH = 6.3. Mutation of any cysteine residue in the zinc binding motif results in a zinc site structure similar to that found for holo-WT-HypA at low pH and is unperturbed by the addition of nickel. Mutation of the histidines that flank the CXXC motifs results in a zinc site structure that is similar to holo-WT-HypA at neutral pH (Zn(Cys)4) and is no longer responsive to nickel binding or pH changes. Using an in vitro urease activity assay, it is shown that the recombinant protein is sufficient for recovery of urease activity in cell lysate from a HypA deletion mutant, and that mutations in the zinc-binding motif result in a decrease in recovered urease activity. The results are interpreted in terms of a model wherein HypA controls the flow of nickel traffic in the cell in response to nickel availability and pH.
Keywords: Helicobacter pylori, XAS, HypA, metallochaperone, zinc, nickel, ITC, NMR
Introduction
Helicobacter pylori is a spiral, gram-negative bacterium that colonizes the human gastric mucosa, causes peptic ulcers, and has been associated with gastric carcinomas.1 It has an optimal growth pH of ca. 7.0, and cannot survive at pHs more acidic than 4.0 or more basic than 8.2 in vitro.2 Its ability to survive in the acid environment of the human stomach (pH < 3.0) requires urea and two nickel-dependent enzymes, urease and NiFe-hydrogenase.3–5 Urease catalyzes the hydrolysis of urea to ammonia, and thus plays a crucial role in modifying the pH of the organism’s environment under acid stress.3,4 Hydrogenase catalyzes the reversible oxidation of hydrogen and may play a role in converting protons to hydrogen and/or in supplying energy to the cell for survival.5 In addition, H. pylori is able to regulate its periplasmic pH at ca. 6.1 under acid stress, and hold its cytoplasmic pH near 7 via the additional use of a membrane linked carbonic anhydrase.6,7
Both urease and hydrogenase feature highly choreographed metallocenter assembly mechanisms8–12 that involve a number of accessory proteins designed to maintain the fidelity of metal insertion in these enzymes. These mechanisms involve two nickel metallochaperones, HypA and UreE, that participate in hydrogenase and urease metallocenter assembly, respectively.9,13,14 Both nickel metallochaperones are found in H. pylori and are known to be low molecular weight (13.2 kDa, 19.4 kDa, respectively),14–16 intrinsic zinc proteins that bind Ni(II) ions with μM affinity (1 μM and 0.15 μM, respectively),14,15 and form homodimers in vitro.14,15 In addition to its role in hydrogenase maturation, H. pylori HypA plays a critical but unknown role in urease maturation.11,17 When hypA was deleted, the ΔhypA cell lysate did not show any urease activity. Activity was restored when Ni(II) was supplied in excess to the lysate from the ΔhypA strain.17 Because of its dual roles in both hydrogenase and urease maturation, HypA appears to stand at a crossroads in nickel trafficking in H. pylori, suggesting interactions with proteins involved in active site assembly in both hydrogenase and urease.
Homodimeric H. pylori HypA has been shown to bind two Zn(II) and two Ni(II) ions per dimer.14 Prior XAS and biophysical studies established the presence of a high-spin Ni(II) center and a tetrahedral Zn(Cys)4 site in holo-WT-HypA.16 The zinc site exhibited a structural change upon nickel binding that implied communication between the metal sites.16,18 Recently, three structures of HypA proteins have been reported. The first was an NMR solution structure of monomeric H. pylori HypA that contained a modified N-terminus.18 The second and third were crystal structures of both monomeric and dimeric forms of HypA from the archaeon Thermococcus kodakaraensis that were chromatographically separated.19 All three structures show the four cysteine residues in the two conserved CxxC motifs coordinating the zinc atom. The NMR structure of monomer HypA reported a nickel-binding site that is near the N-terminus and involves ligation by His2, consistent with prior studies.14,18 However, the structure reported for the Ni(II) site was a planar diamagnetic Ni(II) site that was separated by ~30 Å from the zinc site.18 The crystal structure of the dimeric form of T. kodakaraensis HypA revealed a “domain-swapped dimer” involving a zinc site that is coordinated by the four conserved cysteine residues, but where two cysteine residues arise from one monomer and the other two are from the adjacent subunit.19 The crystal structure was not solved with nickel bound, but provided additional evidence for a structurally flexible zinc site, as well as a possible mechanism for structural communication between the metal binding sites.19
Herein, we show, using a combination of XAS to examine metal site structures, NMR data, and mutagenesis, that the zinc site and protein structure in dimeric HypA is dependent on both the nickel binding status and the pH of the sample, and that the nickel binding properties of the protein vary with both pH and protein structure, as reflected by the zinc site structure, which undergoes unprecedented changes in ligation in response to pH and nickel binding. Further, in vitro assays of urease maturation are used to demonstrate that point mutations in the zinc-binding motif affect the ability of the proteins to produce active urease. These results provide a model by which the nickel bound status of HypA might be communicated to target delivery proteins and, by also acting as pH sensor, suggest a mechanism where the protein might be able to favor nickel delivery to urease or to hydrogenase under different conditions of nickel availability or internal pH of the cell.
Experimental
Protein expression and purification
HypA was over-expressed from the pJI100 plasmid which was prepared previously.16 DL41 (DE3) pLysS (Novagen™) competent cells were transformed with pJI100, and the transformed cells were plated on LB media containing 100 μg/mL ampicillin (amp) and 30 μg/mL chloramphenicol (cam) and grown overnight at 37 °C. Single colonies of cells containing the pJI100 plasmid were grown in 10 mL cultures (LB cam/amp) overnight at 37 °C with shaking, and then added to 1 liter of pre-warmed fresh media. These cultures were grown to an OD of 1.0 at 600 nm and then induced by addition of isopropyl-β-D-thiogalactoside (IPTG) to a final concentration 0.8 mM for 3 hours. Cells (3 L of cells were combined for resuspension) were harvested by centrifugation, resuspended in 50 mL of Q-sepharose buffer I (20 mM Tris•HCl, 1 mM DTT pH = 7.2) and frozen at −80 °C. Cells were lysed upon thawing, and 150 μl of DNAse I solution (10 mg/mL DNAse I, 40 % glycerol) was added. The mixture was incubated at 37 °C until the viscosity of the solution was significantly reduced. A portion of this cell lysate was removed to confirm over expression of HypA using SDS-PAGE.
All chromatographic purification steps were performed at room temperature and employed an AKTA-FPLC system (Amersham Biosciences) monitoring the absorbance at 280 nm. For WT-HypA, the cell lysate was loaded onto a column (Pharmacia HR10) containing Q-sepharose resin (GE Life sciences™) for anion exchange chromatography. To elute HypA from the column resin, a linear gradient was applied from 0 to 30% of Q-sepharose buffer II (20 mM Tris•HCl, 1 M NaCl, 1 mM DTT, pH = 7.2) while collecting fractions every 6 mL. An SDS-PAGE gel was run to determine which fractions contained WT-HypA. Fractions containing WT-HypA were combined and concentrated to 2 mL and then dialyzed against 20 mM HEPES, 200 mM NaCl, 1 mM DTT pH = 7.2 buffer in preparation for size-exclusion chromatography.
The concentrated solution from the Q-sepharose columns was loaded in 200 μL batches onto a superdex 75 10/300 (GE life sciences™) size-exclusion column and eluted using the sample buffer and a flow rate of 0.35 mL/min. HypA eluted with a peak at 12.5 mL after injection, corresponding to a protein of 26 kDa MW based on the elution of known MW standards (albumin (67 kDa), ovalbumin (43 kDa), chymotrypsinogen (25 kDa) and ribonuclease A (13.7 kDa)). Purification of this peak yields dimeric HypA, and excludes the monomeric form that is present in variable amounts, similar to the situation for T. kodakarensis HypA.19 (We have been unable to convert the dimeric form to monomer without denaturing the protein, which is consistent with a chain-swap dimer, vide infra.) An SDS-PAGE gel was run to confirm the purity of WT-HypA in these fractions, and pure fractions were combined and buffer exchanged into 20 mM Tris•HCl, 200 mM NaCl at pH = 7.2 (or pH = 6.3 for low pH studies) for further analysis.
For preparation of HypA samples labeled with 15N and 13C, the cell line was switched from DL41 (DE3) pLysS to BL41 (DE3) pLysS (DL41 is a methionine auxotroph and therefore was not used). HypA was overexpressed in LB media to the same level in both cell lines and there were no differences in purification for either cell line. The competent cells (BL21 (DE3) pLysS) were transformed with pJI100 and plated on LB media containing 100 μg/mL ampicillin (amp) and 30 μg/mL chloramphenicol (cam) and grown overnight at 37 °C. Single colonies of cells containing the pJI100 plasmid were grown in 10 mL cultures (LB cam/amp) overnight at 37 °C with shaking, and then added to 1 liter of supplemented M9 media. One liter of supplemented M9 media was prepared by adding 1 g NH4Cl, 0.5 g NaCl, 3 g KH2PO4, 6 g Na2HPO4, 800 μL 1M MgSO4, 100 μL 1M CaCl2, 100 μL 1M FeCl3, 250 μL 2% thiamine and 20 mL of 20% glucose. To incorporate 15N, 15NH4Cl was used, and for incorporation of 13C, 13C labeled glucose was employed. The cells were then grown at 37 °C to an OD600 of 0.9 and 100 μL of 10 mM ZnSO4 was added and the temperature was dropped to 25 °C. (Additional zinc appears to be required for overproduction of HypA; cells did not grow as well without additional zinc.) After 15 minutes, IPTG was added to a final concentration of 0.8 mM and the cells were grown for another 16 hours and harvested by ultra-centrifugation. The purification of labeled WT-HypA was identical to that as unlabeled WT-HypA (vide supra).
HypA mutagenesis and protein characterization
Point mutations of Cys residues in the two CxxC motifs associated with zinc binding and the two His residues (His79 and His95) that flank these two CxxC motifs were accomplished using the QuikChange (Stratagene™) mutagenesis kit and the pJI100 plasmid. Mutagenic primers and the parental DNA that were used in the PCR reactions are tabulated in the supporting information. The PCR product was used to transform into NovaBlue (Novagen™) competent cells. After transformation, the cells were grown on LB/amp agar plates overnight at 37° C. Three separate colonies were selected from the plate and grown in a 5 mL culture (LB amp media) overnight. The DNA from the resulting cultures was isolated using the Qiagen® miniprep kit and sequenced at the Keck DNA facility (Yale University) to confirm the presence of the desired mutation. This DNA was then used to transform DL41 (DE3) pLysS (Novagen™) competent cells for protein overexpression. Overexpression and purification of the mutant HypA proteins was achieved using the procedures outlined above for WT-HypA (vide supra) as modified below.
Histidine mutants (H79A and H95A) are isolated with zinc bound, but no nickel bound, as was recombinant WT-HypA, and were purified using the same procedure as WT-HypA. Mutation of any one of the four Cys residues in the two CxxC motifs lead to the formation of inclusion bodies, and the cysteine mutant HypA proteins were found in the pellet after cell lysis. In these cases, the protein was refolded by first resuspending the pellet in 20 mM Tris•HCl, 6 M urea, 1mM DTT at pH = 7.2. The resulting solution was then applied to an anion exchange column (Q-sepharose, GE life sciences). The fraction containing mutant HypA was eluted from the column with high salt buffer (20 mM Tris•HCl, 1 mM DTT, 500 mM NaCl at pH = 7.2) and was then slowly refolded out of urea by dialysis at 4 °C using resuspension buffers that contained 200 mM NaCl and 1 mM DTT then decreasing the urea concentration to 0 mM in 0.25 M increments with 6 hour equilibration periods. The last dialysis was against resuspension buffer that contained 1 mM DTT, but no salt or urea, in order to prepare the sample for anion exchange chromatography. The rest of the protein purification was as described for WT-HypA (vide supra).
Mutations of the Cys residues resulted in proteins that did not co-purify with zinc, so the proteins were reconstituted after refolding by first buffer exchanging into 20 mM Tris•HCl and 200 mM NaCl at pH = 7.2 and determining the protein concentration (vide infra). A two-fold excess of ZnSO4 was then added to the protein solution and allowed to incubate at room temperature for one hour. The sample was then treated with Chelex™ beads to remove non-specifically bound zinc. Despite the need for reconstitution of the zinc site in the Cys mutants, CD spectroscopy shows that the protein is folded in a manner that is similar to WT protein, and the urease activity assays show that the Cys mutants maintain some function (vide infra).
The purity of the HypA proteins was determined by SDS-PAGE. The molecular weight of the oligomeric proteins was determined by size exclusion chromatography, as described above. Subunit molecular weights of the proteins expressed (Table 1) were determined by ESI-MS under denaturing conditions (0.1 % formic acid was added to the samples and the skimmer voltage on the instrument was set to 120 V) using a Bruker Esquire instrument equipped with an HP-HPLC for liquid separation to remove the salt present in the solutions.
Table 1.
Physical properties of WT and mutant HypA proteins.
| HypA | Subunit MW (Da) | Oligomer MW (Da)b | Tm(°C) | Tm(°C) | Kd(ITC) | ΔH (ITC) of Ni binding (kcal/mol) | n (ITC) | Zn:apo-HypAe |
|---|---|---|---|---|---|---|---|---|
| Obsa (calc’d) | −Ni | +Ni | (μM) | (Ni/protein) | ||||
| WT | 13,203 (13,202) | 26,600 |
pH = 7.2 58(2)c 59.1(3)d |
pH = 7.2 70(5)c 70.5(4)d |
pH = 7.2 1.08(6) |
pH = 7.2 −18.9(1) |
pH = 7.2 1.050(5) |
pH = 7.2 0.94:1 |
|
pH = 6.3 60(4)d |
pH = 6.3 71(1)d |
pH = 6.3 17(2) |
pH = 6.3 −9.0(6) |
pH = 6.3 0.46(3) |
pH = 6.3 0.92:1 |
|||
|
pH = 8.3 3.7(3) |
pH = 8.3 −9.75(7) |
pH = 8.3 0.936(4) |
||||||
| C74A | 13,169 (13,170) | 28,000 | 63.5(1)d | 69.7(4)d | 54(4) | −13.8(3) | 0.506(8) | 1.03:1 |
| C77A | 13,171 (13,170) | 29,000 | 72.3(1)d | 66.8(5)d | 48(2) | −13.0(2) | 0.478(5) | 1.05:1 |
| C91A | 13,170 (13,170) | 28,300 | 57.6(6)d | 75.8(4)d | 22(3) | −8.6(1) | 0.44(6) | 0.92:1 |
| C94A | 13,171 (13,170) | 29,500 | 66.3(9)d | 73.3(1)d | 16(2) | −16(1) | 0.48(3) | 0.89:1 |
| C94D | 13,213 (13,214) | 73.9(2)d | 79.7(2)d | 22(2) | −22(1) | 0.46(2) | 0.93:1 | |
| H79A | 13,136 (13,136) | 27,000 | 67.1(2)d | 68.6(2)d |
pH = 7.2 3.2(3) |
pH = 7.2 −34.3(3) |
pH = 7.2 0.932(5) |
0.97:1 |
| H95A | 13,136 (13,136) | 30,000 | 66.8(2)d | 68.0(2)d |
pH = 7.2 2.1(1) |
pH = 7.2 −27.6(3) |
pH = 7.2 0.936(6) |
1.06:1 |
|
pH = 6.3 21(2) |
pH = 6.3 −45.7(2) |
pH = 6.3 0.96(3) |
Determined by ESI-MS;
Determined by size-exclusion chromatography;
Melting temperature determined by DSC;
Melting temperature determined by performing a thermal melt and monitoring the CD signature at 222 nm.
Determined using ICP-OES for [Zn] and optical spectrum calibrated with amino acid analysis for [protein].
The concentrations of HypA proteins were determined by measuring the absorption at 280 nm using a HP-8453 diode array, where the extinction coefficient (ε = 6500 M−1cm−1) was determined from a sample whose concentration was determined by amino acid analysis (WM Keck Laboratories at Yale University). Incorporation of Ni(II) into WT-HypA and all mutants was achieved by adding a 2-fold excess of NiCl2 to the Zn-containing apo-HypA proteins in 20 mM Tris•HCl, 200 mM NaCl at pH = 7.2 and allowing them to incubate for 1 hour at room temperature before Chelex™ beads were added to remove excess nickel. Metal contents of apo- and holo-HypA samples were determined using a Perkin-Elmer DV4300 ICP-OES instrument.
Protein Stability
Circular dichroism (CD) spectra were obtained to monitor secondary structure as a function of nickel loading, pH, and mutations using a Jasco J-715 spectropolarimeter. The thermal stabilities of the WT-HypA and the HypA mutants were determined by either differential scanning calorimetry (DSC) or a thermal melt monitored by CD. DSC thermograms were measured with a Microcal VP-DSC with 0.5 mL solutions in the sample and reference cells. HypA samples were concentrated to 100 μM as monomer in buffer (20 mM Tris·HCl, 200 mM NaCl, pH = 7.2, or pH = 6.3), degassed under vacuum and syringed into the sample cell. Degassed buffer was placed in the reference cells. Thermograms were collected at 30 psi between 5–90 °C at a scan rate of 30 °C/hour. Reverse scans were performed, but melting was irreversible in all cases. Baseline correction and normalization was performed with the Microcal interface to the Origin graphing program. Peak maxima were taken as melting temperatures, Tm. The values of Tm obtained are shown in Table 1. (Thermograms are included in supporting information.) Thermal melts determined by CD used HypA samples (5 μM in monomer in 20 mM Tris·HCl, 200 mM NaCl, pH = 7.2, unless otherwise stated) placed in a 1 mm CD cuvette and monitored the change in absorption at 220 nm between 40–95 °C. The data showed one melting event and was fit to a sigmoid, the inflection point of which is reported as the melting temperature of the samples (Table 1). The process was not reversible. Values of Tm obtained using both methods for WT-HypA were in complete agreement. (This rules out aggregation as a cause irreversibility.)
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) was used to determine the binding constant of Ni(II) ions to WT-HypA and mutant HypA proteins. ITC was performed on a Microcal MCS-ITC instrument. HypA (1.5 mL of 30 – 45 μM in 20 mM Tris•HCl, 200 mM NaCl, pH = 7.2, unless otherwise noted) was placed into the sample cell and NiCl2 (0.462 mM in 20 mM Tris•HCl, 200 mM NaCl, pH = 7.2) was placed into the syringe. 5–10 μL of NiCl2 solution was injected 26 times at intervals of 3–4 minutes for equilibration. Data was fit using the one-binding site model in the MicroCal interface to the Origin software. Kitc, ΔHitc and n were all allowed to freely refine. Selected fits obtained are shown in Figure 2, and parameters obtained from the fits are listed in Table 1. The values are uncorrected for buffer effects. Raw titration data and integrated heat data for additional samples are shown in the supporting information.
Figure 2.

Isothermal titration calorimetry of HypA proteins. The integrated heat data for titrations of apo-WT-HypA and selected mutant HypA proteins with NiCl2 solutions is shown for titrations at different pH values. The symbols represent the integrated heat after each injection, while the solid line is the calculated fit of the data used for values in Table 1 (see materials and methods).
UV-vis spectroscopy
In addition to determination of protein concentration, UV-vis absorption spectroscopy was used to examine the structure of the nickel site in holo-HypA. HypA was expressed and purified as above and concentrated to 0.2 mM (in 20 mM Tris pH = 7.2, 200 mM NaCl) to make the apo-HypA sample. To a second apo-HypA sample (0.06 mM), two equivalents a NiCl2 solution were added, and the sample was incubated for 1 hour prior to the addition of Chelex™ beads. After 20 minutes, the sample was concentrated to 0.2 mM to yield the holo-HypA sample. Metal content was determined using ICP-OES (vide supra) and the Zn:Ni ratio was found to be 1:1. Titration of the apo-form of HypA with two and four equivalents of NiCl2 was also performed. Spectra were collected with an HP-8453 spectrometer in a 100 μl quartz cuvette.
NMR data collection
NMR spectra were acquired at 298 K on a Bruker Avance 18.78 T (800 MHz 1H) spectrometer with a 5 mm TCI 1H{13C, 15N} cryogenic probe equipped with pulsed field gradients. NMR samples of appropriately isotopically labeled Zn-containing HypA were prepared in 20 mM Tris, 200 mM NaCl, 1 mM DTT, 10 % D2O, pH = 7.2 and concentrated to the final volume of 0.25 mL and protein concentration of more than 0.1 mM. Nickel and zinc-containing 15N labeled HypA protein sample was prepared starting from isotopically labeled apo-HypA in 20 mM HEPES, 200 mM NaCl, 1 mM DTT, pH = 7.2. Sample was dialyzed overnight against 20 mM Tris, 150 mM NaCl, pH = 7.2 followed by addition of 1.5 mol equivalents of Ni(II) ions in the form of NiCl2. After 1 hour incubation at room temperature, Chelex™ beads were added for 15 minutes at room temperature, in order to bind residual nickel ions in the protein solution, and then removed by centrifugation. To ensure removal of excess nickel ions, the buffer was exchanged by spin-concentration with 20 mM Tris, 150 mM NaCl, 10 % D2O, pH = 7.2. In order to obtain sequence specific backbone assignments a set of sensitivity-enhanced triple resonance experiments was performed, including HNCO, HN(CO)CA, HNCA and HNCACB. In addition, 1H,15N–HSQC, 1H,15N NOESY-HSQC and 1H,15N TOCSY-HSQC data sets were collected, in order to aid backbone assignment procedure and alleviate possible ambiguities. Side chain assignments were made using information contained in HCCH-TOCSY, and two 1H, 13C NOESY-HSQC optimized for detection of 13C resonances in aliphatic and aromatic regions, respectively. All experimental pulse sequences used are standard pulse sequences included in the Bruker Topspin release and were applied without modifications other than parameter calibration.
Data were processed using Topspin, and analyzed using SPARKY (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco). Proton chemical shifts were referenced directly to the water signal at 4.7 ppm, while 15N and 13C shifts were referenced indirectly.20 A computer-assisted manual assignment approach was used to assign backbone resonances. A set of four experiments was used to make sequential backbone resonance assignments: HNCA, HN(CO)CA and HNCACB. Backbone resonance assignments were confirmed by identifying expected sequential inter-residue NOE signals in the 1H,15N NOESY-HSQC. Side chain assignments of aliphatic 1H and 13C resonances were assigned using HCCH-TOCSY experiment, and confirmed by NOEs detected in 1H, 13C NOESY-HSQC spectrum. For aromatic 1H and 13C resonance assignments, a 1H, 13C NOESY-HSQC experiment optimized for aromatic 13C indirect detection was used.
XAS Spectroscopy
X-ray absorption spectroscopy (XAS) data collection and analysis was performed as previously described21 to obtain structural information regarding the metal site structures in HypA and the effects of mutations on the structures. Except where noted, nickel and zinc K-edge XAS data were collected on beamline 9–3 at Stanford Synchrotron Radiation Laboratory (SSRL). Samples were syringed into a polycarbonate sample holder containing a kapton window and then frozen in liquid nitrogen. The sample was analyzed at 10 K using a liquid He cryostat (Oxford Instruments). The ring conditions were 3 GeV and 80–100 mA. Beamline optics consisted of a Si(220) double-crystal monochromator for focusing. X-ray fluorescence was collected using a 30-element Ge detector (Canberra). Scattering was minimized by placing a set of Soller slits with a Z-1 element filter between the sample chamber and the detector. Nickel K-edge XAS data for WT-HypA at pH = 6.3, C74D and C77D as well as zinc XAS for C74D and C77D with and without nickel were collected on beamline X9B at the National Synchrotron Light Source (Brookhaven National Laboratory). Samples run at NSLS were frozen protein solutions (1–2 mM, based on zinc or nickel content, in 20 mM Tris•HCl 200 mM NaCl, pH = 7.2, unless otherwise noted) and placed into polycarbonate holders inserted into aluminum blocks and held near 50 K using a He displex cryostat. The ring conditions for data collection were 2.8 GeV and 120–300 mA. A sagittally focusing Si(111) double crystal monochromator and a 13-channel Ge fluorescence detector (Canberra) were used for data collection.
X-ray absorption near-edge spectroscopy (XANES) data was collected from ± 200 eV relative to the metal edge. The X-ray energy for the K-edge of metal was internally calibrated to 8331.6 eV for nickel and 9660.7 for zinc using transmission data from a metal foil. The data shown is the average of 4 – 8 scans and was analyzed using the EXAFS123 software package.22
Extended X-ray absorption fine structure (EXAFS) was collected to k = 16 Å−1 for the metal being studied and was analyzed over the range k = 2 – 14 Å using the SixPack software package.23 Scattering parameters for EXAFS fitting were generated using the FEFF 8 software package.24 The SixPack fitting software builds on the ifeffit engine24,25 and uses iterative FEFF calculations to fit EXAFS data during model refinement, and is thus an improvement over previous methods that employ a static set of calculated scattering parameters. To compare different models to the same data set, ifeffit uses three goodness of fit parameters: χ2, reduced χ2, and R. χ2 is given by Equation 4, where Nidp is the number of independent data points, Nε2 is the number of uncertainties to minimize, Re(fi) is the real part of the EXAFS fitting function, and Im(fi) is the imaginary part of the EXAFS fitting function. Reduced χ2 = χ2/(Nind−Nvarys), where Nvarys is the number of refining parameters and represents the degrees of freedom in the fit. Ifeffit also calculates the R for the fit, which is given by Equation 5 and is scaled to the magnitude of the data, making it proportional to χ2.
| (4) |
| (5) |
To compare different models (fits), R and reduced χ2 were assessed to determine the best fit, in which case both parameters were minimized. Although the R will always improve with an increasing number of adjustable parameters, reduced χ2 will go through a minimum and then increase, indicating that the model is over-fitting the data.
In vitro activity assay
H. pylori cell lysate for urease activity assays was prepared as described previously.17 Lysates were stored at 4 °C and activity remained constant up to ~ 6 months of storage. To determine if mutations to HypA affected apo-urease maturation, we used an in vitro assay to monitor urease activity. A procedure similar to that of Weatherburn was used.26 In brief, 5 μL of ΔhypA H. pylori lysate is incubated with 7 μg of recombinant HypA and diluted to 15 μL with buffer (20 mM Tris•HCl, 200 mM NaCl at pH = 7.2, unless otherwise stated). The solution is first incubated at 25 °C for 24 hours and then for 14–16 hours at 37 °C. (These conditions were empirically determined and have not been further optimized.) To determine urease activity, 5 μL of the incubated solution was added to 245 μL of reaction buffer (50 mM HEPES, 25 mM urea at pH = 7.0) and incubated for 20 minutes. The assay was then quenched with 375 μL of Buffer A (170 μM Na2[Fe(CN)5(NO)], 106 mM phenol in water) followed by 375 μL of Buffer B (1.25 mM NaOH, 0.044% v/v NaClO in water). The resulting solution was incubated for 30 min. and urease activity (production of ammonia and conversion to indophenol) was monitored by the increase in absorbance at 625 nm. The amount of ammonia that was produced was compared to a standard curve made using ammonium chloride in water and is reported as nmol of ammonia produced in 20 minutes. The assay is reproducible within the errors indicated for different batches of wild-type protein in different batches of lysate following the same protocol. Mutants were assayed using different preparations of lysate in parallel with a wild-type control so all data are, in effect, normalized. Changes in the preincubation protocol (time and temperature) will influence the amount of activity observed, but the relative abilities of the different mutants to complement the activity is preserved.
Results
Protein Characterization
ESI-MS data confirmed that all of the expressed recombinant proteins had the MWs expected from their amino acid sequences, including the N-terminal Met residue (Table 1). Size-exclusion chromatography showed that apo-WT-HypA (containing only zinc), and all of the zinc-containing mutant proteins were dimeric in solution (Table 1 and supporting information). Protein and metal analyses showed that WT-HypA and all of the mutants bound one Zn(II) ion per monomer (Table 1). The nickel binding stoichiometry of WT (pH = 7.2), H79A-, and H95A- (pH = 7.2 and pH = 6.3) HypA was one Ni(II) ion per monomer (based on the Ni:Zn ratio). However, in WT-HypA at pH = 6.3 and in the Cys mutants, the nickel binding stoichiometry was reduced to one per dimer (Table 1).
The secondary structure of HypA was monitored using CD spectroscopy (Figure 1) and is generally similar in all the HypA samples, indicating that no large changes in secondary structure occur as a result of pH change (Figure 1A vs. 1B), nickel binding (apo- vs. holo-HypA), or mutation of ligands in the zinc-binding motif (Figure 1C and supporting information).
Figure 1.

Circular dichroism spectral comparisons of HypA proteins: (A) Apo- (red) vs. Holo- (green) WT-HypA at pH = 7.2. (B) Apo- (red) vs. Holo- (green) WT - HypA at pH = 6.3. (C) Apo-H95A-HypA (blue) vs. apo-C74A-HypA (orange) at pH = 7.2.
The thermal stability of the HypA proteins was determined using a combination of DSC and thermal melts monitored by CD spectroscopy. Table 1 shows the melting temperatures for WT- and mutant HypA proteins. Melting was irreversible in all cases. The addition of nickel to WT-HypA increased the thermal stability by 12 °C from 58 °C to 70 °C. These Tm values were not significantly altered when the pH was lowered from 7.2 to 6.3 (Table 1). However, mutations of two Cys residues in the zinc-binding motif, C77A and C94A, (the residues flanked by His residues) led to apo-proteins with higher thermal stability when compared to apo-WT-HypA, to an extent that was similar to the effect of binding Ni(II) ions to apo-WT-HypA. These mutants did not show a large increase in Tm when nickel was bound. In fact, for C77A, a small decrease in Tm occurred upon nickel binding. Mutations of the other two Cys residues, C74 and C91, did not alter the Tm much from that of apo-WT-HypA, and retained an increase in Tm when nickel was bound. Mutation of the two His residues that flank the CXXC sequences in the zinc binding motif also led to proteins that had higher thermal stability than apo-WT-HypA, and were not stabilized further by binding nickel.
Isothermal Titration Calorimetry
To investigate nickel binding and the possibility that perturbations in the nickel affinity of HypA proteins are responsible for the different activities of the mutants etc. in the in vitro assay, the nickel binding properties of various HypA proteins were investigated using isothermal titration calorimetry (ITC, Figure 2 and Table 1). At pH = 7.2, WT-HypA binds one Ni(II) ion per monomer with a Kd = 1.08(6) μM (Figure 2). At pH = 8.3 the nickel binding characteristics were nearly identical to those at pH = 7.2; one nickel was bound per monomer with Kd = 3.7(3) μM. However, when the pH was lowered to 6.3, the binding affinity was lowered by ~15 fold (Kd = 17(2) μM) and only half as much nickel was bound (ca. one per dimer, Figure 2 and Table 1). This situation could arise if one of the nickel ligands has a pKa below 7.2, or if a pH-dependent structural change occurs that alters the nickel-binding site at low pH (or both).
When the cysteine residues in the zinc-binding motif were mutated, the nickel binding stoichiometry was also reduced to ca. one nickel per dimer. The affinity for Ni(II) in all of the mutated proteins was much weaker than that of WT-HypA at pH = 7.2 (Kd 20 – 50 μM). This result is quite similar to the data from WT-HypA at pH = 6.3, and suggests that a similar conformational change may be involved in the mutants.
Studies of the mutants involving the His residues in the zinc-binding motif showed that at pH = 7.2 both mutants bind nickel with a stoichiometry and affinity similar to WT-HypA. (For H79A-HypA the stoichiometry is one Ni(II) per monomer with Kd = 3.2(3) μM; for H95A-HypA, the stoichiometry is one Ni(II) per monomer with Kd = 2.1(1) μM). When the pH was lowered to 6.3, the dissociation constant for H95A-HypA increased to 21(2) μM, similar to the change observed in WT-HypA when the pH is lowered. However, H95A-HypA was still able to bind one Ni(II) per monomer instead of one Ni(II) per dimer, as is the case with WT-HypA. The data clearly establish that the nickel affinity of HypA is sensitive to pH over the range that is physiologically relevant to H. pylori, and that structural changes in the zinc site affect the nickel binding characteristics of dimeric HypA.
NMR structural information
When treated as originating from a HypA monomer, the nuclear Overhauser effects (NOEs) we observe agree in most respects with the published monomeric structure of H. pylori HypA (PDB entry 2KDX).18 That is, the NOEs that we observe are for the most part those expected based on the published structure, assuming that all the NOEs were between protons on the same molecule. However, NOEs involving the zinc site are not consistent with the 2KDX structure. We observe clear and well-defined NOEs between the β-CH2 protons of Zn ligands Cys 74 and Cys 91, while similar sets of NOEs are observed between the β-CH2 protons of Cys 77 and Cys 94 (Figure 3). This does not agree with the NOEs expected based on the arrangement of these methylene groups in the 2KDX monomer structure, which would predict pair-wise NOEs between the β-CH2 protons of Cys 74 and Cys 77, and no intra-cluster NOEs to the Cys 94 β-CH2 protons. Our observations, however, fit well with the recently published single crystal x-ray diffraction structure of T. kodakarensis HypA, which is a chain-swap dimer (3A44) in which the Zn-ligating Cys ligands homologous with Cys 74 and Cys 77 originate from one monomer unit, while those corresponding to Cys 91 and Cys 94 are from the second monomer. Furthermore, the 2KDX structure exhibits idealized geometry for the Zn-S cluster, with identical Zn-S and S-S distances and bond angles for all atoms in the cluster, while Zn-S bond distances in the 3A44 crystal structure vary between 1.7 and 2.7 Å, and S-S distances vary between 3 and 4 Å. These observations, in combination with observed elution patterns from size exclusion chromatography, are consistent with a dimeric form of the protein that involves polypeptide chain swapping, as is observed for the T. kodakarensis HypA dimer structure, 3A44.19 Our current experiments are aimed at elucidating the exact nature of the dimer interface.
Figure 3.

A plane from 13C-edited three-dimensional 1H,1H NOESY (800.13 MHz 1H) showing β-CH2 NOEs between Zn-ligating Cys residues in HypA as described in the text.
While we are still refining the solution structure of the apo-dimer of H. pylori apo-HypA, we can demonstrate that the binding of Ni(II) to the HypA dimer results in dipolar broadening in the vicinity of the proposed Ni(II) binding site18 (Figure 4) that is consistent with a paramagnetic (S = 1) Ni(II) site, a result that is supported by Ni-ligand bond distances from XAS (vide infra), and prior susceptibility measurements.16 When Ni(II) ions are added to solutions of apo-dimer, correlations corresponding to residues spatially close to the N-terminus in the folded apo-protein are lost from the 1H,15N HSQC spectrum, indicating that high-spin, paramagnetic nickel is bound. The resonances that are lost correspond to residues in the N-terminal helical motif near the expected Ni-binding site (residues 3–5, 8, 9, 31, 34, 36–43, 45, 49 and 51).17 This result is contrary to the results reported for the monomer nickel site, where a planar four-coordinate, diamagnetic Ni(II) site was characterized that involves ligation by His2, but has a modified N-terminal sequence (GSMHE-).18
Figure 4.
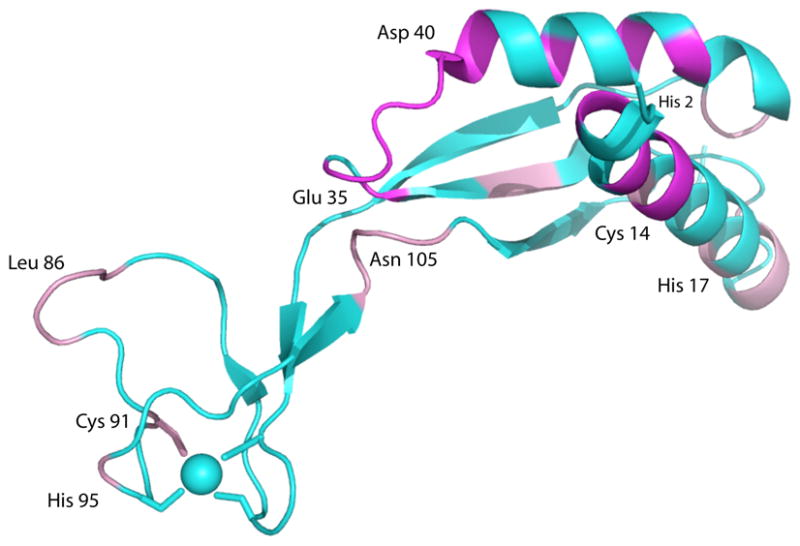
NMR results superimposed on the structure of monomeric HypA (PDB entry 2KDX).18 Resonances that are broadened to invisibility by addition of Ni(II) ions are shown in magenta. Resonances that are perturbed or doubled by binding Ni(II), indicating more than one conformation is present, are shown in pink. The Zn site is indicated by the blue sphere. See text for details.
NMR data also show changes in the protein structure upon Ni(II) binding that support communication between the Zn(II) and Ni(II) binding sites (as defined above, Figure 4), consistent with the zinc-site structural changes observed by XAS (vide infra). In addition to paramagnetic broadening, the presence of Ni(II) ions causes a doubling or other perturbation of some of the resonances, consistent with the presence of more than one conformation of HypA in solution. The residues corresponding to the perturbed resonances include several residues in the zinc binding motif as well as residues in the linker and Ni-binding locus (residues 14, 15, 17, 19, 20, 30, 43, 57, 58 in the Ni-binding locus, residues 85–88, 91 and 95 in the Zn-binding motif, as well as residues 104–106 in the linker region).
X-ray absorption spectroscopy
XAS studies were used to examine the structures of the nickel and zinc sites in WT- and mutant HypA proteins, and to address the effects of pH, point mutations and nickel binding on the structures of the two metal sites. The results are summarized in Figures 5, 6 and 7 and Tables 2 and 3.
Figure 5.
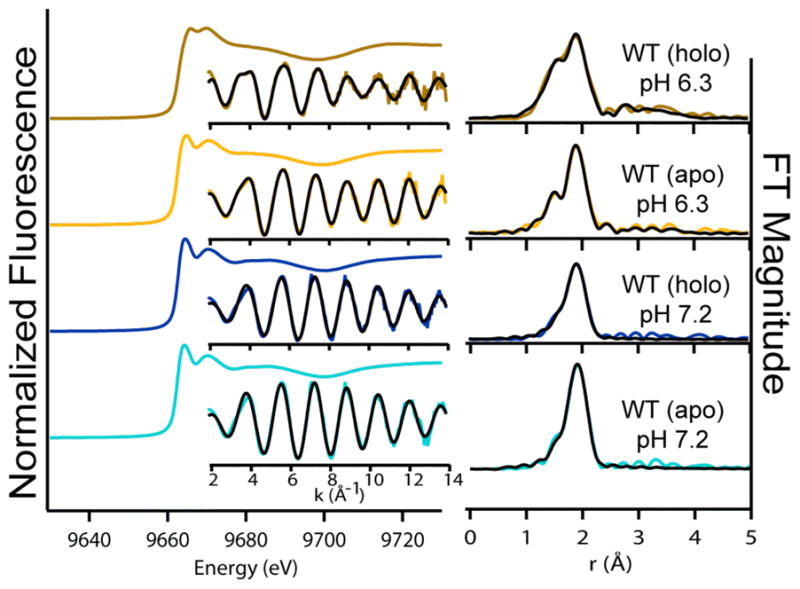
Zinc K-edge XAS of apo- and holo-WT-HypA at pH 7.2 Left: Normalized XANES data with unfiltered k3-weighted EXAFS spectra (colored line) and best fits (black line) from Table 2. Right: Fourier-transformed EXAFS spectra and fits.
Figure 6.

Zinc K-edge XAS data for selected mutant HypA proteins. Left: Normalized XANES spectra with unfiltered k3-weighted EXAFS spectra (colored line) and best fits (black line) from Table 2 inset. Right: Fourier-transformed EXAFS spectra and fits. Data for other Cys → Ala, Cys → Asp and H95A are shown in supporting information.
Figure 7.

Nickel K-edge XAS data for holo-HypA proteins. XANES is shown as a single colored line. Left inset is an expansion of the pre-edge XANES region. Right insets are the unfiltered k3-weighted EXAFS data (colored line) and fits from Table 3 (black line).
Table 2.
Best Fits of Zinc K-edge EXAFS data of HypA Proteins
| −Ni | +Ni | |||||||
|---|---|---|---|---|---|---|---|---|
| HypA | Shell | r (Å) | σ2(x10−2 eV) | R | Shell | r (Å) | σ2(x10−2 eV) | R |
| WT (pH = 6.3) | 1Nimd | 1.96(1) | 2(1) | 0.0263 | 2Nimd | 2.00(2) | 5(1) | 0.0481 |
| 3S | 2.322(5) | 3.5(3) | 2S | 2.33(1) | 3.7(5) | |||
| WT (pH = 7.2) | 3S | 2.32(1) | 2(1) | 0.0277 | 4S | 2.333(7) | 4.0(4) | 0.0270 |
| 1S | 2.42(2) | 2(4) | ||||||
| C74A | 2Nimd | 2.02(2) | 3(1) | 0.0396 | 2Nimd | 1.97(2) | 5(2) | 0.0367 |
| 2S | 2.32(1) | 3.3(6) | 2S | 2.31(2) | 5(3) | |||
| H79A (pH = 6.3) | 4S | 2.332(4) | 3.5(3) | 0.0183 | 4S | 2.334(3) | 3.3(2) | 0.0180 |
| H79A (pH = 7.2) | 4S | 2.32(1) | 4.0(6) | 0.068 | 4S | 2.324(5) | 4.1(3) | 0.0374 |
| C94A | 2Nimd | 1.98(1) | 3.4(9) | 0.0405 | 2Nimd | 1.98(1) | 3.3(8) | 0.0351 |
| 2S | 2.29(1) | 5.9(7) | 2S | 2.30(1) | 6.9(8) | |||
| H95A (pH = 6.3) | 4S | 2.336(4) | 3.6(2) | 0.0170 | 4S | 2.335(4) | 3.3(2) | 0.0176 |
| H95A (pH = 7.2) | 4S | 2.337(5) | 3.4(3) | 0.0114 | 4S | 2.335(4) | 3.5(4) | 0.0217 |
Table 3.
Nickel K-edge XAS of HypA Proteins
| XANES | EXAFS | |||||
|---|---|---|---|---|---|---|
| HypA | 1s → 3d peak area (x 102 eV) | CN/geometry | Shell | r (Å) | σ2 (x10−2 eV) | R |
| WT (pH = 6.3) | 2.3(6) | 6 | 4N | 2.09(2) | 2(1) | 0.0607 |
| 1N (2Im) | 1.93(5) | 6(8) | ||||
| WT (pH = 7.2) | 1.9(7) | 6 | 3N | 2.09(2) | 3(2) | 0.0389 |
| 2N (2Im) | 1.96(3) | 7(5) | ||||
| C74A | 1.8(4) | 6 | 3N | 2.14(2) | 1(3) | 0.0210 |
| 3N (2Im) | 2.092(9) | 1(2) | ||||
| C77A | 6.4(5) | 5 | 3N | 2.09(3) | 6(3) | 0.0823 |
| 2N (2Im) | 1.92(4) | 6(3) | ||||
| C91A | 5.6(4) | 5 | 3N | 2.09(3) | 7(3) | 0.0617 |
| 2N (2Im) | 1.91(3) | 6(2) | ||||
| C94A | 6.2(5) | 5 | 3N | 2.09(3) | 4(2) | 0.0624 |
| 2N (2Im) | 1.93(4) | 8(5) | ||||
| H79A (pH = 6.3) | 2.4(6) | 6 | 3N | 2.04(5) | 10(4) | 0.0146 |
| 2N (2Im) | 2.07(2) | 1(1) | ||||
| H79A (pH = 7.2) | 5.5(7) | 5 | 3N | 2.09(2) | 3(2) | 8(8) |
| 2N (2Im) | 2.24(5) | |||||
| H95A (pH = 6.3) | 2.5(6) | 6 | 3N | 2.08(6) | 9(4) | 0.0247 |
| 2N (2Im) | 2.06(2) | 1(2) | ||||
| H95A (pH = 7.2) | 1.1(3) | 6 | 3N | 2.08(7) | 6(7) | 0.0215 |
| 2N (2Im) | 2.07(5) | 2(5) | ||||
XANES
K-edge XANES analysis can yield information about the coordination number and geometry of the metal site. Nickel has vacancies in the 3d manifold that give rise to transitions in the XANES region of the XAS spectrum below the edge energy that are dependent on the geometry/coordination number of the metal site. These transitions include the 1s → 3d and 1s → 4pz (plus shakedown contributions) that occur near 8331 and 8336 eV, respectively, in Ni(II) complexes.27 Pre-edge XANES analysis with regard to zinc is less informative, as there are no pre-edge transitions due to the closed shell nature of Zn(II). Qualitative information about the types of scattering atoms surrounding the zinc atom can be obtained by observing the ratio of the first and second peaks in the post-edge XANES.28 The presence of mainly sulfur atoms in the coordination sphere results in a strong first peak and, as the number of N/O-donor ligands in coordination sphere increases the second peak will gain intensity and dominates when S-donor ligands are absent.
Zinc Site
Examination of the zinc K- edge XANES spectra for WT-HypA at pH = 7.2 both with and without nickel (Figure 5) reveals that the first peak in the near-edge spectrum is largest, consistent with a zinc site dominated by sulfur donors. When the pH is lowered to 6.3 and no nickel is present, the second peak gains intensity showing that the ratio of N/O- to S-donors in the primary coordination sphere has increased. When nickel is added at pH = 6.3, the two peaks are of equal intensity, indicating a roughly 50/50 mixture of sulfurs and N/O-donor ligands is present. Examination of the XANES data for mutants with point mutations in the two CxxC motifs in the zinc-binding motif (Figure 6) shows that all of the Cys mutants have a zinc site with a mixed NO/S-donor coordination environment, with or without nickel bound, that is similar to the holo-WT-HypA pH = 6.3 sample. This result contrasts with the XANES data from the H79A- and H95A-HypA mutants, where the first peak in the zinc post-edge region dominates the XANES spectra, indicating a coordination sphere composed of predominantly S-donors, similar to the WT-HypA samples at pH = 7.2.
Nickel Site
Examination of the nickel XANES data for WT-HypA (Figure 7, Table 3) reveals a small 1s → 3d transition (~ 0.02 eV2) and no evidence of 1s → 4pz transition at both pH = 7.2 and = 6.3, features that are typical of 6-coordinate nickel centers.27 The 1s → 3d peak areas for the Cys point mutations in the zinc binding motif are higher when compared to WT-HypA (~0.06 eV2), and are typical of five-coordinate nickel centers. The presence of a shoulder associated with the 1s → 4pz transition is consistent with pyramidal geometry.27 The only exception is C74A-HypA, which has a smaller 1s → 3d peak area and no 1s → 4pz transition, similar to WT-HypA and consistent with a six-coordinate nickel environment. The nickel K-edge XANES data for H95A-HypA features a small 1s → 3d peak area and lacks features associated with the 1s → 4pz transition at both pH = 7.2 and = 6.3, consistent with a 6-coordinate nickel site that is similar to WT-HypA. In contrast, in the H79A-HypA mutant, the situation is more like that observed for most of the Cys-mutants. The peak area is larger (~ 0.06 eV2) typical of a 5-coordinate site and is supported by the presence of a small 1s → 4pz transition.
EXAFS
EXAFS analysis was used to provide information about the metal sites regarding the types of atoms in the primary coordination sphere, metric details (bond lengths), and information about the presence of histidine ligands in the primary coordination sphere (from multiple scattering analysis).
Zinc Site
The best fit of the zinc site EXAFS in WT-HypA at pH = 7.2 (Figure 5, Table 2) reveals a site that is composed of an of four CysS-donor ligands, presumably Cys74, Cys77, Cys91 and Cys94, based on sequence homologies of HypA orthologs (vide infra) and the NMR structure of monomeric form of H. pylori HypA.18 One of these Cys ligands has a unique Ni-S distance that is 0.1 Å longer than the other three on average (2.42 Å versus 2.32 Å), and thus the complex has local symmetry that is less than tetrahedral. Upon binding nickel, the zinc site undergoes a subtle structural change yielding a more symmetric Zn(Cys)4 site by shortening the long Zn-S bond. The Zn(Cys)4 site found for WT-HypA at pH = 7.2 is consistent with the results from the XANES analysis that indicated a predominantly S-donor ligand environment.
The effects of pH on the zinc site structure are more dramatic (Figure 5, Table 2). When the pH was lowered to 6.3, the long Zn-S interaction in apo-WT-HypA was replaced with a Zn-N bond from a His imidazole ligand to form a Zn(Cys)3(His) site. This result is fully consistent with the XANES analysis that indicated an increase in the number of N/O donors in the zinc site (vide supra). The unique distance associated with the Zn-N vector is evident by the presence of a new peak in the FT spectrum at a shorter distance than main Zn-S peak (Figure 5). The identity of the new ligand as a His imidazole is indicated by the fit, which includes multiple-scattering from second- and third-coordination sphere C and N atoms that accounts for weak features between ~ 2 and 4 Å (uncorrected for phase shifts) in the FT spectrum, and by the doubling of the first oscillation (k ~ 4 Å−1 in the unfiltered k - space EXAFS spectrum (Figure 5).
The Zn(Cys)3(His) site in WT-HypA at pH = 6.3 undergoes an additional structural change when nickel is added. The best fit indicates that another CysS-donor ligand has been substituted by an additional His imidazole ligand, resulting in a Zn(Cys)2)(His)2 site. Thus, nickel binding to WT-HypA at either pH leads to well-defined and distinct structural changes in the zinc site. Scheme 1 depicts the changes that are observed via XAS around the zinc site based on pH changes and the addition of nickel to HypA.
Scheme 1.

Depiction of the coordination changes around the zinc atom depending on pH and Ni binding status.
Mutations involving the zinc ligands in HypA (Cys → Ala) force structural changes in the zinc site by changing the number of ligands available to bind zinc. Figure 6 and Table 2 show that the zinc site of C74A– and C94A-HypA adopts a N2S2 ligand donor atom set in apo-WT-HypA, consistent with the XANES analysis (vide supra). Multiple-scattering analysis indicates that both nitrogen donors are due to the presence of imidazole ligands. Table 2 shows that all the Cys mutants contain similar Zn(Cys)2(His)2 centers (see supporting information for spectra). This coordination environment is identical to that of WT-HypA at pH = 6.3 with nickel present, and suggest that mutation of any of the Cys residues puts the protein into the same low pH conformation.
To examine whether the structural changes were due simply to the loss of a ligand in the zinc environment, Cys → Asp mutations were also investigated and yielded the same results at both pH = 7.2 and 6.3 that were obtained for Cys → Ala (supplemental information).
Because the pH = 6.3 WT-HypA protein and the Cys mutants involve substitution of Cys residues in the zinc site by His ligands, we examined the structure of the zinc site in the mutants that involve the His residues flanking the CXXC motifs in the zinc-binding site. In the apo-mutants at pH = 7.2, both H79A-HypA and H95A-HypA contained Zn(Cys)4 sites that did not possess the one long Zn-S bond found in apo-WT-HypA under the same conditions (Table 2). When the pH is lowered to 6.3, neither His79A- nor H95A-HypA shows incorporation of an N-donor ligand; the site remains a Zn(Cys)4 site at low pH. This result establishes the essential role played by the flanking histidine residues in the structural changes observed in the zinc site at low pH.
Nickel Site
Analysis of K-edge EXAFS data was also used to examine the structure of the nickel site in HypA proteins, and to address structural changes in the nickel site that occur in response to pH changes or perturbations of the zinc site/protein structure. Figure 7 and Table 3 show the best fits to EXAFS data for the nickel site in both WT- and mutant HypA proteins. In WT-HypA at pH = 7.2 the best fit is for a five-coordinate N/O-donor ligand environment with the second and third coordination spheres modeled as containing two imidazole ligands, Ni(N/O)3(His)2. In the primary coordination sphere, the N/O donors are split into two distinct shells, with two Ni-N/O-donor ligands lying at 1.96 Å and the other three N/O-donors at 2.09 Å.
When the pH is lowered to 6.3 the nickel site remains five-coordinate, with the only apparent change being a lengthening of one of the shorter Ni-N/O bonds to the longer distance. The five coordinate fits are in disagreement with the XANES analysis for WT-HypA, which clearly indicates a six-coordinate ligand environment. Although fits to six-coordinate models are possible (see supporting information) the values of both R and reduced χ2 are lower for the five-coordinate EXAFS fits. This situation could arise if one of the ligands is weakly bound, or highly disordered, as in the case of the Met ligand in the blue Cu center of plastocyanin that is not observed by EXAFS.29
The structural changes that occur upon mutation of the Cys ligands at the zinc site are not reflected in the structure of the nickel site. With the exception of C74A-HypA, the best fit is a five-coordinate site with all N/O-donors, almost identical with the structure of the WT-HypA nickel site structure. In the case of C74A-HypA, the best fit is for a six-coordinate model, and the additional Ni-N/O vector and two shorter Ni-N/O bonds average 2.14 Å, as compared with 1.96 Å in WT-HypA. The mutations of Cys → Asp were also analyzed and all four presented a site that was nearly identical to that of WT-HypA (supporting information). C74D-HypA was similar to WT-HypA and did not fit to the six-coordinate site like C74A-HypA.
When His residues flanking the CXXC motifs in the zinc site were mutated, more pronounced changes in the nickel site structure were observed. For example, in H79A-HypA at pH = 7.2 the average Ni-N/O distance is longer than in WT-HypA, although the nickel site remains five-coordinate and the data are still fit best by two shells of scattering atoms. When the pH is lowered to 6.3, the nickel site becomes more symmetric, with all five ligands lying at an average Ni-N/O distance of 2.05 Å. In the case of H95A-HypA, the EXAFS data for the nickel site is best fit by a five-coordinate model with an average Ni-N/O distance of 2.06 Å at both pHs. The site is indistinguishable from the nickel site in the H79A-HypA mutant at pH = 6.3.
Activity Assay
The functional properties of WT- and mutant HypA proteins were tested in a urease activity assay. In this experiment, lysate from an H. pylori hypA deletion strain was incubated with HypA protein. The feasibility of this previously untested approach was demonstrated by the ability of WT HypA to restore urease activity after prolonged incubation (Figure 8). All of the Cys mutants and the His mutants of HypA showed lower levels of activity, with mutations involving Cys77 and Cys94 having a particularly deleterious effect. These two cysteines are the second Cys residues in each CxxC motif in the zinc binding site, and each is flanked by a His residue, H79 and H95, respectively. Although mutation of H79 resulted in more of a decrease in activity than did mutation of H95, neither His mutation destroyed the ability of the protein to provide nickel for urease biosynthesis. These results establish that the recombinant HypA used in these studies is functional, that dimeric HypA is sufficient for activity, and that mutations in the zinc site affect the ability of HypA to function in urease maturation.
Figure 8.

In vitro urease activity assay for WT- and mutant HypA proteins. Activity is shown as the amount of ammonia produced per 20 minutes. The amount of ammonia produced was calibrated to a standard curve created using NH4Cl.
Discussion
Metallochaperones function to “bind and deliver” the cognate metal to apo-enzymes.30 In doing so, it seems desirable to have a mechanism by which metallochaperones with the correct metal ion bound could be recognized by their delivery targets. Prior XAS studies have shown that the zinc site in HypA is able to change structure upon binding nickel to the protein, indicating that distinct HypA structures that affect the zinc site structure exist.16 The data presented here detail these structural changes and clearly establish that structural changes in the zinc site affect the nickel binding characteristics of dimeric HypA, as well the ability of HypA to participate in urease maturation. The results also show that the nickel binding properties of HypA are sensitive to pH over the range that is physiologically relevant to H. pylori.
Structure and Function
The NMR data shows that the overall protein structure in the dimer is similar to the NMR structure obtained on the monomeric construct that featured a non-native N-terminal extension (Gly-Ser) after N-terminal His-tag cleavage.18 However, the Zn(Cys)4 site displays NOEs that are consistent with the dimeric crystal structure,19 but are inconsistent with those expected for the tetrahedral site reported in the monomer structure.18 Also in contrast to results reported with the monomeric NMR structure, the data presented here show that the Ni(II) site in dimeric HypA with the native N-terminus is high-spin and paramagnetic (S = 1). These data are supported by prior Evans susceptibility measurements,16 and are consistent with the Ni-ligand distances measured by XAS (vide infra). Further, we do not observe the optical absorption near 420 nm that has been attributed to a planar Ni(II) site (see supporting information).18 The residues affected by the dipolar line broadening include His2, which is in common with nickel site in the prior NMR stucture.18
The nickel site structure was examined by XAS analysis, which reveals a five- or six-coordinate Ni(His)2(N/O)3–4 site with metal-ligand distances appropriate for a high-spin structure (modeled as two shells of ligands with 2 M-N/O distances of 1.96(3) Å and 3 M-N/O distances of 2.09(2) Å in WT-HypA, Table 3). There is no evidence of the distinctive XANES signature27 or short Ni-ligand bonds in the EXAFS fit that are typical of a planar, four-coordinate site27 like that found for the N-terminally modified protein.18 Furthermore, there is no optical transition near 420 nm (see supporting information) like that seen for modified monomeric protein.18 This is perhaps not surprising since the nickel site includes His2 and thus lies near the N-terminus, and the position of the His residue with respect to the N-terminal amine may very well affect the nature of the metal complex formed.31,32 It is unknown if the modified HypA protein is functional in nickel-enzyme active site assembly.
Scheme 1 summarizes the data from zinc K-edge XAS analysis (Table 2). At neutral pH (pH = 7.2) the zinc site is best modeled a distorted tetrahedral ZnS4 structure that is in agreement with data from both HypA structures and in a prior work.16,18,19 The distorted Zn(Cys)4 site features 3 Zn-S interactions at 2.32(1) Å and one longer Zn-S bond at 2.42(2) Å. Upon binding Ni(II), the site becomes a regular tetrahedron with four Zn-S bonds at 2.333(7) Å. ITC (Table 2) establishes that the binding stoichiometry under these conditions is one Ni(II) ion per monomer with a Kd = 1.08(6) μM. This result differs from a previous study of Ni(II) binding to H. pylori HypA that used equilibrium dialysis measurements, where two binding events with a Hill coefficient of 2.0 were observed.14 The first Ni(II) ion bound with Kd = 1.3 μM, similar to that observed here by ITC and typical of other metallochaperones.15 The second Ni(II) ion was much more weakly bound with a Kd of 58 μM.14 The latter value was similar to the Kd observed for Ni(II) binding to E. coli HypA, which binds stoichiometric nickel with a Kd of 60 μM.33 The relatively subtle change in the zinc site structure that occurs upon Ni(II) binding is not accompanied by a significant change in protein secondary structure as monitored by CD spectroscopy (Figure 1), but Ni(II) binding does increase the stability of the protein as the Tm value increases by 12 °C upon binding Ni(II).
While no significant changes were noted at neutral (pH = 7.2) and higher pH (pH = 8.3), at lower pH (pH = 6.3) a ligand substitution occurred in the apo-WT-HypA zinc site wherein one of the Zn-S bonds was replaced with a Zn-N bond that was best modeled using multiple-scattering EXAFS analysis (Table 2) as arising from an imidazole ligand. Since the unique long Ni-SCys bond found in the neutral-pH zinc site structure would be expected to be the most basic, protonation and loss of this ligand under acidic conditions could open a coordination position for an incoming ligand to facilitate forming the Zn(Cys)3(His) site. When Ni(II) is bound to the low-pH apo-HypA protein, a second ligand substitution results in the formation of a Zn(Cys)2(His)2 site. It should be noted that the structure derived from EXAFS analysis is an average structure, and the Zn(NHis)(SCys)3 site observed in the absence of nickel may not be distinguishable from a 1:1 mixture of the high- (Zn(SCys)4) and low-pH (Zn(NHis)2(SCys)2) nickel bound forms solely on the basis of EXAFS analysis. This raises the possibility that there are two conformations of the protein; a form that predominates at neutral pH and features a Zn(Cys)4 site and a form that predominates at acid pH that contains a Zn(His)2(Cys)2 site, and that the equilibrium between the two forms is perturbed by nickel binding. In any case, binding Ni(II) at low and high pH leads to two distinct and well-characterized zinc site structures. Mutations of the His residues that flank the CxxC motifs in the zinc binding motifs (H79A and H95A), show that the His ligands are supplied by these residues. This result establishes the essential role played by the flanking histidine residues in the structural changes observed in the zinc site at low pH, and suggests that a protonated His-dependent switch is involved in the conformational change. The potential physiological relevance of this switch is discussed below.
There are only a few examples of proteins that have zinc sites that undergo ligand substitutions, and we are aware of no metallochaperone other than H. pylori HypA that does so. One example is the enzyme farnesyltransferase, which transfers a farnesyl group from a farenyldiphosphate to a protein substrate that contains a C-terminal CAAX sequence. The resting state of the protein has a zinc atom bound in a (O/N)3S coordination environment.34,35 There is debate over the exact mechanism, but it is clear that the substrate peptide is able to bind to the zinc center, creating a new Zn-S bond during turnover.34–36 Farnesyltransferase is part of a small group of proteins that are able to undergo ligand substitutions or modify the metal ligands during catalysis.37 Most of these enzymes add a sulfur-donor during turnover and lose an O/N-donor ligand.
Under acidic conditions, ITC (Table 1) shows that the Ni(II) binding stoichiometry is altered from one Ni(II) ion per monomer to one Ni(II) ion per dimer. This change in stoichiometry is accompanied by a decrease in the Ni(II) binding affinity, Kd = 17 μM (pH = 6.3). The structure of the Ni(II) site from EXAFS analysis at pH = 6.3 differs only in the change of one bond distance (modeled as two shells of ligands with 1 M-N/O distances of 1.93(5) Å and 4 M-N/O distances of 2.09(2) Å in WT-HypA, Table 3). Although the nickel stoichiometry and affinity are altered, the stability of the protein is not. The Tm values measured are the same as at pH = 7.2, ~ 60° for the apo protein and ~ 70° for holo-HypA. Again, only minor perturbations in secondary structure are observed by CD spectroscopy (Figure 1).
To further probe the connection between the zinc site structure, nickel binding, and metallochaperone functions of HypA, single point mutations in all four Cys residues in the zinc binding motif and their flanking His residues were characterized. XAS data (Figure 6, Table 2) taken at pH = 7.2 on the four Cys → Ala point mutations show that the zinc site adopts a Zn(SCys)2(His)2 structure that is indistinguishable from the zinc site in the low-pH nickel-bound WT-protein regardless of which cysteine residue is mutated or whether Ni(II) is present. Since Cys → Ala mutations force a change in the ligand environment of the zinc site because of ligand loss, Cys → Asp mutants were also made. These mutations resulted in the formation of the same Zn(SCys)2(His)2 site by EXAFS analysis, and the properties of the Cys → Asp mutants were essentially identical to those of the corresponding Cys → Ala mutations (see Figure 6 and supporting information). Mutation of any Cys residue led to a general increase in the stability of the apo-Cys-HypA mutants over apo-WT-HypA, and the increase in Tm associated with Ni(II) binding was diminished. (The only exception being C91A, which has ca. the same Tm as WT-HypA and a Tm increase similar to WT-HypA upon Ni(II) binding). Such an observation is consistent with a model wherein increased stability is linked to zinc-site structural changes independent of nickel binding.
The nickel binding properties of the Cys mutants are also altered in a way that is similar to that observed for the low-pH form of HypA. ITC shows that the Ni(II) binding stoichiometry is one Ni(II) ion per dimer, with lower affinity (Kd ~ 20 – 50 μM). Thus, disrupting the Zn(SCys)4 site leads to a protein that is no longer able to sense Ni(II) or pH changes, and appears to adopt the specific conformation of WT-HypA at low pH with Ni(II) bound. Although EXAFS models do not indicate a significant change from the WT-HypA nickel site structure (Figure 7, Table 3) the Cys mutants were all impaired in their ability to restore urease activity in lysate from the ΔhypA mutant, despite the weaker affinity of the chaperone for Ni(II) ions. Mutations of either Cys residue with a flanking His residue (Cys77 or Cys94) were particularly deleterious to urease maturation (Figure 8). This suggests that interactions between trafficking proteins have been altered by the change in protein structure.
Mutations of the His residues flanking the CXXC motifs in the zinc binding motif also lead to a protein that no longer senses Ni(II) binding or pH change, but have a generally opposite effect from Cys mutations on nickel binding and metallochaperone function. Zinc K-edge XAS on H79A- and H95A-HypA reveals a zinc site structure that is indistinguishable from the symmetric Zn(SCys)4 site found in nickel-bound WT-HypA protein at pH = 7.2 (Figures 5 and 6 and Table 2). Further, the structure is the same at both pH = 7.2 and pH = 6.3 regardless of whether Ni(II) is bound or not. Thus, it appears that mutation of either His residue results in a structure that closely resembles the neutral pH with nickel bound form. The properties of the His mutant HypA proteins are consistent with this notion. The Tm values found for the His mutants are more similar to the nickel bound WT form (~ 67°) regardless of nickel loading, and no longer shift upon Ni(II) binding. ITC reveals that one Ni(II) ion per monomer is bound with an affinity that is similar to WT-HypA at pH = 7.2 (kd = 2 – 3 μM). Further, upon dropping the pH to 6.3, the stoichiometry is not altered (one Ni(II) per monomer), although the affinity is reduced by an amount that is similar to that associated with low-pH in WT-HypA (kd = 21 μM), thus providing a measure of how much the nickel site affinity is reduced by pH in the absence of a conformation change, and indicating that most of the changes in nickel site affinity observed are due largely to pH change. EXAFS analysis reveals that the nickel sites in the two His mutants are very similar and altered only slightly from that found in holo-WT-HypA (the ligand bonds are more similar in the two shells of O/N donors, Figure 7, Table 3). These changes result in a modest loss in the ability of His79A- and His95A-HypA to restore urease activity in lysate from the ΔhypA mutant.
The data from the His79- and His95-HypA mutations confirm their role as ligands in forming the low-pH apo-WT-HypA (Zn(Cys)3(His)) and holo-WT-HypA Zn(Cys)2(His)2 zinc site structures, their involvement in sensing nickel loading at low pH, and thus suggest a role in pH sensing. Metals bound by CxxC motifs are common in biology38–40 and there are examples where a histidine residue close to the CxxC motif is critical in relaying information from or to the metal site.41 One good example of a flanking histidine residue relaying information is in adrenodoxin.41,42 Adrenodoxin is a 2Fe2S protein that is responsible for shuttling electrons from NADPH dependent reductases to cytochrome P450 enzymes.41 Studies have shown that His56, which flanks the CxxC motif involved in iron binding, is involved in an H-bonding network that interacts with the redox partners of adrenodoxin. Mutations to this key histidine residue leads to an enzyme that can no longer communicate redox changes at the metal site to the rest of the protein.41 Another example of a flanking histidine residue playing a critical sensing role is in T7 DNA primase. T7 DNA primase consists of two domains, the RNA polymerase domain and the zinc binding domain (ZBD), which are connected by a flexible linker.42 The zinc ion in the ZBD is coordinated by two CxxC motifs and the second motif has a flanking histidine residue (His33 near Cys36 on the N-terminal side). The zinc site acts as a sensor for DNA binding: When no DNA is present the zinc site is a symmetric site with all four cysteine ligands around 2.3 Å.43 However in the presence of DNA, the site changes to a site where two of the cysteine residues become protonated and the Zn-SCys bond length increases to ~ 2.7 Å.43 Mutation of the flanking histidine residue (His33) to alanine has two pronounced effects: First, the recognition sequence, 5′-GTC-3′, is not longer recognized and DNA primers are no longer made.44 Secondly, when the ZBD domain is studied alone the affinity for zinc decreases, showing that His33 is required to properly orient the zinc center.43 It is believed that His33 is essential to help protonate the proper cysteine residues in the presence of DNA as well as a play a role in DNA sequence specificity.43 In the case of H. pylori HypA protein, the histidine residues actually substitute for the Cys ligands and appear to play a crucial role in communicating nickel loading and pH changes to the zinc site/protein conformation.
Role of HypA in H. pylori nickel trafficking and nickel enzyme assembly
Sensing of pH is important in H. pylori, where the organism must survive rapid exposure and adapt to the low pH environment of the stomach. H. pylori requires two nickel enzymes for survival in the acidic environment of the human stomach, urease and NiFe-hydrogenase.3–5 Under acid stress, H. pylori is able to regulate its internal pH and modify its local environment using the enzyme urease, which converts urea to ammonia.7 One level of urease regulation is via control of the amount of Ni(II) ions available for incorporation into apo-urease. The bacterium is constantly producing apo-urease, even when essential urease accessory proteins are deleted.13 Scott et al. have shown that activation of urease increases under acidic conditions via a process that involves association of the structural proteins UreA and UreB with the accessory proteins UreE, UreF, UreG and UreH on the cytopasmic side of the inner membrane, and is dependent on HP0244, a cytoplasmic histidine kinase.7 This study provides evidence for another mechanism of post-translational regulation of urease activity.
While HypA is known to serve as a nickel metallochaperone in the maturation of NiFe-hydrogenases in a number of microorganisms,9,45,46 maturation of urease in H. pylori also requires HypA.11,17 Similarly, in H. hepaticus both hydrogenase and urease activities are lost when the hypA gene is deleted.47 One possible basis for this dual function is that HypA preferentially acquires nickel ions made available form import or storage proteins in order to control the flow of Ni(II) ions between the two enzymes by serving as a metallochaperone for hydrogenase. In this regard, it is noteworthy that UreE-HypA heterodimers are known to form48 and thus HypA may play a pivotal role in controlling the flow of nickel between two target enzymes in response to environmental changes, such as nickel flux in the cell and pH. A rapid conformational response to reduced pH could either serve to facilitate rapid Ni(II) incorporation into urease under acid stress by increased interaction of HypA and UreE, or help maintain a supply of Ni(II) ions for hydrogenase maturation under conditions that are geared for rapid production of urease. The latter is suggested by the decrease in urease activity obtained using Cys-mutant HypA proteins, which resemble the WT protein under acidic conditions, in the in vitro assay. In either event, the trafficking of Ni(II) ions for the assembly of two different enzymes could be controlled by conformational changes in HypA that favor or disfavor interaction with UreE, or affect the interaction with apo-hydrogenase or other proteins involved in hydrogenase maturation, such as HypB.
Delineating the role of conformational changes in HypA function will require further studies of interactions with nickel-enzyme assembly proteins. The specific role of His79 and His95 in pH sensing is consistent with species-specific sequence variations in HypA homologues (Figure 9). HypA from H. hepaticus and T. kodakaraensis both contain the first flanking histidine residue (T. kodakaraensis possess an insertion loop that is not present in any other HypA or HypA homologue) but the second flanking histidine residue is shifted by several amino acids down toward the C-terminus. Whereas, HypA and HybF from E. coli contain the second flanking histidine residue but the first histidine residue is replaced by a glutamine. Since both flanking His residues are required to form the low-pH holo-HypA zinc site structure, neither of these proteins can sense pH in the same manner although these residues may still participate in H-bond interactions.
Figure 9.

Sequence alignment of HypA and HypA homologue proteins from various bacteria. Black boxes represent strictly conserved residues, red boxes show the two conserved CxxC motifs and blue boxes depict the alignment with the flanking His residue from H. pylori. H. pylori HypA is 53%, 22%, 25%, 20% identical and 77%, 48%, 51%, 56% similar to HypA homologues from H. hepaticus, E. coli, and T. kodakaraensis, respectively.
The three HypA proteins from H. pylori, H. hepaticus47 and E. coli33 all perform similar functions in that they bind Ni(II) and deliver it to NiFe-hydrogenase, but all three organisms occupy different niches. H. pylori colonizes the human stomach, which requires the bacterium to be able to survive at low pH. H. hepaticus, which colonizes the livers of mice, does not need to acclimate to an acid environment. While H. hepaticus HypA is clearly associated with both urease and hydrogenase maturation,47 the fact that H. hepaticus does not have to survive chronic low pH conditions means that HypA in this organism may not need a pH sensor. In contrast, E. coli K-12 expresses only NiFe hydrogenases, which can be induced under acid stress conditions.49 HypA has been shown to deliver nickel to hydrogenase 3, while HybF is the nickel metallochaperone associated with hydrogenases 1 and 2.9,50–52 The differences in nickel trafficking requirements in these organisms likely contribute both the importance of a pH-dependent switch mechanism, as well as the potential interacting protein partners that would might be sensitive to a conformational change.
Supplementary Material
Acknowledgments
This work was supported by NIH grants GM-69696 (MJM) and GM-44191 (TCP). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. Stanford Synchrotron Radiation Laboratory, is a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural and Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program. The U.S. Department of Energy, Division of Materials Sciences and Division of Chemical Sciences supported XAS collection at the National Synchrotron Light Source at Brookhaven National Laboratory. The NIH supports Beamline X3B at NSLS. We thank Dr. Erin Benanti for the generous gift of H. pylori lysates for the urease activity assays.
Footnotes
Supporting Information Available: Figures of CD spectra for zinc-site cysteine mutants of HypA, Thermal melts of WT- and zinc-site cysteine and histidine mutants, molecular weight determinations by size-exclusion chromatography, ITC thermograms for zinc-site cysteine and histidine mutants, raw ITC titration data, zinc K-edge XANES and EXAFS data and fits for Cys → Asp and His95A mutations, nickel K-edge XANES and EXAFS data and fits for Cys → Asp zinc-site mutations, and UV-vis spectra of HypA with nickel bound. Tables of mutagenic primers, best EXAFS fits to Zn K-edge data for Cys → Asp mutations, best EXAFS fits to Ni K-edge data for zinc-site Cys → Asp mutations, alternate fits for zinc and nickel K-edge EXAFS (39 pages). This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Marshall BJ, Warren JR. Lancet. 1984;1:1311. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 2.Scott D, Weeks D, Melchers K, Sachs G. Gut. 1998;43(Suppl 1):S56. doi: 10.1136/gut.43.2008.s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eaton KA, Brooks CL, Morgan DR, Krakowka S. Infect Immun. 1991;59:2470. doi: 10.1128/iai.59.7.2470-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eaton KA, Krakowka S. Infect Immun. 1994;62:3604. doi: 10.1128/iai.62.9.3604-3607.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olson JW, Maier RJ. Science. 2002;298:1788. doi: 10.1126/science.1077123. [DOI] [PubMed] [Google Scholar]
- 6.Wen Y, Marcus EA, Matrubutham U, Gleeson MA, Scott DR, Sachs G. Infect Immun. 2003;71:5921. doi: 10.1128/IAI.71.10.5921-5939.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott DR, Marcus EA, Wen YSS, Feng J, Sachs G. J Bacterol. 2010;192:94. doi: 10.1128/JB.00848-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hausinger RP. Journal of Biological Inorganic Chemistry. 1997;2:279. [Google Scholar]
- 9.Hube M, Blokesch M, Bock A. J Bacteriol. 2002;184:3879. doi: 10.1128/JB.184.14.3879-3885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGlynn SE, Mulder DW, Shepard EM, Broderick JB, Peters JW. Dalton Trans. 2009:4274. doi: 10.1039/b821432h. [DOI] [PubMed] [Google Scholar]
- 11.Olson JW, Mehta NS, Maier RJ. Mol Microbiol. 2001;39:176. doi: 10.1046/j.1365-2958.2001.02244.x. [DOI] [PubMed] [Google Scholar]
- 12.Quiroz S, Kim JK, Mulrooney SB, Hausinger RP. Nickel and its Surprirsing Impact in Nature. John Wiley & Sons, Ltd; Chichester: 2007. [Google Scholar]
- 13.Benoit S, Maier RJ. J Bacteriol. 2003;185:4787. doi: 10.1128/JB.185.16.4787-4795.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehta N, Olson JW, Maier RJ. J Bacteriol. 2003;185:726. doi: 10.1128/JB.185.3.726-734.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bellucci M, Zambelli B, Musiani F, Turano P, Ciurli S. Biochem J. 2009;422:91. doi: 10.1042/BJ20090434. [DOI] [PubMed] [Google Scholar]
- 16.Kennedy DC, Herbst RW, Iwig JS, Chivers PT, Maroney MJ. J Am Chem Soc. 2007;129:16. doi: 10.1021/ja066958x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benanti EL, Chivers PT. J Bacteriol. 2009;191:2405. doi: 10.1128/JB.01657-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia W, Li H, Sze KH, Sun H. J Am Chem Soc. 2009;131:10031. doi: 10.1021/ja900543y. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe S, Arai T, Matsumi R, Atomi H, Imanaka T, Kunio Miki. Journal of Molecular Biology. 2009;394:448. doi: 10.1016/j.jmb.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 20.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. J Biomol NMR. 1995;6:135. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 21.Herbst RW, Guce A, Bryngelson PA, Higgins KA, Ryan KC, Cabelli DE, Garman SC, Maroney MJ. Biochemistry. 2009;48:3354. doi: 10.1021/bi802029t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Padden KM, Krebs JF, MacBeth CE, Scarrow RC, Borovik AS. J Am Chem Soc. 2001;123:1072. doi: 10.1021/ja003282b. [DOI] [PubMed] [Google Scholar]
- 23.Webb SM. Physica Scripta. 2005;T115:1011. [Google Scholar]
- 24.Ankudinov ALRB, Rehr JJ. Physical Review B. 1998;58:7565. [Google Scholar]
- 25.Zabinsky SI, Rehr JJ, Ankudinov A, Albers RC, Eller MJ. Phys Rev B Condens Matter. 1995;52:2995. doi: 10.1103/physrevb.52.2995. [DOI] [PubMed] [Google Scholar]
- 26.Weatherburn MW. Anal Chem. 1967;39:971. [Google Scholar]
- 27.Colpas GJ, Maroney MJ, Bagyinka C, Kumar M, Willis WS, Suib SL, Mascharak PK, Baidya N. Inorg Chem. 1991;30:920. [Google Scholar]
- 28.Clark-Baldwin K, Tierney DL, Govindaswamy N, Gruff ES, Kim C, Berg J, Koch SA, Penner-Hahn JE. Journal of the American Chemical Society. 1998;120:8401. [Google Scholar]
- 29.Penner-Hahn JE, Murata M, Hodgson KO, Freeman HC. Inorganic Chemistry. 1989;28:1826. [Google Scholar]
- 30.Rosenzweig AC. Chem Biol. 2002;9:673. doi: 10.1016/s1074-5521(02)00156-4. [DOI] [PubMed] [Google Scholar]
- 31.Harford C, Sarkar B. Accounts Chem Res. 1997;30:123. [Google Scholar]
- 32.Kozlowski H, Bal W, Dyba M, Kowalik-Jankowska T. Coordination Chemistry Reviews. 1999;184:319. [Google Scholar]
- 33.Atanassova A, Zamble DB. J Bacteriol. 2005;187:4689. doi: 10.1128/JB.187.14.4689-4697.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Long SB, Casey PJ, Beese LS. Nature. 2002;419:645. doi: 10.1038/nature00986. [DOI] [PubMed] [Google Scholar]
- 35.Tobin DA, Pickett JS, Hartman HL, Fierke CA, Penner-Hahn JE. J Am Chem Soc. 2003;125:9962. doi: 10.1021/ja035927o. [DOI] [PubMed] [Google Scholar]
- 36.Sousa SF, Fernandes PA, Ramos MJ. Biophys J. 2005;88:483. doi: 10.1529/biophysj.104.048207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Penner-Hahn J. Curr Opin Chem Biol. 2007;11:166. doi: 10.1016/j.cbpa.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 38.Lee JW, Helmann JD. J Biol Chem. 2006;281:23567. doi: 10.1074/jbc.M603968200. [DOI] [PubMed] [Google Scholar]
- 39.Lucarelli D, Russo S, Garman E, Milano A, Meyer-Klaucke W, Pohl E. J Biol Chem. 2007;282:9914. doi: 10.1074/jbc.M609974200. [DOI] [PubMed] [Google Scholar]
- 40.Traore DA, El Ghazouani A, Ilango S, Dupuy J, Jacquamet L, Ferrer JL, Caux-Thang C, Duarte V, Latour JM. Mol Microbiol. 2006;61:1211. doi: 10.1111/j.1365-2958.2006.05313.x. [DOI] [PubMed] [Google Scholar]
- 41.Kostic M, Bernhardt R, Pochapsky TC. Biochemistry. 2003;42:8171. doi: 10.1021/bi034500r. [DOI] [PubMed] [Google Scholar]
- 42.Kato M, Ito T, Wagner G, Richardson CC, Ellenberger T. Mol Cell. 2003;11:1349. doi: 10.1016/s1097-2765(03)00195-3. [DOI] [PubMed] [Google Scholar]
- 43.Akabayov B, Lee SJ, Akabayov SR, Rekhi S, Zhu B, Richardson CC. Biochemistry. 2009;48:1763. doi: 10.1021/bi802123t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mendelman LV, Beauchamp BB, Richardson CC. Embo J. 1994;13:3909. doi: 10.1002/j.1460-2075.1994.tb06702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gubili J, Borthakur D. Journal of Applied Phycology. 1998;10:163. [Google Scholar]
- 46.Tibelius KH, Du L, Tito D, Stejskal F. Gene. 1993;127:53. doi: 10.1016/0378-1119(93)90616-b. [DOI] [PubMed] [Google Scholar]
- 47.Benoit SL, Zbell AL, Maier RJ. Microbiology. 2007;153:3748. doi: 10.1099/mic.0.2007/010520-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benoit SL, Mehta N, Weinberg MV, Maier C, Maier RJ. Microbiology. 2007;153:1474. doi: 10.1099/mic.0.2006/003228-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hayes ET, Wilks JC, Sanfilippo P, Yohannes E, Tate DP, Jones BD, Radmacher MD, BonDurant SS, Slonczewski JL. BMC Microbiol. 2006;6:89. doi: 10.1186/1471-2180-6-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jacobi A, Rossmann R, Bock A. Arch Microbiol. 1992;158:444. doi: 10.1007/BF00276307. [DOI] [PubMed] [Google Scholar]
- 51.Lutz S, Jacobi A, Schlensog V, Bohm R, Sawers G, Bock A. Mol Microbiol. 1991;5:123. doi: 10.1111/j.1365-2958.1991.tb01833.x. [DOI] [PubMed] [Google Scholar]
- 52.Maier T, Lottspeich F, Bock A. Eur J Biochem. 1995;230:133. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.